

Abstract
Alzheimer's disease (AD) drug development is limited by the presence of the blood-brain barrier (BBB). More than 98% of all small molecule drugs, and ∼100% of all large molecule drugs, do not cross the BBB. Despite the fact that the vast majority of AD drug candidates do not cross the BBB, the present-day AD drug development effort is characterized by an imbalance, whereby >99% of the drug development effort is devoted to CNS drug discovery, and <1% of drug development is devoted to CNS drug delivery. Future AD drug development needs a concerted effort to incorporate the BBB sciences early in the CNS drug discovery process. This can be accomplished by a reallocation of resources, and an expansion of the effort in the pure science of BBB biology and the applied science of brain drug targeting technology.
Keywords: blood-brain barrier, brain drug targeting, endogenous transporters, biopharmaceuticals
1. Introduction
The drug development mission for Alzheimer's disease (AD), or for any other brain disorder, suffers from an imbalance in CNS drug discovery and CNS drug delivery. Owing to the presence of the blood-brain barrier (BBB), an effective CNS drug development program cannot endure without equal efforts in discovery and delivery. The BBB problem is illustrated in Figure 1, which lists the incongruities in the CNS drug development process [1]:
Figure 1.
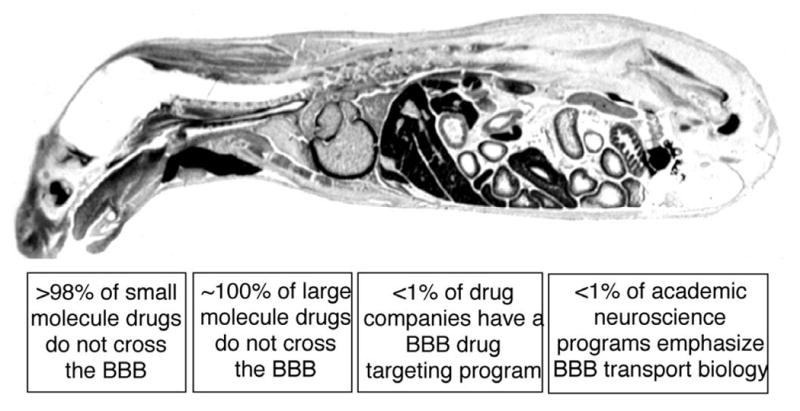
Whole body autoradiogram of a mouse sacrificed after the intravenous injection of a small molecule, histamine, which has a molecular weight of 111 Da. The histamine readily distributes to the extra-vascular space of all organs of the body, except for the brain and spinal cord. Histamine does not penetrate the CNS, owing to the presence of the BBB.
>98% of small molecule drugs do not cross the BBB
∼100% of all large molecule drugs, i.e., the products of biotechnology, do not cross the BBB
No Big Pharma in the world today has a BBB drug targeting program
Even if Big Pharma wanted to start a BBB drug targeting program, there would be few personnel trained in the BBB to hire, because no academic neuroscience program in the U.S. emphasizes BBB transport biology, much less BBB drug targeting.
The decades of chronic under-development of BBB drug targeting technology are a major cause of clinical trial drug failures in brain disorders. Despite the massive effort in CNS drug discovery in both academia and industry, there has been no parallel effort in CNS drug delivery, which is peculiar given the existence of the BBB. Owing to the under-development of BBB delivery research, and to the fact that so few drugs cross the BBB, there are predictable failures in CNS drug development. Clinical trials have been performed on drugs that do not cross the BBB; the trial fails, which is attributed to the drug, and the issue of brain penetration is not considered. In other trials, the BBB is considered before the clinical trial is initiated. However, since no BBB drug targeting technology has been co-developed, the only recourse is to administer the drug via a trans-cranial drug delivery system. As discussed below, trans-cranial drug delivery is not effective. The clinical trial fails, and the company not infrequently terminates its involvement in CNS drug development. The challenges of the CNS drug development process is a natural outcome of 2 opposing forces: (a) the vast majority of CNS drug candidates do not cross the BBB, and (b) there is little, if any, BBB drug targeting technology being developed by either academia or industry.
2. Invasive Brain Drug Delivery
Trans-cranial brain drug delivery is the only option for the CNS drug developer that carries out a drug discovery program in the absence of a parallel BBB drug delivery effort. It is difficult to envision the treatment of millions of people suffering from AD with a delivery system that requires a neurosurgical intervention. Nevertheless, the point of emphasis is that trans-cranial drug delivery to the brain is not effective, owing to the limitations of diffusion within the 1.2 kg human brain. Intra-cerebroventricular drug administration is not effective, as the drug only distributes to the ependymal surface of the brain [2,3]. Diffusion decreases with the square of the diffusion distance. In contrast, cerebrospinal fluid (CSF) moves rapidly through the CSF flow tracts within the brain. The 140 mL of CSF in the human brain turns over 4-5 times per day and is drained into the systemic circulation at the superior sagittal sinus [3]. Drug injection into the CSF is similar to a slow intravenous injection, as noted by Fishman and Christy in 1965 [4]. The intra-cerebral injection of drug or polymeric implant is not effective as the concentration of drug within the brain decreases exponentially from the injection site, and is only a fraction of the original concentration at distances <1 mm from the depot site [5,6]. In an attempt to overcome the limitations of diffusion, convection-enhanced diffusion (CED) has been tried. In this approach, a reservoir is implanted in the abdomen, and a catheter is inserted into the brain parenchyma. Fluid is infused into the brain via the reservoir pump. However, once the fluid exits the catheter within the brain, the bulk flow is hindered by the resistance of brain tissue. The delivery to the human brain of subjects with Parkinson's disease (PD) was attempted for a neurotrophin, glial-derived neurotrophic factor (GDNF). The clinical trial was aborted owing to lack of efficacy [7], and subsequent primate studies demonstrated poor penetration of GDNF into brain parenchyma with CED [8].
3. Small Molecules and AD Drug Development
It is assumed that BBB drug targeting technology is not needed when the aim of drug development is the discovery of small molecules. It is believed that small molecules cross the BBB via lipid-mediated free diffusion. In fact, only a small fraction of small molecules cross the BBB via lipid-mediation, and these molecules have the following molecular properties:
Molecular weight (MW) <400 Daltons (Da)
Hydrogen bonding (N) <8-10
If the MW of the drug is >400 Da and/or the drug forms more than 8 hydrogen bonds, than the likelihood of the drug crossing the BBB via lipid-mediation in pharmacologically significant amounts is low [3].
Molecular weight
The MW threshold of BBB transport is a more prominent issue in modern day CNS drug discovery, which is driven by high throughput screening (HTS) technology. HTS approaches increasingly select for drugs with higher MW [9]. The MW threshold is a property of all biological membranes [10]. A screen of CNS acting drugs showed that all brain drugs have a MW < 426 Da [11]. The BBB permeability decreases 100-fold when the surface area of the drug is increased from 50 angstroms (A)2 to 100 A2 [11], which would be the case when the MW of a drug is increased from 300 Da to 450 Da. The dependence of solute diffusion through biological membranes on MW has been known for many years. Lieb and Stein [12] noted the diffusion of solutes through biological membranes is different from diffusion through water, and that a MW threshold existed for diffusion through membranes. Trauble [13] has proposed a molecular model to explain the effect of molecular size on membrane permeation of solutes. Inquiry into the MW is the first step in the evaluation of the potential for brain penetration of a lead CNS candidate.
Hydrogen bonding
The lipid solubility of a drug is inversely related to the number of hydrogen bonds the drug forms with solvent water, which is 55 M. The hydrogen bonding of any given drug can be determined by inspection of the molecular structure, and should be considered in tandem with the MW of the drug. The role of hydrogen bonding is illustrated in the review by Rishton and co-workers [14], who report the following data on hydrogen bonding of AD drugs:
Current acetylcholinesterase (ACE) inhibitors form 3-5 hydrogen bonds, which predicts effective BBB transport
Widely used CNS drugs form 1-5 hydrogen bonds, which predicts effective BBB transport
γ-secretase inhibitors for AD have higher degrees of hydrogen bonding and half of the drugs in this class form 8-10 hydrogen bonds, which predicts restricted BBB transport
β-secretase inhibitors for AD are the most polar and all members of this class form 8-12 hydrogen bonds, which predicts restricted BBB transport
A CNS drug discovery effort for the identification of lead compounds for inhibitors of either γ-secretase or β-secretase for AD will most likely end in termination. Although effective inhibitors will be identified, the polarity of these compounds will preclude effective brain penetration in vivo. Termination ensues because the CNS drug discovery process was allowed to mature without a parallel effort in CNS drug delivery.
The difficulties in identification of small molecule drugs for AD that also have effective BBB permeation are not unique to AD drug development. In a survey of >7,000 drugs in the Comprehensive Medicinal Chemistry (CMC) database, only 5% of all drugs were effective in the CNS, and these drugs only treated a restricted number of brain disorders: depression, schizophrenia, and insomnia [15]. In another survey of small molecule drugs, only 12% of all drugs were active in the brain; however, if affective disorders were excluded, only 1% of all drugs were active in the CNS [16].
Small molecules, medicinal chemistry, and BBB transporters
The current approach to small molecules and brain penetration is to use medicinal chemistry to increase the lipid solubility of promising CNS drug leads, which are too polar to penetrate the brain. However, despite the very large world-wide medicinal chemistry effort supported by the pharmaceutical industry, there is not a single example of a current FDA-approved polar drug that was made brain-penetrating by medicinal chemistry. What is needed is a re-direction of medicinal chemistry away from attempts to increase the lipid-mediated transport of the compound and toward the use of medicinal chemistry to increase the carrier-mediated transport (CMT) of the drug.
Dopamine is therapeutic in PD, but does not cross the BBB. The α-carboxylation of dopamine yields dihydroxyphenylalanine (DOPA), which does cross the BBB via carrier-mediated transport on the large neutral amino acid transporter type 1, which is the protein product of the LAT1 gene [17]. Similar to LAT1, there are many other endogenous BBB CMT systems that are potential portals of entry into the brain for drugs that have been modified into a molecular structure that is recognized by the endogenous BBB transporter [1]. For such an effort to take hold within the pharmaceutical industry, the following would have to take place:
Expand the knowledge base of the multitude of endogenous BBB transporters
Discover new endogenous BBB transporters
Develop structure-transport relationships (STR) to the same extent as structure-activity relationships (SAR) are developed for drug receptors
Build transporter-based HTS systems following the cloning of the transporter genes, and the engineering of transfected cell lines over-expressing the BBB transporter
4. Amyloid Antibody Drug Development in AD
Abeta and blood-brain barrier transport
The amyloid plaque of AD is formed by a 42-43 amino acid peptide, Aβ1-42/43. Carboxyl truncated forms include Aβ1-40. It is generally assumed that Aβ peptides are transported bi-directionally between blood and brain. If significant Abeta influx from blood to brain did occur, then Abeta peptide radiopharmaceuticals would have been developed for the in vivo imaging of brain amyloid, but such efforts were not successful. The apparent influx of Aβ from blood to brain is due to non-specific absorption to the brain vasculature, without measureable transport of Abeta from blood to brain [18]. With respect to Abeta efflux from brain, this may take place for Abeta monomers, but Aβ dimers do not efflux from brain [19]. The efflux of Abeta from blood to brain across the BBB underlies the sink hypothesis of immune therapy of AD. However, anti-Aβ antibodies in the plasma cannot disaggregate plaque in brain behind the BBB, unless the antibody is physically transported into brain.
Active immunization
The active immunization of AD transgenic mice with Aβ peptides mixed with adjuvant leads to the formation of anti-Abeta amyloid antibodies (AAA) in blood and to the dissolution of amyloid plaque in brain [20]. These results were in accord with other findings that the physical association of AAAs with amyloid plaque in vitro causes the disaggregation of Abeta amyloid [21]. The intra-cerebral injection of AAAs bypassed the BBB, allowed the AAA to physically interact with amyloid plaque, and this was associated with plaque disaggregation in vivo, and with the repair of dystrophic neurites [22]. If the disaggregation of amyloid plaque in AD by AAAs requires the physical association of the AAA and the plaque, then how could active immunization work to disaggregate the plaque? How could the AAAs in blood reach the plaque behind the BBB? It is likely that the BBB plays a role in both the efficacy and the toxicity of active immunization.
The immune adjuvant used in the active immunization of AD mice was Freund's adjuvant [20], which contains the mannan polysaccharide. Antibodies against mannan are formed in parallel with the formation of the AAAs in active immunization. Any mechanism of action of active immunization in AD models should consider the following facts about Freund's adjuvant and anti-mannan antibodies:
The administration of Freund's adjuvant to mice results in BBB disruption and leakage into brain of circulating IgG molecules [23].
The intravenous administration of anti-mannan antibodies results in the immediate disruption of the BBB [24].
In active immunization, it is likely the adjuvant causes BBB disruption, which allows the circulating AAAs to enter brain and access the amyloid plaque. However, BBB disruption leads to the brain uptake of plasma proteins, and plasma albumin is toxic to astrocytes [25]. A single BBB disruption leads to chronic neuropathologic changes in the brain [26]. If the mechanism of action of active immunization requires BBB disruption, then it would follow that active immunization would be associated with toxicity. Active immunization was ultimately terminated following the findings of an encephalitis-like clinical syndrome [27], which may have been caused by adjuvant-mediated BBB disruption.
Passive immunization
The BBB may also play a role in both the efficacy and the toxicity of passive immunization of AD. The systemic administration of large doses, e.g. >10 mg/kg, of AAAs to non-immunized AD mice results in the disaggregation of Abeta amyloid plaque in brain behind the BBB [28,29]. What is the mechanism of this disaggregation assuming it is necessary for there to be physical contact between the AAA in blood and the amyloid plaque behind the BBB? The following facts are known about passive immunization:
A consistent side effect of passive immunization is BBB disruption and cerebral micro-hemorrhage [30,31].
The systemic administration of a therapeutic dose of an AAA results in an enourmous increase in the plasma concentration of the Abeta peptide from low nM concentrations to nearly 1μM [32].
The markedelevation of the plasma Aβ peptide concentration results in BBB disruption, non-specific IgG uptake by brain from blood, and cerebral micro-hemorrhage [33]. This finding replicated prior work showing that the intra-carotid arterial administration of the Aβ peptide results in marked BBB disruption to plasma protein [34].
If the mechanism of action of passive immunization requires BBB disruption to enable MAb entry into the brain, then the passive immunization of patients with AD may be associated with MRI changes consistent with brain edema caused by BBB disruption.
AD, AAAs, and BBB transporters
An alternative approach to the immune therapy of AD is the re-engineering of AAAs to enable receptor-mediated transport (RMT) of the AAA across the BBB in both the blood-to-brain and the brain-to-blood directions [35]. In order for an AAA to cause a net decrease in the Abeta amyloid burden of AD brain, it is necessary for the AAA to enter brain from blood, bind/disaggregate plaque, and then the AAA:Abeta peptide complex must exit brain and return to blood. Moreover, it is important that the AAA be rapidly cleared by peripheral tissues so that a high plasma concentration of AAA:Abeta complex is not generated. As discussed above, the high plasma concentration of the AAA:Abeta complex may cause the cerebral micro-hemorrhage that is typical of passive immunization. It is possible to re-engineer an AAA so that the antibody exhibits these properties, and this is accomplished with BBB drug targeting technology [36].
Similar to the CMT systems for small molecules, the BBB also expresses endogenous RMT systems that transport some large molecules in blood such as insulin or transferrin (Tf) [3]. Similarly, the BBB insulin receptor or Tf receptor transports certain peptidomimetic monoclonal antibodies (MAb), which are directed against exofacial epitopes on the BBB receptor. Such MAb's cross the BBB via RMT, and may act as molecular Trojan horses to ferry across the BBB an attached drug, including AAAs. The most potent molecular Trojan horse known to date is a MAb against the human insulin receptor (HIR). The HIRMAb has been genetically engineered and both chimeric and humanized HIRMAb's rapidly cross the BBB in vivo in Old World primates such as the Rhesus monkey [37]. HIRMAb fusion proteins have been genetically engineered, whereby the neurotherapeutic protein, which does not cross the BBB, is fused to the carboxyl terminus of the heavy chain of the HIRMAb, and IgG fusion proteins have been engineered for neurotrophins [38,39], enzymes [40,41], and single chain Fv (ScFv) antibodies [36].
The genes encoding the variable region of the heavy chain (VH) and the variable region of the light chain (VL) of an AAA were cloned for expression of an anti-Abeta ScFv [36]. The ScFv gene was then fused to the gene encoding the heavy chain (HC) of the HIRMAb to produce the HIRMAb-ScFv fusion protein. In this formulation, an anti-Abeta ScFv was fused to the carboxyl terminus of each HC of the HIRMAb. The HIRMAb-ScFv fusion protein bound with high affinity both to the HIR, to mediate influx from blood to brain across the BBB, and to the Abeta peptide, to mediate plaque disaggregation. In addition, the CH2-CH3 interface of the HIRMAb-ScFv fusion protein bound the BBB Fc receptor (FcR), which mediates the selective, asymmetric reverse transcytosis of IgG molecules from brain back to blood across the BBB. The BBB FcR does not mediate the transport of IgG molecules in the blood-to-brain direction. In vivo experiments demonstrated the HIRMAb-ScFv fusion protein retained the 3 required functionalities [36]:
Influx from blood to brain across the BBB via transport on the HIR
Efflux from brain to blood across the BBB via transport on the FcR
Abeta plaque disaggregation
The re-engineering of an AAA for brain penetration would not be possible without the development of a BBB drug targeting technology for biopharmaceuticals such as MAb-therapeutics.
5. Neurotrophin Drug Development in AD
Future drug therapy of AD with biopharmaceuticals may follow a 2-step process where a course of AAA therapy, to disaggregate amyloid plaque, is followed by a course of neurotrophin drug therapy, to accelerate the repair of dystrophic neurites. One neurotrophin that may be particularly suitable for AD is brain-derived neurotrophic factor (BDNF), although multiple neurotrophins are neuroprotective in chronic neurodegeneration. The intra-cerebral administration of a retrovirus expressing BDNF is therapeutic in AD transgenic mice [42].
BDNF does not cross the BBB [43], which is why the clinical trials of amyotrophic lateral sclerosis (ALS) with subcutaneous BDNF failed [44]. Attempts have been made to identify small molecule BDNF-mimetics, but such compounds invariably have a MW>400 Da, and/or have high hydrogen bonding [45]. Such small molecules do not penetrate the BBB in pharmacologically significant amounts, and would still require re-engineering with a small molecule BBB drug targeting technology.
Neurotrophins, AD, and BBB transporters
Neurotrophins, such as BDNF or GDNF, have been re-engineered as a HIRMAb-neurotrophin fusion protein [38,39]. The fusion protein retains high affinity binding to both the HIR, for the mediation of the transport into the brain via the BBB HIR, and to the neurotrophin receptor, for mediation of neural repair in brain, once the fusion protein crosses the BBB. The re-engineering of neurotrophins for brain penetration would not be possible without the development of a BBB drug targeting technology for biopharmaceuticals.
6. Conclusions
The elderly population in the U.S. will increase by 50% by 2020, and the cost of caring for the large number of patients with AD could approach $0.5 trillion per year. Thus, there is great urgency for a national program in AD drug development to produce a cure, or at least a drug that slows the progression of amyloid deposition in the brain of patients with AD. With regard to the BBB and future AD drug development, the neuroscience body politic can continue the status quo, and the role of the BBB in AD drug development can continue to be under-developed. The future would continue present trends whereby >99% of the AD drug development enterprise would be devoted to CNS drug discovery, and <1% of the effort would be focused on CNS drug delivery. However, what the future needs is a more balanced AD drug development program, where a concerted effort is made to expand the pure science of BBB biology and the applied science of BBB drug targeting technology. Advances in the BBB field are difficult, and future success will require a substantial re-allocation of neuroscience research priorities and resources away from drug discovery and toward drug delivery. Such a reallocation is needed. Future advances in the development of AD drugs that actually work in clinical trials will be directly proportional to the future progress in the BBB sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blasberg RG, Patlak C, Fenstermacher JD. Intrathecal chemotherapy: brain tissue profiles after ventriculocisternal perfusion. J Pharmacol Exp Ther. 1975;195:73–83. [PubMed] [Google Scholar]
- 3.Pardridge WM. Brain Drug Targeting: the Future of Brain Drug Development. Cambridge University Press; Cambridge, United Kingdom: 2001. pp. 1–370. [Google Scholar]
- 4.Fishman RA, Christy NP. Fate of adrenal cortical steroids following intrathecal injections. Neurology. 1965;15:1–6. doi: 10.1212/wnl.15.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Dykstra KH, Arya A, Arriola DM, Bungay PM, Morrison PF, Dedrick RL. Microdialysis study of zidovudine (AZT) transport in rat brain. J Pharmacol Exp Ther. 1993;267:1227–36. [PubMed] [Google Scholar]
- 6.Fung LK, Shin M, Tyler B, Brem H, Saltzman WM. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res. 1996;13:671–82. doi: 10.1023/a:1016083113123. [DOI] [PubMed] [Google Scholar]
- 7.Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459–66. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 8.Salvatore MF, Ai Y, Fischer B, Zhang AM, Grondin RC, Zhang Z, et al. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp Neurol. 2006;202:497–505. doi: 10.1016/j.expneurol.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 10.Cohen BE, Bangham AD. Diffusion of small non-electrolytes across liposome membranes. Nature. 1972;236:173–4. doi: 10.1038/236173a0. [DOI] [PubMed] [Google Scholar]
- 11.Fischer H, Gottschlich R, Seelig A. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J Membr Biol. 1998;165:201–11. doi: 10.1007/s002329900434. [DOI] [PubMed] [Google Scholar]
- 12.Lieb WR, Stein WD. Non-Stokesian nature of transverse diffusion within human red cell membranes. J Membr Biol. 1986;92:111–9. doi: 10.1007/BF01870701. [DOI] [PubMed] [Google Scholar]
- 13.Träuble H. The movement of molecules across lipid membranes: a molecular theory. J Membr Biol. 1971;4:193–208. doi: 10.1007/BF02431971. [DOI] [PubMed] [Google Scholar]
- 14.Rishton GM, LaBonte K, Williams AJ, Kassam K, Kolovanov E. Computational approaches to the prediction of blood-brain barrier permeability: A comparative analysis of central nervous system drugs versus secretase inhibitors for Alzheimer's disease. Curr Opin Drug Discov Devel. 2006;9:303–13. [PubMed] [Google Scholar]
- 15.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 16.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 17.Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci U S A. 1999;96:12079–84. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito Y, Buciak J, Yang J, Pardridge WM. Vector-mediated delivery of [125I]- labeled β-amyloid peptide Aβ1-40 through the blood-brain barrier and binding to Alzheimer's disease amyloid of the Aβ1-40/vector complex. Proc Natl Acad Sci USA. 1995;92:10227–10231. doi: 10.1073/pnas.92.22.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T. Cerebral clearance of human amyloid –β peptide (1-40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J Neurochem. 2007;103:2482–2490. doi: 10.1111/j.1471-4159.2007.04938.x. [DOI] [PubMed] [Google Scholar]
- 20.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 21.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94:4109–12. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, et al. Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–83. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund's adjuvant in mice provoke leakage of serum proteins through the blood-brain barrier without inducing reactive gliosis. Brain Res. 1999;832:84–96. doi: 10.1016/s0006-8993(99)01479-1. [DOI] [PubMed] [Google Scholar]
- 24.Namer IJ, Steibel J. Antibody directed against mannan of the Mycobacterium tuberculosis cell envelope provokes blood-brain barrier breakdown. J Neuroimmunol. 2000;103:63–8. doi: 10.1016/s0165-5728(99)00236-2. [DOI] [PubMed] [Google Scholar]
- 25.Nadal A, Fuentes E, Pastor J, McNaughton PA. Plasma albumin is a potent trigger of calcium signals and DNA synthesis in astrocytes. Proc Natl Acad Sci U S A. 1995;92:1426–30. doi: 10.1073/pnas.92.5.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salahuddin TS, Johansson BB, Kalimo H, Olsson Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. An electron microscopic study. Acta Neuropathol. 1988;77:5–13. doi: 10.1007/BF00688236. [DOI] [PubMed] [Google Scholar]
- 27.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 28.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 29.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 31.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su GC, Arendash GW, Kalaria RN, Bjugstad KB, Mullan M. Intravascular infusions of soluble beta-amyloid compromise the blood-brain barrier, activate CNS glial cells and induce peripheral hemorrhage. Brain Res. 1999;818:105–17. doi: 10.1016/s0006-8993(98)01143-3. [DOI] [PubMed] [Google Scholar]
- 34.Jancso G, Domoki F, Santha P, Varga J, Fischer J, Orosz K, et al. Beta-amyloid (1-42) peptide impairs blood-brain barrier function after intracarotid infusion in rats. Neurosci Lett. 1998;253:139–41. doi: 10.1016/s0304-3940(98)00622-3. [DOI] [PubMed] [Google Scholar]
- 35.Pardridge WM. Re-engineering biopharmaceuticals for delivery to brain with molecular Trojan horses. Bioconjug Chem. 2008;19:1327–38. doi: 10.1021/bc800148t. [DOI] [PubMed] [Google Scholar]
- 36.Boado RJ, Zhang Y, Xia CF, Pardridge WM. Fusion antibody for Alzheimer's disease with bidirectional transport across the blood-brain barrier and abeta fibril disaggregation. Bioconjug Chem. 2007;18:447–55. doi: 10.1021/bc060349x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boado RJ, Zhang Y, Pardridge WM. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol Bioeng. 2007;96:381–91. doi: 10.1002/bit.21120. [DOI] [PubMed] [Google Scholar]
- 38.Boado RJ, Zhang Y, Pardridge WM. Genetic engineering, expression, and activity of a fusion protein of a human neurotrophin and a molecular Trojan horse for delivery across the human blood-brain barrier. Biotechnol Bioeng. 2007;97:1376–86. doi: 10.1002/bit.21369. [DOI] [PubMed] [Google Scholar]
- 39.Boado RJ, Zhang Y, Wang Y, Pardridge WM. GDNF fusion protein for targeted-drug delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;100:387–96. doi: 10.1002/bit.21764. [DOI] [PubMed] [Google Scholar]
- 40.Boado RJ, Zhang Y, Xia CF, Wang Y, Pardridge WM. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;99:475–84. doi: 10.1002/bit.21602. [DOI] [PubMed] [Google Scholar]
- 41.Boado RJ, Zhang Y, Wang Y, Pardridge WM. IgG-paraoxonase-1 fusion protein for targeted drug delivery across the human blood-brain barrier. Mol Pharm. 2008;5:1037–43. doi: 10.1021/mp800113g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–7. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakane T, Pardridge WM. Carboxyl-directed pegylation of brain-derived neurotrophic factor markedly reduces systemic clearance with minimal loss of biologic activity. Pharm Res. 1997;14:1085–91. doi: 10.1023/a:1012117815460. [DOI] [PubMed] [Google Scholar]
- 44.The BDNF Study Group. A controlled trial of recombinant methionyl human BDNF in ALS: The BDNF Study Group (Phase III) Neurology. 1999;52:1427–33. doi: 10.1212/wnl.52.7.1427. [DOI] [PubMed] [Google Scholar]
- 45.Wilkie N, Wingrove PB, Bilsland JG, Young L, Harper SJ, Hefti F, et al. The non-peptidyl fungal metabolite L-783,281 activates TRK neurotrophin receptors. J Neurochem. 2001;78:1135–45. doi: 10.1046/j.1471-4159.2001.00504.x. [DOI] [PubMed] [Google Scholar]