

Abstract
Yersinia pestis, the agent of plague, uses a type III secretion injectisome to deliver Yop proteins into macrophages to counteract phagocytosis and induce apoptosis. Additionally, internalized Y. pestis can survive in the phagosomes of naïve or gamma interferon (IFN-γ)-activated macrophages by blocking vacuole acidification. The Y. pestis LcrV protein is a target of protective antibodies. The binding of antibodies to LcrV at the injectisome tip results in neutralization of the apoptosis of Y. pestis-infected macrophages and is used as an in vitro correlate of protective immunity. The cytokines IFN-γ and tumor necrosis factor alpha can cooperate with anti-LcrV to promote protection against lethal Y. pestis infection in mice. It is not known if these phagocyte-activating cytokines cooperate with anti-LcrV to increase the killing of the pathogen and decrease apoptosis in macrophages. We investigated how anti-LcrV and IFN-γ impact bacterial survival and apoptosis in cultured murine macrophages infected with Y. pestis KIM5. Y. pestis KIM5 opsonized with polyclonal or monoclonal anti-LcrV was used to infect macrophages treated with or without IFN-γ. The phagocytosis and survival of KIM5 and the apoptosis of macrophages were measured at different time points postinfection. The results show that anti-LcrV reduced apoptosis at an early time point (5 h) but not at a later time point (24 h). Polyclonal anti-LcrV was unable to inhibit apoptosis at either time point in IFN-γ-activated macrophages. Additionally, anti-LcrV was ineffective at promoting the killing of KIM5 in naïve or activated macrophages. We conclude that Y. pestis can bypass protective antibodies to LcrV and activation with IFN-γ to survive and induce apoptosis in murine macrophages.
There are three human pathogenic Yersinia species: Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica (27). All three species contain an approximately 70-kb virulence plasmid (pCD1 in Y. pestis and pYV in Y. pseudotuberculosis and Y. enterocolitica) that encodes a type III secretion system (T3SS) and effector proteins termed Yersinia outer proteins or Yops. Y. pestis is the causative agent of pneumonic and bubonic plague and the latter two cause gastroenteritis. Y. pestis is thought to be closely related to Y. pseudotuberculosis (1). In addition to carrying pCD1, Y. pestis harbors two additional plasmids, pMT1 and pPCP1, that give it increased virulence compared to Y. pseudotuberculosis (27). Historically, Y. pestis has had a major impact on society, killing large numbers of people worldwide. Today, with the development of antibiotics and increased sanitary conditions, bubonic and pneumonic plague are no longer major public health concerns. However, there are still rodent populations infected with plague, and small numbers of humans within the population are infected annually (27). It is important to further study Y. pestis to create a safe and effective vaccine, both because there is still a natural reservoir and because there is the potential danger that pneumonic plague may be used for acts of bioterrorism.
The pCD1 plasmid encodes a T3SS composed of the secretion apparatus, chaperones, Yops (9), and the translocator proteins (YopB, YopD, and LcrV). Six effector Yops have been identified: YopH, YopO/YpkA, YopP/YopJ, YopE, YopM, and YopT. YopJ (YopP in Y. enterocolitica) inhibits mitogen-activated protein kinase signaling cascades as well as the activation of NF-κB, a transcription factor important for the expression of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), by preventing the phosphorylation and subsequent degradation of the NF-κB inhibitor (IκB) (25). As a result, YopJ inhibits the expression of TNF-α, diminishing the inflammatory response. YopJ has also been shown to induce apoptosis, due to the loss of mitogen-activated protein kinase and NF-κB activity (25).
The phagocytosis of an opsonized pathogen by a macrophage is mediated by surface receptors, such as the Fc receptor. During the process of phagocytosis several signaling cascades are activated. Rho GTPase family members (Rac-1, RhoA, and Cdc42) that are localized in the plasma membrane become activated through the binding of GTP. These GTPases regulate the polymerization of actin to drive phagocytosis (2). Several of the Yops have antiphagocytic effects, through the direct inhibition of Rho GTPases, including YopO (also known as Yersinia protein kinase A), YopT, and YopE (25, 30). YopH has been shown to inhibit phagocytosis and the expression of monocyte chemoattractant protein 1, a chemokine involved in macrophage recruitment, and diminish the Fc-mediated oxidative burst in neutrophils and macrophages (6, 25).
The expression of the T3SS and the regulation of Yop translocation are dependent on temperature, calcium levels, and host cell contact. At 28°C, the expression of the T3SS is downregulated. At 37°C, the T3SS is maximally induced (9), and a needle-like surface structure, the Ysc injectisome, is formed. Upon contact with a host cell, the T3SS is systematically activated. The translocators YopB and YopD are believed to form a channel in the host cell membrane, allowing the delivery of the effector Yops. The effector Yops are translocated into the host cell cytoplasm, where they disrupt host cell signaling (9).
In addition to YopB and YopD, the LcrV protein is necessary to deliver the effector Yops into the host cell (28). The mechanism by which LcrV mediates translocation is not fully understood, but it appears to be important for the correct assembly of the translocation channel (23). LcrV has been shown to localize to the tip of the injectisome (23). LcrV, also known as V antigen, has many other important roles. It has a regulatory role in Yop secretion within the bacterium (27). LcrV is also a soluble protein and is an important protective antigen (24, 42).
Y. pestis is efficiently phagocytosed and survives within the phagosomes of naïve murine macrophages when the bacteria are grown at 28°C prior to in vitro infection (13, 14, 34, 41). Y. pestis can block phagosome acidification, which may be important for survival in macrophages (34). The growth of Y. pestis at 37°C prior to infection promotes Yop delivery during phagocytosis, and as a result, the efficiency of bacterial uptake by macrophages is reduced. However, ∼20 to 35% of 37°C-grown Y. pestis bacteria that associate with macrophages are internalized (10, 43). Yop-expressing Y. pestis that are internalized by naïve macrophages are able to survive intracellularly (21). In addition, macrophages infected with 37°C-grown Y. pestis die of YopJ-induced apoptosis (12, 21, 43). Thus, Yop-expressing Y. pestis can counteract the antibacterial functions of naïve macrophages by intracellular survival and the induction of apoptosis if they are unable to avoid phagocytosis. Lukaszewski et al. showed that naïve mice infected with Y. pestis could harbor Y. pestis within CD11b+ spleen macrophages for several days postinfection (p.i.) and that a significant percentage of these phagocytes died of apoptosis during this time period (22).
Mice can be protected against lethal Y. pestis infection by passive immunization with anti-LcrV antibodies (15-17, 19, 38, 39, 42, 44). Opsonization with anti-LcrV antibodies increases the phagocytosis of Y. pestis by macrophages (10, 29, 43). The increased phagocytosis of Y. pestis mediated by anti-LcrV antibody opsonization is associated with reduced Yop translocation (10, 29) and reduced apoptosis (10, 29, 43). The ability of anti-LcrV antibodies to inhibit apoptosis in macrophages infected with Y. pestis is commonly used as a measure of neutralizing activity (3, 43-45).
In addition to antibodies, the cytokines gamma interferon (IFN-γ) and TNF-α are important for protective immune responses against Y. pestis infection (11, 19, 24, 26, 38, 39). Y. pestis can survive in macrophages activated with IFN-γ (22, 32-34), but a significantly reduced intracellular persistence of the bacteria is observed when macrophages are exposed to both IFN-γ and TNF-α (22).
Macrophages are important for anti-LcrV-mediated protection against Y. pestis in the livers of infected mice (10, 29). In addition to protecting macrophages from apoptosis, it is possible that opsonization with anti-LcrV decreases the survival of Y. pestis in phagocytes, due to the ability of Fc receptors to activate bactericidal processes. The activation of macrophages with IFN-γ is known to upregulate Fc receptor expression and function, which could further increase intracellular killing, following the uptake of anti-V-opsonized Y. pestis (19, 38, 39). It is also possible that activation per se could decrease the sensitivity of Y. pestis-infected macrophages to undergo apoptosis.
In this study, we investigated how opsonization with anti-LcrV antibodies, and activation with IFN-γ, impacts the ability of Y. pestis to survive within and kill macrophages. The findings have implications for understanding how anti-LcrV antibodies and cytokines function to protect against plague.
MATERIALS AND METHODS
Bacterial strains.
The strains used are shown in Table 1. Y. pestis pgm mutants KIM5 and KIM5/GFP (where GFP is green fluorescent protein) contain pCD1Ap. KIM5/GFP also contains an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible plasmid encoding GFP (pMMB207gfp3.1). The KIM5 yopB mutant contains an in-frame deletion of yopB in pCD1Ap as described by Lilo et al. (21). The KIM5 phoP mutant contains pCD1Ap and an in-frame deletion of phoP as described by Grabenstein et al. (13). Y. pseudotuberculosis serogroup I strain 32777, formerly known as IP2777, contains pYV.
TABLE 1.
Yersinia strains used in this study
| Strain | Characteristic(s)a | Reference |
|---|---|---|
| Y. pestis | ||
| KIM5 | pCD1Ap, pMT1+, pPCP1+, pgm mutant, Ampr | 21 |
| KIM5/GFP | KIM5/pMMB207gfp3.1, Ampr, Camr | 13 |
| KIM5 phoP | pCD1Ap phoP in-frame deletion | 13 |
| KIM5 yopB | pCD1Ap yopB in-frame deletion of nucleotides 496 to 774, Ampr | 21 |
| Y. pseudotuberculosis | ||
| 32777 | pYV+ | 37 |
Ampr, ampicillin resistance; Camr, chloramphenicol resistance.
Sera and antibodies.
Anti-LcrV serum and control serum were obtained from LcrV-immunized or adjuvant-only injected mice, respectively, as described by Ivanov et al. (18). The monoclonal antibody (MAb) specific for LcrV (MAb 7.3) has been described previously (17). A hybridoma producing anti-YopD MAb 248.19 was generated by the Hybridoma Facility at Stony Brook University. BALB/c mice were immunized with YopBDE antigen (17). Following the fusion of spleen cells from immunized mice, hybridoma clones producing anti-YopD MAb were identified by enzyme-linked immunosorbent assay (ELISA) and immunoblotting. One subclone producing anti-YopD MAb was designated 248.19, and the MAb was isotyped immunoglobulin G1 (IgG1). The MAb was purified by the ammonium sulfate precipitation of hybridoma supernatants. The precipitated protein was dissolved in phosphate-buffered saline (PBS) and dialyzed against PBS.
Primary macrophage cell culture.
Bone marrow-derived macrophages (BMDMs) were isolated from the femurs of C57BL/6 mice (Jackson Laboratory) and prepared as previously described (31). BMDMs were seeded in 24-well cell culture plates at 1.5 × 105 cells per well for 24 h prior to infection in Dulbecco's modified Eagle's medium (Gibco) with 10% fetal bovine serum (HyClone), 1 mM sodium pyruvate, 2 mM glutamate, and 15% L-cell-conditioned medium.
Infection conditions.
Bacterial cultures were grown in heart infusion broth (HI) containing ampicillin (Amp) (25 μg/ml) and chloramphenicol (30 μg/ml) to select for pCD1Ap or pMMB207gfp3.1, respectively, overnight with aeration at 28°C. Overnight cultures were diluted 1:40 in HI with Amp and 2.5 mM CaCl2 (and with chloramphenicol and 0.05 mM IPTG to induce the expression of GFP when necessary) and incubated with aeration for 2 h at 37°C. Ten microliters of KIM5 (1.5 × 106 CFU per 1 ml) suspended in PBS was incubated with 10 μl of LcrV or control serum or 10 μl PBS (KIM5 nonopsonized) and incubated at 37°C with 5% CO2 for 10 min. The volume was then increased to 1 ml with cell culture medium and added to the macrophages (multiplicity of infection of 10 bacteria per macrophage). The plates were centrifuged for 5 min at 95 × g to facilitate contact between the macrophages and the bacteria and incubated for 20 min at 37°C with 5% CO2.
Phagocytosis assay.
RAW 264.7 mouse macrophage-like cells (ATCC TIB-71) were grown in Dulbecco's modified Eagle's medium plus Glutamax (Gibco BRL) with 10% heat-inactivated fetal bovine serum (HyClone) and 1 mM sodium pyruvate at 37°C with 5% CO2. Macrophages were seeded on glass coverslips at 1.5 × 105 cells in 1 ml medium in 24-well cell culture plates and incubated overnight. Macrophages were infected as described above with opsonized or nonopsonized KIM5/GFP expressing GFP. The plates were centrifuged at 95 × g for 5 min to facilitate contact between the macrophages and the bacteria and incubated for 20 min at 37°C with 5% CO2. The wells were then washed with prewarmed PBS once and fixed with 2.5% paraformaldehyde (PFA) in PBS at room temperature for 30 min. After being washed with PBS, coverslips were blocked with 3% bovine serum albumin in PBS for 20 min. Extracellular bacteria were labeled with rabbit anti-Yersinia antiserum SB349 (5) and with goat anti-rabbit antibody conjugated to Alexa Fluor 594 (Molecular Probes). The coverslips were washed and mounted with ProLong Gold antifade reagent (Invitrogen) onto slides. The slides were examined by epifluorescence microscopy using a Ziess Axioplan2 microscope. Pictures of three fields per coverslip in at least three independent experiments were taken. The number of macrophage-associated intracellular bacteria (green only) and the number of macrophage-associated extracellular bacteria (red and green overlay) were counted, and the percent internalization was determined as follows: [no. of intracellular bacteria/(no. of intracellular bacteria + no. of extracellular bacteria)] × 100.
LDH release assay.
Lactate dehydrogenase (LDH) release from the supernatants collected from Y. pestis-infected BMDMs at 5 and 24 h p.i. was determined using the CytoTox-96 nonradioactive cytotoxicity assay (Promega). The supernatants were collected and centrifuged to remove cellular debris, and LDH levels were determined in triplicate. Total LDH release was determined from supernatants from freeze-thaw-lysed BMDMs. Spontaneous LDH release was determined from supernatants obtained from uninfected cells. The percentage of LDH release was calculated as follows: % LDH release = [(infected cell LDH release - spontaneous LDH release)/(total LDH release - spontaneous LDH release)] × 100.
CFU assay.
At 25 min, 1.5 h, and 5 h p.i., Y. pestis-infected BMDMs were washed with PBS and lysed with 500 μl of 0.1% Triton X-100 in PBS. The plate was then incubated at 37°C with 5% CO2 for 10 min. Wells were scraped and lysates collected in microcentrifuge tubes. A total of 500 μl PBS were used to wash the wells. Lysates and washes were combined and used for serial dilutions. Dilutions were plated on HI-plus-Amp plates and incubated at 28°C for 2 days. The plates were enumerated, and the log10 CFU per ml were determined from the results of three independent experiments.
TNF-α and IL-1β ELISA.
At 24 h p.i., the supernatants from wells containing infected BMDMs were collected, centrifuged for 10 min at 200 × g to remove cellular debris, and transferred to new tubes. The supernatants were diluted appropriately, and 50 μl of each diluted sample was analyzed. The concentration of TNF-α and interleukin-1β (IL-1β) in the supernatant was measured using the Quantikine mouse TNF-α and IL-1β immunoassay kits (R&D Systems).
Phagosome acidification.
J774A.1 murine macrophage-like cells (ATCC TIB-67) seeded in 24-well plates on glass coverslips at 1.5 × 105 cells/well were infected with KIM5 expressing GFP as described above. Determination of the colocalization of phagosomes containing GFP-positive Y. pestis with LysoTracker Red DND 99 was performed by fluorescence microscopy (34). As a positive control for the colocalization with LysoTracker Red DND 99, we used KIM5/GFP fixed in 2.5% PFA (34). One hour prior to fixation, the cell culture medium was removed from the wells, and cell culture medium containing 50 nM LysoTracker Red DND 99 was added. Coverslips were fixed with 2.5% PFA and mounted onto the slides. The percentage of KIM5/GFP that colocalized with LysoTracker Red was determined as described previously (34).
Statistical analysis.
Experiments were performed at least three times. The results were subjected to analysis of variance and the Tukey posttest using Prism (GraphPad). The results were considered significantly different if the probability (P) values were less than 0.05.
RESULTS AND DISCUSSION
Opsonization with anti-V serum increases the phagocytosis of Y. pestis by naïve but not activated macrophages.
In order to study how anti-LcrV antibodies and IFN-γ impact the ability of Y. pestis to survive within and kill macrophages, we utilized mouse anti-LcrV immune serum (anti-V), previously shown to be protective (18), and cultured mouse macrophages (RAW 264.7 macrophage-like cell line or primary BMDMs). To confirm that the anti-V used in this study contained antibodies that could increase the uptake of Y. pestis by macrophages, a phagocytosis assay was performed. A culture of Y. pestis KIM5/GFP (Table 1) was shifted from 28°C to 37°C in the presence of IPTG to induce the expression of the T3SS and GFP. Samples of the bacteria were preincubated with normal mouse control serum, anti-V or PBS (nonopsonized) for 10 min prior to infection. Naïve RAW 264.7 cells were then infected with the bacteria at a multiplicity of infection of 10 for 25 min and then fixed. Phagocytosis was measured by fluorescence-based microscopy. The percent internalization of nonopsonized KIM5/GFP was ∼30%, which was significantly lower than that under the control serum condition (∼45%), likely due to the absence of complement proteins in the nonopsonization condition (Fig. 1A). KIM5/GFP preincubated with anti-V were phagocytosed to a significantly higher level (∼65%) than bacteria preincubated with control serum (Fig. 1A), showing that this serum contained anti-LcrV antibodies that could increase the phagocytosis of Y. pestis by macrophages.
FIG. 1.
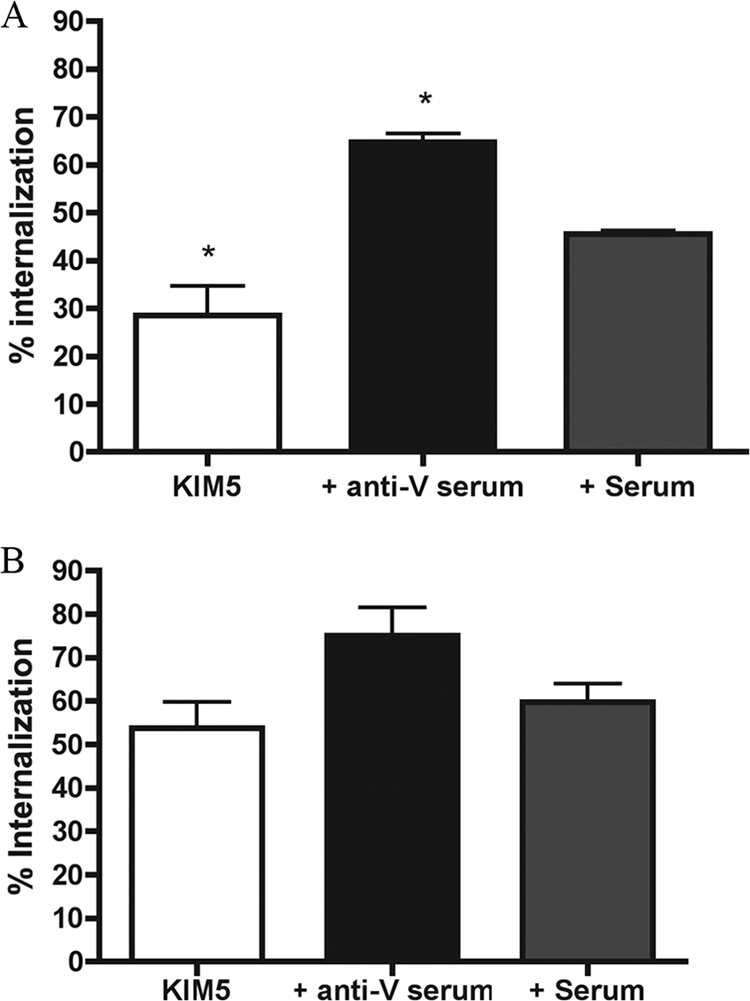
Phagocytosis assay with KIM5-infected naïve or IFN-γ-stimulated RAW 264.7 macrophages. KIM5/GFP was grown under T3SS-inducing conditions and incubated with PBS (KIM5), anti-V (+ anti-V serum), or control serum (+ Serum) prior to infecting naïve macrophages (A) or IFN-γ-stimulated macrophages (B) on coverslips. Twenty-five minutes p.i., the cells were fixed and extracellular bacteria were labeled for detection by immunofluorescence. Extracellular and intracellular bacteria from three fields per experiment were counted by fluorescence microscopy, and the percentage of internalization was determined. Results were taken from three independent experiments and averaged. Error bars show standard deviations. *, P < 0.05 compared to the control serum condition.
We next examined the interaction between IFN-γ-stimulated macrophages and KIM5/GFP opsonized with or without anti-V. The phagocytosis assay was performed as above except that RAW 264.7 cells were activated with IFN-γ (100 U/ml) for 24 h before infection. With IFN-γ-activated macrophages, there was an overall increase in bacterial uptake, but the increase was most dramatic for the nonopsonized and control serum-opsonized conditions (Fig. 1B). Approximately 55 to 60% of the nonopsonized or control-opsonized KIM5/GFP was internalized. Anti-V-opsonized KIM5/GFP was internalized at a slightly higher level (∼75%), but this increase was not significant compared to that of the control. These results showed that the activation state of the macrophage was important for the ability of anti-V to increase the phagocytosis of Y. pestis. The fact that an increased phagocytosis of Y. pestis was observed following exposure to IFN-γ indicated that the cytokine treatment was upregulating antibacterial functions in macrophages.
The opsonization of Y. pestis with anti-V serum decreases apoptosis in naïve but not in activated macrophages at an early stage of infection.
We next examined the effect of anti-V sera on YopJ-induced macrophage death using an LDH release assay. Naïve BMDMs were infected at a multiplicity of infection of 10 with KIM5 (Table 1) grown and opsonized as described above. BMDMs were also infected in parallel with the noncytotoxic KIM5 yopB mutant (Table 1) as a control. After phagocytosis was allowed to occur for 25 min, the survival of extracellular bacteria was prevented by the addition of a low concentration of gentamicin to the tissue culture medium. This infection protocol was used because it allows for the measurement of intracellular survival and apoptosis under the same infection conditions. KIM5, being a pgm mutant, lacks the ripCBA genes required for the growth of Y. pestis in activated macrophages (33), and therefore, to avoid decreases in survival due to the absence of the ripCBA genes, the CFU-based survival assays (see Fig. 3) were limited to a short time period (5 h). LDH release was initially determined at 5 h p.i. The results showed that macrophages infected with the nonopsonized yopB mutant, deficient in Yop translocation, released very small amounts of LDH, about 2.5% of the total, by 5 h of infection (Fig. 2A). This was a significantly lower LDH release than that of BMDMs infected with control serum-opsonized KIM5 (∼18%). Compared to BMDMs infected with control serum-opsonized KIM5, LDH was also significantly lower when KIM5 was opsonized with anti-V (3%) but not when the bacteria were left unopsonized (12.5%). These results were consistent with previous studies, which showed that rabbit polyclonal anti-LcrV antibodies could reduce Yop translocation and decrease apoptosis in naïve J774A.1 macrophage-like cells infected with Y. pestis (10, 29, 43).
FIG. 3.

Comparison of the intracellular survival of Y. pestis and Y. pseudotuberculosis 32777 in BMDMs. Naïve BMDMs (A, B) and IFN-γ-stimulated BMDMs (C) were infected with the indicated strains, with or without prior opsonization with serum. Intracellular bacterial survival was determined at 25 min, 1.5 h, and 5 h p.i. by CFU assay. Results were taken from three independent experiments and averaged. Error bars show standard deviations. ***, P < 0.001 compared to the level at 25 min.
FIG. 2.

Examination of YopJ-mediated cell death in Y. pestis-infected macrophages through determination of LDH release. Naïve BMDMs (A and B) and IFN-γ-stimulated BMDMs (C and D) were left uninfected or infected with nonopsonized KIM5 or KIM5 yopB or KIM5 opsonized with the indicated serum. LDH levels in the supernatants of the BMDMs at 5 h (A and C) and 24 h (B and D) p.i. were measured. Results were normalized by subtracting background levels of LDH from uninfected macrophages and are shown as percentages of the total LDH. Results were taken from three independent experiments and averaged. Error bars show standard deviations. *, P < 0.05; **, P < 0.01 compared to the control serum.
When the LDH assay was performed with IFN-γ-activated macrophages, only the BMDMs infected with the nonopsonized yopB mutant released significantly lower levels of LDH than the control serum (2.3% versus 18%) (Fig. 2C). Thus, the ability of anti-V to inhibit apoptosis was diminished in activated macrophages.
Opsonization with anti-V does not decrease the survival of Y. pestis in activated macrophages.
To determine if the opsonization of Y. pestis with anti-V would result in increased killing within BMDMs, we performed a CFU assay at various times p.i. Naïve BMDMs were infected with opsonized or nonopsonized bacteria as above for the LDH assay. As a preliminary test to confirm that the BMDMs used were bactericidal under the conditions of the assay, a CFU assay was performed with Y. pseudotuberculosis grown alongside KIM5 at 37°C. The internalization of nonopsonized 37°C-grown Y. pseudotuberculosis into BMDMs triggers a bactericidal process that requires macrophage sensing of the T3SS (35, 48). As shown in Fig. 3A, by 5 h p.i there was a significant decrease in the number of CFU for Y. pseudotuberculosis 32777. As reported previously (21), there was no significant decrease in the number of CFU over the same time period in BMDMs infected with nonopsonized KIM5 (Fig. 3A). Although it is unclear why the T3SS of Y. pestis KIM5 did not stimulate intracellular killing in BMDMs, these results confirmed that the macrophages used were bactericidal. Furthermore, because the T3SS of KIM5 did not stimulate intracellular killing in BMDMs, we did not face the problem of multiple bactericidal processes occurring simultaneously, which could complicate the analysis of the potential role of anti-V opsonization and IFN-γ activation in the decreasing survival of Y. pestis in macrophages.
Next, CFU assays were carried out using opsonized or nonopsonized KIM5. A KIM5 phoP mutant (Table 1), shown previously to be highly defective for intracellular survival (13), was included as a control. As shown in Fig. 3B, by 5 h p.i., there was a significant (∼2-log) decrease in the number of intracellular CFU that could be recovered from the macrophages infected with nonopsonized KIM5 phoP. In contrast, no significant decrease in the number of CFU was observed for the nonopsonized KIM5 yopB mutant, or KIM5 under any condition (nonopsonized, anti-V, or control serum) (Fig. 3B). Based on these results, we concluded that opsonization with anti-V does not lead to intracellular killing of KIM5 by naïve BMDMs.
Seeing no evidence for the intracellular killing of anti-V-opsonized KIM5 in naïve BMDMs, a CFU assay was performed with macrophages activated with IFN-γ (Fig. 3C). As with the naïve BMDMs, we used KIM5 phoP as a control for intracellular killing. The results obtained with IFN-γ-stimulated BMDMs were similar to those seen with naïve BMDMs, as only KIM5 phoP showed a significant decrease in the number of CFU by 5 h p.i. (Fig. 3C). Therefore, Y. pestis that was opsonized with anti-V prior to uptake was able to survive in macrophages that were activated with IFN-γ.
The opsonization of Y. pestis with anti-V serum does not decrease apoptosis in naïve or activated macrophages at a late stage of infection.
Under the infection conditions used in this study, in which internalized Y. pestis survives within macrophages (Fig. 3) (21), the kinetics of apoptosis is extended, such that LDH release is first detected by 5 h p.i. (early stage) (Fig. 2), and continues to increase over the next 19 h (late stage) (21). To determine if opsonization with anti-V would affect apoptosis at a late stage of infection, an LDH release assay was performed on Y. pestis-infected naïve or activated macrophages at 24 h p.i. Interestingly, only macrophages infected with the KIM5 yopB mutant released significantly lower levels of LDH than the control (Fig. 2B and D). Thus, there was no significant decrease in apoptosis at 24 h in either naïve or activated macrophages when KIM5 was opsonized with anti-V prior to infection (Fig. 2B and D).
The result with naïve macrophages was unexpected, since we had observed an ∼sixfold decrease in apoptosis in BMDMs infected with anti-V-opsonized KIM5 compared to that with control-opsonized KIM5 at 5 h p.i. (Fig. 2A) and assumed this proportional difference would be maintained to the 24-h time point (Fig. 2B). Instead, the results suggested that apoptosis was accelerated in BMDMs infected with anti-V-opsonized Y. pestis between 5 and 24 h compared to that with the control infection. Since anti-V-opsonized KIM5 was internalized by naïve macrophages at a higher level than control-opsonized KIM5 (Fig. 1A), it was possible that larger numbers of internalized bacteria allowed for higher levels of cell death between 5 and 24 h p.i.
Macrophages treated with IFN-γ appeared to be less sensitive to YopJ-dependent cell death, as the overall levels of LDH released from KIM5-infected BMDMs were lower in activated cells than the naïve phagocytes (compare Fig. 2B and D). Activation with lipopolysaccharide desensitizes macrophages to YopJ/YopP-induced apoptosis during infection with Y. pseudotuberculosis or Y. enterocolitica (4, 36), and a similar phenomenon may be operating in IFN-γ-activated BMDMs infected with Y. pestis.
The opsonization of Y. pestis with anti-V serum does not alter the secretion of TNF-α by infected macrophages.
TNF-α has been shown to cooperate with IFN-γ to promote protective immunity against Y. pestis (39). In addition, macrophages activated with TNF-α and IFN-γ are more bactericidal against Y. pestis than macrophages treated with IFN-γ alone (22). Previous studies have shown that Y. pestis can partially suppress the secretion of TNF-α from infected macrophages and that YopJ is required for this suppression (21, 46). Lilo et al. showed that after 24 h of infection, ∼2,000 pg/ml TNF-α was released by KIM5-infected BMDMs (21). We wanted to determine if TNF-α was secreted during our infections with KIM5 and to what degree opsonization affected TNF-α levels. In parallel, we determined levels of another cytokine (IL-1β) that is secreted from KIM5-infected macrophages in a YopJ-dependent manner (21). BMDMs were infected with opsonized or nonopsonized KIM5 or nonopsonized KIM5 yopB (Fig. 4). Supernatants were collected at 24 h, and ELISA was performed to measure TNF-α and IL-1β levels. We found that with BMDMs infected with KIM5 yopB, there was ∼5,000 pg/ml TNF-α released (Fig. 4A). Consistent with the concept that YopJ can interfere with the production of TNF-α, the level of TNF-α secreted was diminished to ∼2,500 pg/ml in KIM5-infected macrophages. When KIM5 was opsonized with control serum or anti-V, the levels of TNF-α that were secreted from the infected macrophages were similar to those secreted from macrophages infected with nonopsonized KIM5 (Fig. 4A), showing that anti-V did not significantly affect the secretion of TNF-α. Additionally, opsonization with anti-V did not affect the secretion of IL-1β from BMDMs infected with KIM5 (Fig. 4B).
FIG. 4.
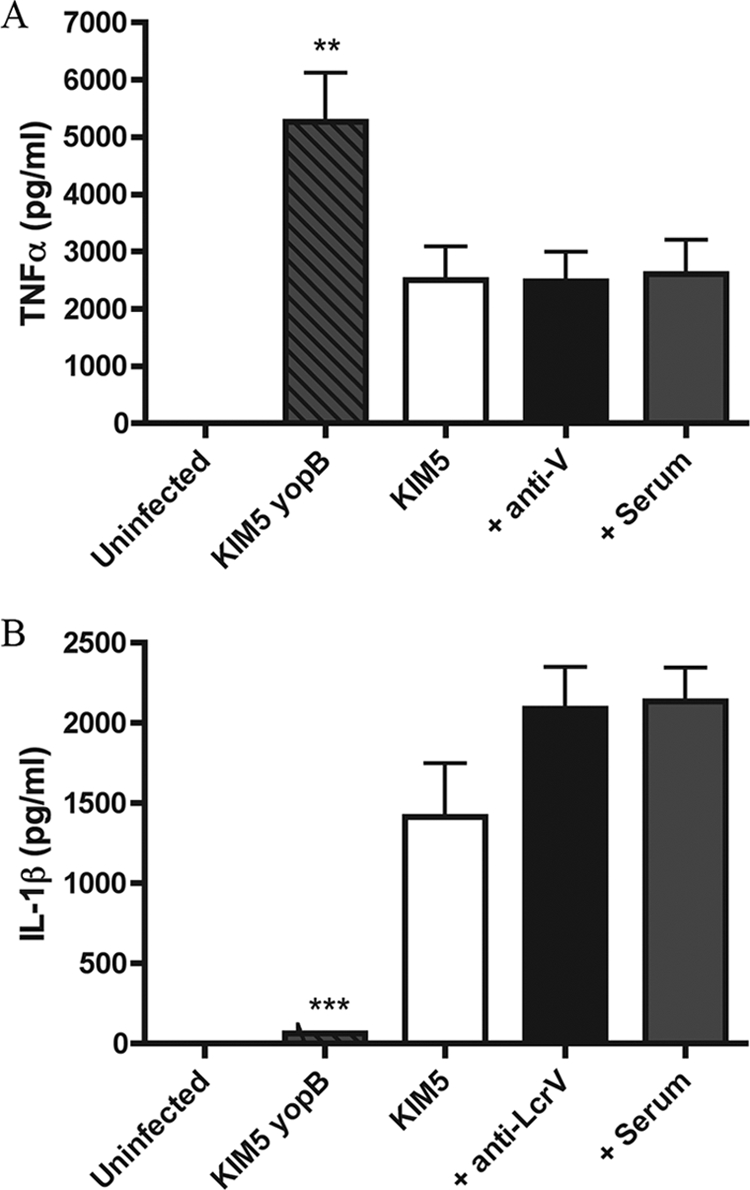
Monitoring TNF-α and IL-1β levels in the supernatants of Y. pestis-infected BMDMs. BMDMs were left uninfected or infected with the indicated strains with or without prior opsonization with serum. Supernatants were collected after 24 h of infection, and TNF-α (A) and IL-1β (B) levels were measured by ELISA. Results were taken from three independent experiments and averaged. Error bars show standard deviations. **, P < 0.01; ***, P < 0.001 compared to serum.
The opsonization of Y. pestis with anti-LcrV MAb 7.3 does not decrease survival in macrophages or inhibit late-stage apoptosis.
Polyclonal antibodies and MAbs that are specific for the same antigen can exhibit different activities with respect to neutralizing function (7). The anti-V used in these experiments had been shown to passively protect mice against Y. pestis KIM5, a conditionally virulent strain, in an intravenous infection model (18). However, it was desirable to repeat the above experiments using a well-characterized protective MAb specific for LcrV. The opsonization of Y. pestis with anti-LcrV IgG1 MAb 7.3 (17) increases phagocytosis and decreases apoptosis in naïve J774A.1 cells (43). In addition, MAb 7.3 is known to provide protection against fully virulent Y. pestis in murine bubonic and pneumonic plague infections (15-17). We opsonized KIM5/GFP or KIM5 with MAb 7.3 and repeated the phagocytosis, intracellular survival, and LDH release assays. In parallel, macrophages were infected with Y. pestis that was left nonopsonized or incubated with an IgG1 isotype control MAb specific for YopD (MAb 248.19). Figure 5 shows that MAb 7.3 significantly increased the phagocytosis of KIM5/GFP by naïve but not activated RAW 264.7 cells compared to the control. Opsonization with MAb 7.3 did not result in the significant killing of Y. pestis in either naïve or activated BMDMs (Fig. 6A and B). The opsonization of KIM5 with MAb 7.3 significantly reduced apoptosis at the early infection time point (5 h) in naïve BMDMs (Fig. 7A) but not at the late time point (24 h) in either naïve or activated macrophages (Fig. 7B and D, respectively). These results were similar to those obtained with anti-V (Fig. 1 to 3). The only difference between MAb 7.3 and anti-V was that opsonization with the former significantly reduced apoptosis in activated macrophages at the 5-h time point (Fig. 7C).
FIG. 5.
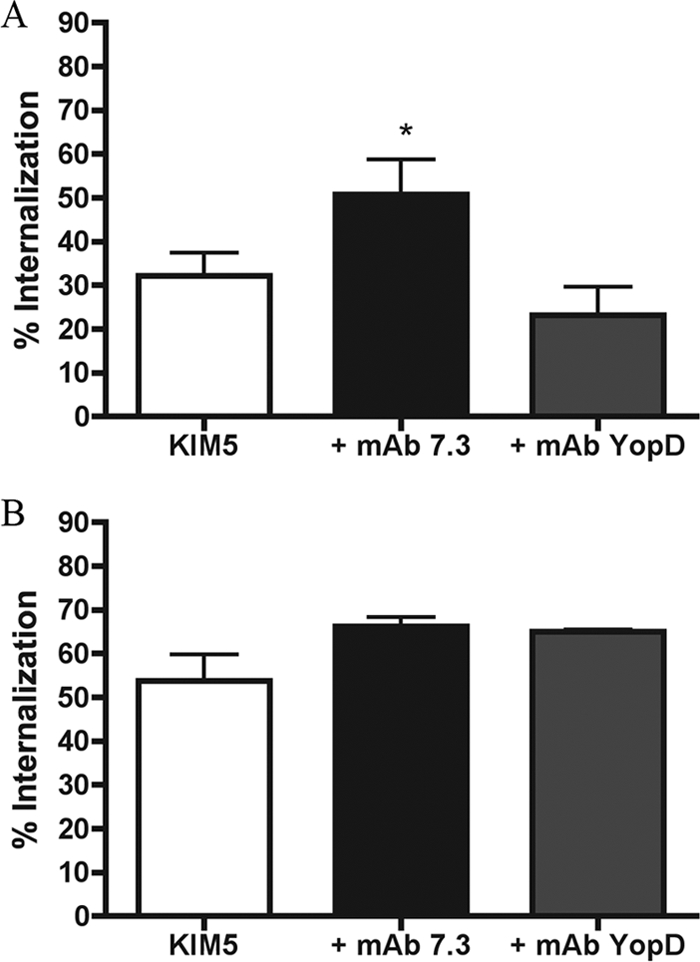
Phagocytosis assay with KIM5-infected naïve or IFN-γ-stimulated RAW 264.7 macrophages. KIM5/GFP was left unopsonized or incubated with anti-LcrV MAb 7.3 or anti-YopD MAb prior to infecting naïve macrophages (A) or IFN-γ-stimulated macrophages (B). Twenty-five minutes p.i., the cells were fixed and the percentage of internalization was determined as described in the legend to Fig. 1. Results were taken from three independent experiments and averaged. Error bars show standard deviations. *, P < 0.05 compared to MAb YopD.
FIG. 6.
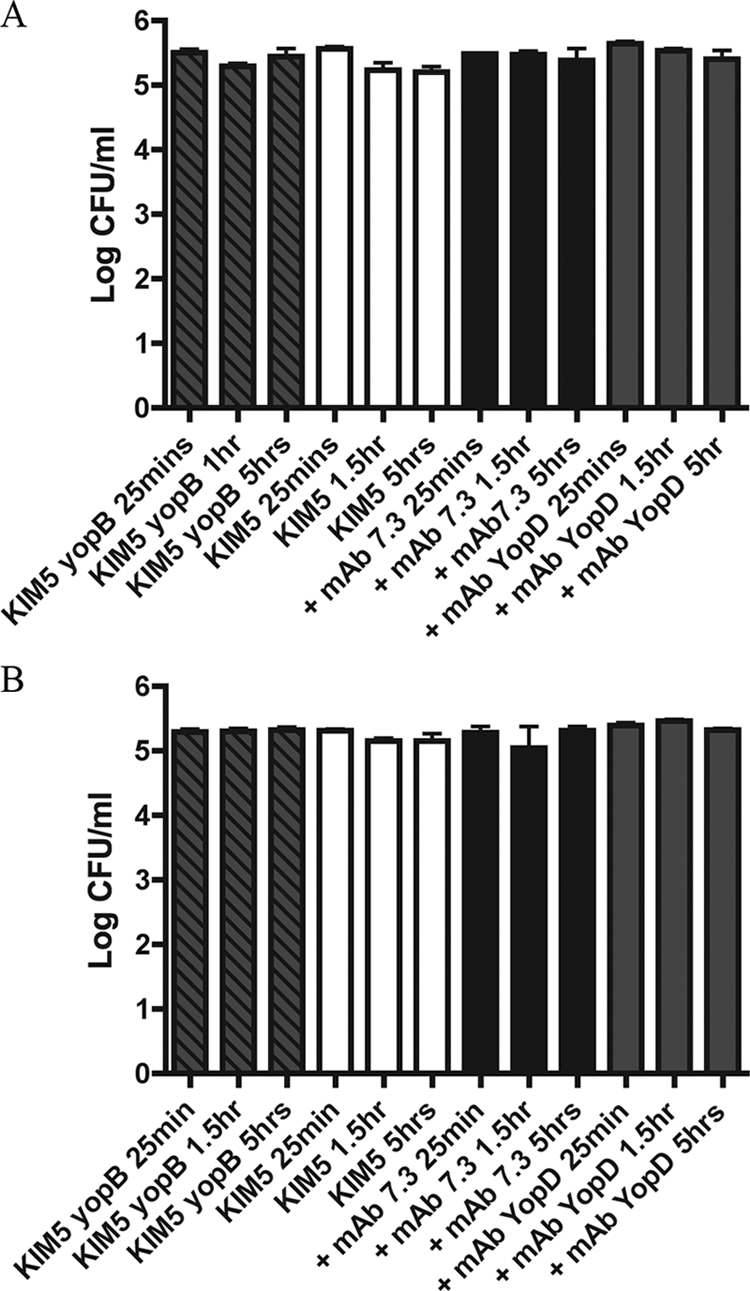
Effect of MAbs on the intracellular survival of Y. pestis in BMDMs. Naïve BMDMs (A) and IFN-γ-stimulated BMDMs (B) were infected with the indicated strains with or without preincubation with MAbs, and intracellular bacterial survival was determined at 25 min, 1.5 h, and 5 h p.i. as indicated in the legend to Fig. 3. Results were taken from three independent experiments and averaged. Error bars show standard deviations.
FIG. 7.

Effect of MAbs on YopJ-mediated cell death through determination of LDH release. Naïve BMDMs (A and B) or IFN-γ-stimulated BMDMs (C and D) were infected with the indicated strains of KIM5 with or without preincubation with MAbs or left uninfected. LDH levels in the supernatants of the BMDMs at 5 h (A and C) and 24 h (B and D) p.i. were measured as indicated in the legend to Fig. 2. Results were taken from three independent experiments and averaged. Error bars show standard deviations. *, P < 0.05; **, P < 0.01 compared to MAb YopD.
Anti-V opsonization does not overcome the block in acidification of Y. pestis-containing phagosomes.
To date, studies that have shown a block in the acidification of Y. pestis-containing phagosomes in macrophages have used 28°C-grown nonopsonized bacteria (34). To determine if opsonization with anti-V would increase phagosome acidification, we infected J774A.1 mouse macrophage-like cells and used microscopy to measure the colocalization of Y. pestis-containing phagosomes with the acidotropic probe LysoTracker Red DND-99 (34). Prior to infection, KIM5/GFP was grown at 28°C for 2 h or at 37°C for 2 h and then not opsonized or opsonized with control serum or anti-V. The colocalization of GFP-expressing KIM5 and LysoTracker Red was determined at 1.25 h p.i. As a positive control for phagosome acidification, we used KIM5/GFP fixed with PFA. Only phagosomes containing fixed KIM5/GFP (both grown at 28°C and 37°C) showed significantly increased colocalization with LysoTracker Red (>90%) than the standard condition (nonopsonized, 28°C) (Fig. 8). We concluded that opsonization with anti-V does not overcome the block in phagosome acidification imposed by Y. pestis in macrophages.
FIG. 8.

Effect of bacterial growth, temperature, and serum opsonization on phagosome acidification within naïve J774A.1 cells. J774A.1 cells were infected with fixed or live KIM5/GFP pregrown at 28°C or 37°C and left unopsonized or opsonized as indicated. LysoTracker Red DND 99 was used to label acidic compartments, and colocalization with KIM5/GFP was determined at 1.25 h p.i. by fluorescence microscopy. Results were taken from three independent experiments and averaged. Error bars show standard deviations. ***, P < 0.001 compared to the unopsonized KIM5 28°C subculture.
Conclusions.
This study focused on investigating the potential cooperative effect of anti-V opsonization and IFN-γ activation in allowing macrophages to protect themselves from being colonized and killed by Y. pestis. We conclude that Y. pestis can efficiently evade the combined effects of anti-V opsonization and IFN-γ activation in macrophages due to its ability to block phagosome acidification. In fact, we obtained evidence that activation with IFN-γ could be counterproductive for antibody-mediated protection, as anti-V was unable to significantly reduce apoptosis at the early time point in activated macrophages infected with KIM5 (although this effect was not seen with MAb 7.3). On the other hand, activation with IFN-γ increased overall levels of bacterial phagocytosis and decreased apoptosis at late stages, illustrating a protective activity of this cytokine.
We also obtained evidence that the ability of anti-V to inhibit apoptosis decreased as the time of the infection was extended. This suggests that intracellular bacteria can contribute to apoptosis and that anti-V is not neutralizing once the bacteria are internalized. It is not clear if the intracellular survival of Y. pestis is important for late-stage apoptosis. KIM5 phoP does not survive intracellularly (Fig. 3B and C); however, BMDMs infected with this mutant released levels of LDH at 5 h and 24 h p.i. similar to those of BMDMs infected with KIM5 (data not shown). In the case of Y. pseudotuberculosis, the results suggest that protein synthesis is required for intracellular bacteria to induce apoptosis (48). Perhaps internalized KIM5 causes increased apoptosis by continuing to synthesize and translocate YopJ for a limited period of time within nascent phagosomes, but bacterial survival is not required per se. The possibility that Y. pestis can synthesize and translocate small amounts of YopJ from within the phagosome is not necessarily in conflict with a previous study showing that no delivery of YopH could be detected by immunoblotting after the first 30 to 45 min of infection of macrophages (10). Immunoblotting may not be sensitive enough to detect the translocation of biologically significant amounts of a Yop. Alternatively, some Yops may be more efficiently translocated from an intracellular location (e.g., YopJ) than other Yops (e.g., YopH).
In our studies, we activated macrophages using IFN-γ only. There is evidence that TNF-α cooperates with IFN-γ to kill Y. pestis in macrophages in the absence of anti-V (22). Perhaps the addition of TNF-α is required along with anti-V and IFN-γ in macrophages to increase the killing of Y. pestis. While we did not add exogenous TNF-α in our CFU assays, we note that KIM5-infected macrophages secrete detectable levels of TNF-α by 4 h p.i. (21) and that ∼2,500 pg/ml TNF-α was detected in supernatants by 24 h p.i. (Fig. 4). Therefore, we can conclude that Y. pestis can bypass protection mediated by anti-V antibodies, preactivation with IFN-γ, and autocrine activation mediated by endogenous TNF-α produced during infection with KIM5. An additional consideration is that the experiments described here utilized the KIM5 pgm mutant, which lacks the ripCBA genes needed for the long-term survival and growth of Y. pestis in IFN-γ-activated macrophages (33). Because of this, our intracellular survival assays were restricted to a maximum of a 5-h time period. Use of a fully virulent pgm+ strain would have allowed for the use of longer time points in CFU assays, and a role for anti-V in the killing of Y. pestis in IFN-γ-activated macrophages might be revealed at later time periods.
These results have implications for understanding the mechanism of protective immunity to plague mediated by anti-V and phagocyte-activating cytokines. As our results suggest that opsonization with anti-V does not dramatically increase the killing of Y. pestis by activated macrophages, it is possible that neutrophils may be more critical for protection than macrophages (10, 29). Consistent with this idea is the demonstration that macrophages are not essential for anti-V-mediated protection against Y. pestis in the spleens of infected mice (10, 29), while neutrophils appear to be important for anti-V-mediated protection in both the spleens and livers of mice challenged with plague (10). It is possible that TNF-α cooperates with IFN-γ to upregulate microbicidal processes in neutrophils. Anti-V increases the phagocytosis of Y. pestis by murine neutrophils (10). Y. pestis is unable to survive in rodent (8) or human (40) neutrophils. It has been shown that Yop delivery does not protect intracellular Y. pestis from neutrophil killing (40). However, whether anti-V opsonization accelerates the killing of Y. pestis in neutrophils has not been examined.
The neutralization of apoptosis in Y. pestis-infected macrophages by anti-V is commonly used as an in vitro correlate of protective immunity (3, 43-45). Although this assay has many positive features, including ease of use, reproducibility, and a quantifiable readout, we feel the following points should be considered with respect to improving the in vitro correlate of protective immunity assay. First, our results showing that anti-V can promote the uptake of Y. pestis into macrophages without killing the bacteria, and that anti-V may not inhibit the translocation of YopJ by the intracellular population, indicate that the time point at which apoptosis levels are quantified is critically important. In addition, the activation status of the macrophage is an important variable, as we found that IFN-γ-activated macrophages are less sensitive to apoptosis. Finally, Y. pestis strains differ in their ability to induce apoptosis (KIM has high activity [21]) and not all strains require apoptosis for virulence (20, 47). Therefore, there continues to be a need to develop an anti-LcrV neutralization assay that does not rely on macrophage apoptosis as a readout.
Acknowledgments
We wish to thank Rebecca Rowehl, Anne Savitt, Maya Ivanov, Gloria Viboud, and Edison Mejia for help with the production, characterization, and purification of the anti-YopD MAb. Galina Romanov provided excellent technical assistance with BMDM production, and Kathryn Klein provided guidance with the phagosome acidification assays. Hana Fukuto, Maya Ivanov, Kathryn Klein, Joseph McPhee, and Gloria Viboud provided thoughtful review of the manuscript.
This work was supported by grants from the NIH (R01 AI043389, P01 AI055621, and Northeast Biodefense Center U54 AI057158-Lipkin) awarded to J.B.B.
Footnotes
Published ahead of print on 26 August 2009.
REFERENCES
- 1.Achtman, M., K. Zurth, G. Morelli, G. Torrea, A. Guiyoule, and E. Carniel. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 96:14043-14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aderem, A., and D. M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593-623. [DOI] [PubMed] [Google Scholar]
- 3.Bashaw, J., S. Norris, S. Weeks, S. Trevino, J. J. Adamovicz, and S. Welkos. 2007. Development of in vitro correlate assays of immunity to infection with Yersinia pestis. Clin. Vaccine Immunol. 14:605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergsbaken, T., and B. T. Cookson. 2007. Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 3:e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black, D. S., and J. B. Bliska. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37:515-527. [DOI] [PubMed] [Google Scholar]
- 6.Bliska, J. B., and D. S. Black. 1995. Inhibition of the Fc receptor-mediated oxidative burst in macrophages by the Yersinia pseudotuberculosis tyrosine phosphatase. Infect. Immun. 63:681-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casadevall, A. 2003. Antibody-mediated immunity against intracellular pathogens: two-dimensional thinking comes full circle. Infect. Immun. 71:4225-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavanaugh, D. C., and R. Randall. 1959. The role of multiplication of Pasteurella pestis in mononuclear phagocytes in the pathogenesis of fleaborne plague. J. Immunol. 85:348-363. [PubMed] [Google Scholar]
- 9.Cornelis, G. R. 2002. Yersinia type III secretion: send in the effectors. J. Cell Biol. 158:401-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cowan, C., A. V. Philipovskiy, C. R. Wulff-Strobel, Z. Ye, and S. C. Straley. 2005. Anti-LcrV antibody inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect. Immun. 73:6127-6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elvin, S. J., and E. D. Williamson. 2004. Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microb. Pathog. 37:177-184. [DOI] [PubMed] [Google Scholar]
- 12.Goguen, J. D., W. S. Walker, T. P. Hatch, and J. Yother. 1986. Plasmid-determined cytotoxicity in Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 51:788-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabenstein, J. P., H. S. Fukuto, L. E. Palmer, and J. B. Bliska. 2006. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect. Immun. 74:3727-3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabenstein, J. P., M. Marceau, C. Pujol, M. Simonet, and J. B. Bliska. 2004. The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect. Immun. 72:4973-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill, J., C. Copse, S. Leary, A. J. Stagg, E. D. Williamson, and R. W. Titball. 2003. Synergistic protection of mice against plague with monoclonal antibodies specific for the F1 and V antigens of Yersinia pestis. Infect. Immun. 71:2234-2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill, J., J. E. Eyles, S. J. Elvin, G. D. Healey, R. A. Lukaszewski, and R. W. Titball. 2006. Administration of antibody to the lung protects mice against pneumonic plague. Infect. Immun. 74:3068-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill, J., S. E. Leary, K. F. Griffin, E. D. Williamson, and R. W. Titball. 1997. Regions of Yersinia pestis V antigen that contribute to protection against plague identified by passive and active immunization. Infect. Immun. 65:4476-4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov, M. I., B. L. Noel, R. Rampersaud, P. Mena, J. L. Benach, and J. B. Bliska. 2008. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1− Yersinia pestis. Infect. Immun. 76:5181-5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kummer, L. W., F. M. Szaba, M. A. Parent, J. J. Adamovicz, J. Hill, L. L. Johnson, and S. T. Smiley. 2008. Antibodies and cytokines independently protect against pneumonic plague. Vaccine 26:6901-6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemaitre, N., F. Sebbane, D. Long, and B. J. Hinnebusch. 2006. Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not required for virulence in a rat model of bubonic plague. Infect. Immun. 74:5126-5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lilo, S., Y. Zheng, and J. B. Bliska. 2008. Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect. Immun. 76:3911-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukaszewski, R. A., D. J. Kenny, R. Taylor, D. G. Rees, M. G. Hartley, and P. C. Oyston. 2005. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect. Immun. 73:7142-7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mueller, C. A., P. Broz, S. A. Muller, P. Ringler, F. Erne-Brand, I. Sorg, M. Kuhn, A. Engel, and G. R. Cornelis. 2005. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310:674-676. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima, R., and R. R. Brubaker. 1993. Association between virulence of Yersinia pestis and suppression of gamma interferon and tumor necrosis factor alpha. Infect. Immun. 61:23-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navarro, L., N. M. Alto, and J. E. Dixon. 2005. Functions of the Yersinia effector proteins in inhibiting host immune responses. Curr. Opin. Microbiol. 8:21-27. [DOI] [PubMed] [Google Scholar]
- 26.Parent, M. A., L. B. Wilhelm, L. W. Kummer, F. M. Szaba, I. K. Mullarky, and S. T. Smiley. 2006. Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infect. Immun. 74:3381-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersson, J., A. Holmstrom, J. Hill, S. Leary, E. Frithz-Lindsten, A. von Euler-Matell, E. Carlsson, R. Titball, A. Forsberg, and H. Wolf-Watz. 1999. The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol. Microbiol. 32:961-976. [DOI] [PubMed] [Google Scholar]
- 29.Philipovskiy, A. V., C. Cowan, C. R. Wulff-Strobel, S. H. Burnett, E. J. Kerschen, D. A. Cohen, A. M. Kaplan, and S. C. Straley. 2005. Antibody against V antigen prevents Yop-dependent growth of Yersinia pestis. Infect. Immun. 73:1532-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prehna, G., M. I. Ivanov, J. B. Bliska, and C. E. Stebbins. 2006. Yersinia virulence depends on mimicry of host Rho-family nucleotide dissociation inhibitors. Cell 126:869-880. [DOI] [PubMed] [Google Scholar]
- 31.Pujol, C., and J. B. Bliska. 2003. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 71:5892-5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pujol, C., and J. B. Bliska. 2005. Turning Yersinia pathogenesis outside in: subversion of macrophage function by intracellular yersiniae. Clin. Immunol. 114:216-226. [DOI] [PubMed] [Google Scholar]
- 33.Pujol, C., J. P. Grabenstein, R. D. Perry, and J. B. Bliska. 2005. Replication of Yersinia pestis in interferon gamma-activated macrophages requires ripA, a gene encoded in the pigmentation locus. Proc. Natl. Acad. Sci. USA 102:12909-12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pujol, C., K. A. Klein, G. A. Romanov, L. E. Palmer, C. Cirota, Z. Zhao, and J. B. Bliska. 2009. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 77:2251-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy, D., D. R. Liston, V. J. Idone, A. Di, D. J. Nelson, C. Pujol, J. B. Bliska, S. Chakrabarti, and N. W. Andrews. 2004. A process for controlling intracellular bacterial infections induced by membrane injury. Science 304:1515-1518. [DOI] [PubMed] [Google Scholar]
- 36.Ruckdeschel, K., and K. Richter. 2002. Lipopolysaccharide desensitization of macrophages provides protection against Yersinia enterocolitica-induced apoptosis. Infect. Immun. 70:5259-5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonet, M., and S. Falkow. 1992. Invasin expression in Yersinia pseudotuberculosis. Infect. Immun. 60:4414-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smiley, S. T. 2008. Current challenges in the development of vaccines for pneumonic plague. Expert Rev. Vaccines 7:209-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smiley, S. T. 2008. Immune defense against pneumonic plague. Immunol. Rev. 225:256-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spinner, J. L., J. A. Cundiff, and S. D. Kobayashi. 2008. Yersinia pestis type III secretion system-dependent inhibition of human polymorphonuclear leukocyte function. Infect. Immun. 76:3754-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Straley, S. C., and P. A. Harmon. 1984. Growth in mouse peritoneal macrophages of Yersinia pestis lacking established virulence determinants. Infect. Immun. 45:649-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Titball, R. W., and E. D. Williamson. 2004. Yersinia pestis (plague) vaccines. Expert Opin. Biol. Ther. 4:965-973. [DOI] [PubMed] [Google Scholar]
- 43.Weeks, S., J. Hill, A. Friedlander, and S. Welkos. 2002. Anti-V antigen antibody protects macrophages from Yersinia pestis-induced cell death and promotes phagocytosis. Microb. Pathog. 32:227-237. [DOI] [PubMed] [Google Scholar]
- 44.Williamson, E. D., H. C. Flick-Smith, E. Waters, J. Miller, I. Hodgson, C. S. Le Butt, and J. Hill. 2007. Immunogenicity of the rF1+rV vaccine for plague with identification of potential immune correlates. Microb. Pathog. 42:11-21. [DOI] [PubMed] [Google Scholar]
- 45.Zauberman, A., S. Cohen, Y. Levy, G. Halperin, S. Lazar, B. Velan, A. Shafferman, Y. Flashner, and E. Mamroud. 2008. Neutralization of Yersinia pestis-mediated macrophage cytotoxicity by anti-LcrV antibodies and its correlation with protective immunity in a mouse model of bubonic plague. Vaccine 26:1616-1625. [DOI] [PubMed] [Google Scholar]
- 46.Zauberman, A., S. Cohen, E. Mamroud, Y. Flashner, A. Tidhar, R. Ber, E. Elhanany, A. Shafferman, and B. Velan. 2006. Interaction of Yersinia pestis with macrophages: limitations in YopJ-dependent apoptosis. Infect. Immun. 74:3239-3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zauberman, A., B. Velan, E. Mamroud, Y. Flashner, A. Shafferman, and S. Cohen. 2007. Disparity between Yersinia pestis and Yersinia enterocolitica O:8 in YopJ/YopP-dependent functions. Adv. Exp. Med. Biol. 603:312-320. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, Y., J. Murtha, M. A. Roberts, R. M. Siegel, and J. B. Bliska. 2008. Type III secretion decreases bacterial and host survival following phagocytosis of Yersinia pseudotuberculosis by macrophages. Infect. Immun. 76:4299-4310. [DOI] [PMC free article] [PubMed] [Google Scholar]