

Abstract
Dermatan sulfate epimerase 1 (DS-epi1) and DS-epi2 convert glucuronic acid to iduronic acid in chondroitin/dermatan sulfate biosynthesis. Here we report on the generation of DS-epi1-null mice and the resulting alterations in the chondroitin/dermatan polysaccharide chains. The numbers of long blocks of adjacent iduronic acids are greatly decreased in skin decorin and biglycan chondroitin/dermatan sulfate, along with a parallel decrease in iduronic-2-O-sulfated-galactosamine-4-O-sulfated structures. Both iduronic acid blocks and iduronic acids surrounded by glucuronic acids are also decreased in versican-derived chains. DS-epi1-deficient mice are smaller than their wild-type littermates but otherwise have no gross macroscopic alterations. The lack of DS-epi1 affects the chondroitin/dermatan sulfate in many proteoglycans, and the consequences for skin collagen structure were initially analyzed. We found that the skin collagen architecture was altered, and electron microscopy showed that the DS-epi1-null fibrils have a larger diameter than the wild-type fibrils. The altered chondroitin/dermatan sulfate chains carried by decorin in skin are likely to affect collagen fibril formation and reduce the tensile strength of DS-epi1-null skin.
Chondroitin sulfate (CS) is an unbranched polymer chain composed of alternating glucuronic acid (GlcA) and N-acetylgalactosamine (GalNAc) units (36, 49). In dermatan sulfate (DS), d-glucuronic acid is converted to its epimer l-iduronic acid (IdoA) (25). The extent of this modification, which varies from a few percent of the glucuronic acid being epimerized to a predominant presence of iduronic acid, depends on the variable epimerase activity in tissues and on the core protein attached to the chain in CS/DS proteoglycans (PGs) (41, 47). The same CS/DS PG has a different iduronic acid content, depending on the cell type and tissue of origin (4, 5). The name CS/DS denotes the hybrid GlcA-IdoA nature of the chain. It has long been known that the distribution of iduronic acids within the chain is not random but follows two patterns: either they are clustered together, forming long iduronic acid blocks, or they are isolated, i.e., interspersed among surrounding glucuronic acids (11). DS epimerase 1 (DS-epi1) and DS-epi2, encoded in mouse by the Dse and Dsel (Dse-like) genes, respectively, are present in organisms ranging from Xenopus tropicalis to humans but not in worms and flies (23, 34). During DS biosynthesis, epimerization is followed by the action of eight C-specific O-sulfotranferases, which transfer a sulfate group to C-2 of both IdoA and GlcA and to C-4, C-6, and C-4/C-6 of GalNAc (18). These modification reactions, individually affecting only part of the available substrate, produce structural variability in the CS/DS chain. Considerable efforts have been made to characterize specific sequences in the CS/DS chains responsible for binding to protein and the subsequent mediation of a biological effect (28). For instance, (IdoA-2OS-GalNAc-4OS)3- and GalNAc-4/6-diOS-containing structures bind and activate heparin cofactor II, which is the major antithrombotic system in the subendothelial layer (48). IdoA/GlcA-2OS-GalNAc-6OS-containing structures bind to pleiotrophin, mediating neuritogenic activity (3, 44). IdoA-GalNAc-4OS-containing structures bind to basic fibroblast growth factor, and the complex has been shown to be active in wound healing (46).
CS/DS PGs are mainly found in the extracellular matrix. They belong to four families: lecticans, e.g., versican, aggrecan, brevican, and neurocan; collagens, e.g., collagen IX; basement membrane PGs, e.g., SMC3, collagen XV, and perlecan, containing both heparan sulfate (HS) and CS/DS; and small leucine-rich repeat PGs. Some PGs of the first three groups are referred to as CS PGs. The actual presence of iduronic acid, depending on the tissue examined and on the developmental stage, has been overlooked in many cases (37, 44). The archetypical small leucine-rich repeat PG family members decorin, biglycan, fibromodulin, and lumican bind fibrillar collagens and affect collagen fibril and scaffold formation in connective tissues (15). Decorin and biglycan are substituted with one and two CS/DS chains, respectively. Decorin is involved in collagen type I fibril formation and matrix assembly in a wide range of connective tissues and binds near the C terminus of collagen monomers, delaying their accretion to the growing fibrils. We have identified an SYIRIADTNIT sequence in decorin as essential for binding to collagen (16). The role of the decorin CS/DS chain in vivo has not been explored, although in vitro studies suggest that IdoA promotes the binding of CS/DS to collagen (31) and is required for self-association of CS/DS chains (6, 10, 22).
Here the function of DS-epi1 in mice was disrupted. DS-epi1-deficient mice show CS/DS with a marked deficiency in iduronic acid-containing structures. The deletion of DS-epi1 is likely to affect many types of PGs and to result in a complex phenotype. We focus on skin alterations presumably caused by altered decorin/biglycan CS/DS chains.
MATERIALS AND METHODS
Materials.
Cell culture reagents were from Invitrogen. Red-Sepharose gel, Superose 6 HR 10/30, Superdex Peptide 10/300 GL, PD-10 columns, and ECL-Plus reagent were from GE Healthcare. DE52 anion-exchange resin was from Whatman. 35SO4 (1,500 Ci/mmol) was from Perkin-Elmer. Chondroitinases ABC, B, and AC-I were from Seigakaku. Brilliant blue G colloidal gel-staining solution was from Sigma.
Construction of the targeting vector and generation of chimeric mice.
A genomic Dse/Sart2 clone was isolated from a mouse genomic P1 artificial chromosome library (RPCI21; Geneservice, Cambridge, United Kingdom) (33). A 6.3-kb SphI-StuI fragment was ligated into pBluescript II KS (Stratagene), and a phosphoglycerate kinase-neomycin resistance cassette (pGKNeo) (26) was inserted in the reverse transcriptional orientation into a unique XhoI site located in the second exon of Dse/Sart2 (Fig. 1A). The targeting vector was linearized at a NotI site and used to transfect R1 mouse embryonic stem (ES) cells at the Lund Transgenic Core Facility as described previously (45). Clones were picked and analyzed by Southern blotting after NcoI digestion with a 420-bp external probe. This probe hybridizes with a 6.3-kb wild-type fragment and a 4.8-kb NcoI fragment (Fig. 1B). Three individually targeted clones were injected into C57BL/6 blastocysts to generate chimeric mice. Chimeric males were obtained from two clones and mated with C57BL/6 females. F1 mice with germ line transmission were intercrossed to produce all genotypes in a mixed C57BL/6-129/SvJ genetic background. All experiments in this study were conducted with littermates of this mixed genetic background. Mice derived from two ES cell clones were characterized for (i) Dse allelic distribution at weaning, (ii) Dse expression by quantitative reverse transcription (qRT)-PCR and Western blotting, (iii) shape of the tail at birth, and (iv) weight measured between birth and weaning. Given the complete identity of the Dse−/− mutant mice from the two ES cell clones, according to the above four criteria, only mice derived from one ES cell clone were further characterized. Mice were genotyped by PCR from tail DNA with the following primers: forward primer for both alleles, 5′-AGCACATTGCAGCTCGGCTTAC-3′; reverse primer for the wild-type allele, 5′-GCTGCCATCCTCTCCATGTAGTC-3′; reverse primer for the neomycin cassette-mutated allele, 5′-TGGATGTGGAATGTGTGCGAGG-3′. The use of animals for research complied with national guidelines, and permission was given by the regional ethical board.
FIG. 1.

Targeted disruption of the Dse gene. (A) First line, schematic view of the Dse genomic structure. Second line, restriction enzyme map of the targeted Dse locus in exon 2; the location of the PCR product obtained from the wild-type allele is indicated. Third line, targeting vector; the location of the PCR product obtained from the mutated allele is indicated. The origin of the 6.3- and 4.8-kb NcoI fragments and the location of the common probe are shown in the last two lines. (B) Southern blotting of DNA isolated from ES cells. (C) PCR genotyping of mice.
qRT-PCR.
Total RNA was extracted from tissues with an RNeasy kit (Qiagen) and DNase treated with a DNA-free kit (Applied Biosystems), and cDNA synthesis was performed with a SuperScript VILO cDNA synthesis kit (Invitrogen). The samples were mixed with primers and SYBR green Master Mix (Applied Biosystems) and amplified in an ABI Prism instrument (Applied Biosystems, Foster City, CA) starting with an initial 2-min heating at 50°C and 10-min heating at 95°C, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. The data were analyzed with SDS 2.1 software (Applied Biosystems). The calculated threshold cycle (CT) values were normalized to the CT value for glyceraldehyde-3-phosphate dehydrogenase. The primers used were as follows: for Dse, 5′-TGTGTGCTGTATCCTGAGAACA-3′ and 5′-CAAGGCGCATCTTTCACCAAC-3′; for glyceraldehyde-3-phosphate dehydrogenase, 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Establishment of primary fibroblast cell cultures.
Lung and skin fibroblasts were derived from 2-month-old animals. Small pieces of the organs were allowed to adhere to plastic and cultivated in minimal essential medium-10% fetal bovine serum. Outcoming cells were grown for 10 days and split (passage 1). Cells were utilized for the experiments between passage 4 and passage 7.
Western blotting and immunohistochemistry.
Cells were lysed in a lysis buffer containing 20 mM morpholinepropanesulfonic acid (pH 6.5), 150 mM NaCl, 10% glycerol, 2 mM dithiothreitol, 1 mM EDTA, 1% Triton X-100, and protease inhibitors, i.e., 1 mM phenylmethylsulfonyl fluoride and aprotinin, leupeptin, and pepstatin at 1 μg/ml each. Spleens, lungs, kidneys, skin, and brains were homogenized in an approximately threefold excess (vol/wt) of lysis buffer with a Potter homogenizer. Protein content was determined in cleared lysates by the Bradford method (Bio-Rad), with bovine serum albumin as the standard. Alternatively, 5-mg organ lysates were bound in batch to 50 μl of Red-Sepharose gel, incubated for 60 min, washed five times with lysis buffer, and finally eluted with reducing Laemmli sample buffer.
DS-epi1 was detected with 1 μg/ml of an immunopurified anti-DS-epi1 rabbit polyclonal antibody obtained after antigen peptide immunization (KWSKYKHDLAAS, corresponding to amino acids 509 to 520 of the human/murine sequence; Innovagen, Sweden). ECL-Plus was used as a horseradish peroxidase substrate. Immunohistochemistry was done as described previously (45), with antisera against decorin and biglycan.
DS epimerase activity.
A 200-μl volume of organ or cell lysate was desalted by using as the dialysis buffer a mixture of 20 mM morpholinepropanesulfonic acid (pH 5.5 at 37°C), 10% glycerol, 0.5 mM EDTA, 0.1% Triton X-100, 1 mM dithiothreitol, and protease inhibitors. Protein content was determined after dialysis, and equal amounts of proteins of samples from the different genotypes were assayed for epimerase activity. The final assay volume was 100 μl of a mixture of 0.8× dialysis buffer, 2 mM MnCl2, 0.5% NP-40, and 30,000 dpm of the labeled chondroitin substrate ([5-3H]defructosylated K4 prepared according to reference 14). Incubations were carried out at 37°C for 20 h, and the released tritium was quantified as described in reference 23.
Preparation of in vivo-labeled CS/DS.
Ten-day-old mice were intraperitoneally injected with 50 μl phosphate-buffered saline (PBS) containing 200 μCi 35SO4 and kept alive for 2 h before sacrifice. To assess the structure of the CS/DS chains from the whole body, entire mice were cut into pieces and ground in 30 ml of buffer containing 100 mM Tris (pH 8.5 at 55°C), 200 mM NaCl, and 5 mM EDTA. The mixture was made to contain 0.1% sodium dodecyl sulfate (SDS), proteinase K was added to a final concentration of 100 μg/ml, and the mixture was incubated at 55°C overnight. Further proteinase was added to cleared material, and the incubation was continued for an additional 6 h. The material was diluted 1:4 with 200 mM acetate (pH 5.5), bound in batch with 1 ml DE-52 anion-exchange resin, washed with 50 mM acetate (pH 5.5)-200 mM NaCl, eluted with 50 mM acetate (pH 5.5)-1.5 M NaCl-10 μg sulfated CS-6 (added as a carrier), and desalted with a PD-10 column run in water. GAG chains were cleaved by β elimination in 50 mM KOH-100 mM NaBH4 at 45°C for 16 to 18 h and reisolated on DE-52 resin in principle as described above. HS chains were depolymerized by deamination reaction at pH 1.5 (43), followed by reisolation of CS/DS chains on Superose 6 run in 0.2 M NH4HCO3.
Preparation of labeled skin decorin/biglycan- and versican-CS/DS chains.
Skin from 10-day-old mice, labeled as described above, was extracted with a Potter homogenizer in 10 volumes (vol/wt) of 4 M guanidine-50 mM acetate (pH 5.8)-10 mM EDTA-5 mM N-ethylmaleimide-protease inhibitors. Cleared extracted material was dialyzed versus an excess of 6 M urea-50 mM acetate (pH 5.5)-0.2 M NaCl-1 mM EDTA-protease inhibitors. After dialysis, Triton was added to 0.1% and PGs were bound to DE-52 resin, washed with dialysis buffer-0.1% Triton, and eluted with 4 M guanidine-50 mM acetate (pH 5.8)-10 mM EDTA. One hundred micrograms of dextran T500 was added as a carrier, and PGs were size separated on a Superose 6 column run in 4 M guanidine-50 mM acetate (pH 5.8)-10 M EDTA-0.1% Triton. Ten micrograms of sulfated CS-6 was added as a carrier, the isolated PGs were desalted with PD-10 columns run in water, and the GAG chains were cleaved from the core protein by β elimination, repurified on DE-52 resin, freed from HS, and finally recovered as described above.
Preparation of unlabeled skin decorin/biglycan.
Skin from 3- to 4-month-old mice was extracted in guanidine-containing buffer, dialyzed versus urea-containing buffer, and DE-52 resin purified as described above. DE-52 resin-purified material, without addition of any carrier, was ethanol precipitated and run on Superose 6 as described above with no addition of any carrier. Elution fractions were analyzed by Western blotting with antibiglycan and antidecorin antibodies. Pooled material was dialyzed stepwise versus 50 mM HEPES (pH 7.4)-0.15 M NaCl solutions containing decreasing concentrations of urea (6, 3, and 0 M) and finally extensively versus water. Samples were lyophilized and resuspended in PBS. Most of the material was soluble, as it was recovered in the supernatant after centrifugation at 20,000 × g. Purity was verified by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (see Fig. 9).
FIG. 9.

Biophysical properties of Dse−/− skin and effect of skin decorin/biglycan on collagen fibril formation in vitro. (A) Differential scanning calorimetry of intact skin. Skin samples from 3-month-old mice were immersed in PBS in a calorimetric cell and heated over a temperature range of 30 to 90°C. Thermograms (no peaks after 70°C) were normalized to a buffer blank run without skin. Wt, wild type. (B) Five hundred nanograms of extracted decorin/biglycan from adult skin was subjected to SDS-PAGE and stained with Brilliant blue G colloidal solution. (C) PGs were extracted from identical amounts (fresh weight) of tail skin of 4-month-old mice. PGs were purified on a DE52 anion-exchange column, and identical volumes of the eluates were Western blotted and stained with antidecorin antibody. (D) Differential scanning calorimetry of in vitro-formed collagen fibrils. Prior to calorimetry, 500 μg acid-solubilized collagen was incubated for 4 h at 37°C with the addition of 10 μg wild-type or Dse−/− mutant decorin/biglycan extract from skin (one preparation is shown in panel B). Collagen fibrils were then heated in a calorimeter over a temperature range of 30 to 60°C. Thermograms are normalized to a buffer blank run without collagen. (E) Turbidimetric measurements of in vitro collagen fibrilization. Fifty micrograms of acid-solubilized collagen was mixed with 1 μg of decorin/biglycan extracts either left untreated or pretreated with chondroitinase ABC. The samples were then incubated at 37°C in a spectrophotometer, and turbidity readings were taken every 4 min and plotted.
Chondroitinase treatment of CS/DS and analysis of the cleavage products.
Cleavage of CS/DS chains by lyases (Seikagaku) and their analyses were done according to reference 34.
Collagen staining.
Tissues were fixed in 95% ethanol-1% acetic acid and decalcified in 6% EDTA in PBS for 1 week prior to dehydration and embedding. Sections were stained with Chromotrope 2R and Sirius Red (Sigma).
Electron microscopy.
Tissues from 3-month-old wild-type and Dse−/− mutant mice were analyzed by transmission electron microscopy (TEM). After fixation in 0.15 M sodium cacodylate-buffered 2.5% glutaraldehyde and postfixation in 0.1 M s-collidine-buffered 2% osmium tetroxide, samples were embedded in epoxy resin. Ultrathin sections were analyzed in a Philips CM-10 electron microscope, and collagen fibril diameters were quantified with ImageJ software (NIH). To calculate fibril density, the area occupied by collagen fibrils was measured by converting the image into black (collagen fibrils)-and-white (empty area) pixels, and their concurrent quantification was performed with ImageJ software (NIH). The Mann-Whitney U test was used to calculate significance, with a P value of <0.05 considered statistically significant (32).
Differential scanning calorimetry.
Differential scanning calorimetry measurements were performed either with dissected skin or with acid-solubilized collagen (Vitrogen) preincubated for 4 h in 37°C in PBS in the presence of extracted decorins from wild-type or Dse−/− mutant mice. The thermograms were recorded in VP-DSC (MicroCal) at a scan rate of 0.25°C/min and medium feedback. Each thermogram was corrected by subtraction of a linear baseline based on a blank buffer sample and normalized for collagen concentration.
Collagen fibrillogenesis assay.
Acid-solubilized collagen (Vitrogen) was neutralized and diluted (100 μg/ml) in 150 mM NaCl-20 mM HEPES (pH 7.4) with decorin extracted from wild-type or Dse−/− mutant mice (2 μg/ml). Solutions were degassed and then incubated at 37°C in a spectrophotometer, where the absorbance was continuously recorded at 400 nm.
Tensile strength determination.
Dorsal skin samples from 5-month-old Dse-deficient (n = 5) and wild-type (n = 5) mice were harvested immediately after sacrifice. Skin samples were cut with a template 4 mm wide by 20 mm long. Two skin samples identical in width and length were harvested from each mouse. The long axis of all samples coincided with the anterior-posterior direction of the mice. The thickness of each sample was determined with a micrometer at two consecutive locations along the middle 5-mm part of the skin specimens, and an average of the two measurements was used for analysis. Approximately 7 mm of each end of the skin samples was subsequently allowed to air dry at room temperature while keeping the middle 5-mm part moist with PBS-soaked gauze. Thereafter, the dried sample ends were glued to the uncoated aluminum endplates of the mechanical rig with cyanoacrylate glue. When the glue had hardened, the skin samples and the endplates were attached to the mechanical test frame and submerged in a petri dish with PBS. Subsequently, the mechanical test frame was placed under a stereoscopic microscope (SMZ1000; Nikon, Tokyo, Japan) and the failure test was initiated. All failure tests were conducted with a constant plate-plate elongation velocity of 2.0 mm/min, corresponding to 40% strain/min.
The microtensile mechanical rig was equipped with a 50-Newton load cell with 1% accuracy (Deben UK Ltd., Suffolk, United Kingdom), a specimen chamber, and two specimen clamps driven by a Maxon 118516 motor with a 241062 encoder fitted that achieved and measured changes in grip displacement.
The cross-sectional areas of the skin samples were calculated based on the measured width and thickness of the individual samples (width times thickness) on the assumption that the samples had a square cross-section. Sample stress was calculated as the tensile force (in Newtons) divided by the cross-sectional area (in square meters) of the samples and reported in megapascals. Peak stress was defined as the greatest magnitude of stress prior to tissue failure. All samples failed between the endplates of the mechanical rig, approximately in the middle of the specimen.
RESULTS
Generation of Dse knockout mice.
In order to functionally disrupt the Dse gene, a neomycin resistance cassette was inserted after the ATG translational start in exon 2 (Fig. 1A). The resulting 6.3-kb targeting vector was electroporated into 129/Sv ES cells. Of 62 ES cell clones screened for homologous recombination by Southern blotting, 10 contained the mutated allele (Fig. 1B). Three positive ES cell clones were injected into blastocysts, of which two yielded chimeric mice. Dse+/− mice were generated by mating a chimeric male with a C57BL/6 female. Breeding of the resulting heterozygous 129/Sv-C57BL/6 mice, i.e., with a mixed genetic background, resulted in Dse-deficient and wild-type littermates, which were analyzed in this report (Fig. 1C). The mice (n = 198) were 18% Dse−/−, 33% Dse+/+, and 49% Dse+/−.
Dse−/− mutant mice are smaller and have a kinked tail.
At birth, Dse−/− mutant mice were smaller, with a 20 to 30% reduced body weight compared to that of their wild-type littermates (Fig. 2A and B). All Dse−/− mutant pups had a tail with a kink that was not present after 4 weeks of age. At 80 days of age, the Dse-deficient mice remained 5 to 10% shorter (crown-to-rump length) and 10% lighter (Fig. 2B). The adrenal glands, brains, intestines, kidneys, lungs, and other major organs of 6-month-old Dse-deficient mice were necroscopically and histologically examined, and no gross changes were observed. Skin (see below) showed histological alterations. Dse−/− mutant mice had reduced fertility; intercrosses of Dse−/− mutant mice gave litters of 2 to 4 pups (9 litters), compared to the 8 to 12 pups obtained from heterozygous or wild-type parents. Six Dse−/− mutant mice were followed until 16 months of age and did not have increased mortality.
FIG. 2.

Macroscopic appearance of Dse−/− mutant mice. (A) Dse−/− mutant newborn. The arrow points to the kinked tail. (B) Eight wild-type and Dse−/− mutant males were weighed (± 2 standard deviations). Similarly, female Dse−/− mutant mice were smaller than wild-type (Wt) mice (data not shown). KO, knockout.
In Dse−/− mutant mice, DS-epi1 expression is abolished and epimerase activity is reduced.
DS-epi1 is highly expressed in the spleen. A qRT-PCR product of the expected size was obtained from wild-type spleen (CT = 27 or a ΔCT of 6.5 compared with the reference 18S rRNA gene) but was absent, even after 40 PCR cycles, in Dse−/− mutant mouse organs. Spleen was also analyzed for DS-epi1 protein expression by Western blotting. Analysis of 100 μg of spleen lysates revealed an ∼100-kDa band present in wild-type samples and absent in Dse−/− mutant samples (Fig. 3A). This band comigrated with the band derived from DS-epi1 overexpressed in 293HEK cells. To increase the sensitivity of the analysis, DS-epi1 was enriched from 5 mg of lysates by Red-Sepharose gel and Western blotted. The ∼100-kDa DS-epi1 band was reduced to about 50% in Dse+/− mutant compared to wild-type mouse spleens and was missing in Dse−/− spleens derived from two mice, each originating from two different ES cell clones. A less predominant band of ∼78 kDa was present only in the enriched sample from spleen and might represent a degradation product. Identical results were generated from lung tissue (data not shown). DS-epi1 expression was similarly analyzed in skin fibroblasts derived from adult wild-type and Dse−/− mutant mice (Fig. 3A). A single ∼100-kDa band was detected in wild-type cells but not in Dse−/− mutant cells. Similar results were obtained with adult lung fibroblasts and mouse embryonic fibroblasts derived from wild-type and Dse−/− mutant mice (data not shown). Epimerase activity was assayed in five organ lysates from 4-week-old mice, which showed greatly diversified epimerase specific activities: skin released 1,170 dpm of tritium from the substrate/h/mg of protein, spleen released 753, lung released 466, kidney released 127, and brain released 41. The Dse+/− mutant lysates contained activity that spanned 47 to 107% of that of the corresponding wild-type organs (Fig. 3B). Dse−/− mutant spleens and kidneys contained 4% of the activity of the wild-type samples, lung contained 18%, skin contained 35%, and brain contained 86% (Fig. 3B). The remaining activity is most likely catalyzed by DS-epi2. The epimerase specific activity was 2,074 dpm/h/mg in wild-type adult skin fibroblasts and decreased 35% in the corresponding Dse−/− mutant cells.
FIG. 3.

DS-epi1 expression and epimerase activity in Dse−/− mutant mice. (A) (Left gel) Spleen homogenates were directly Western blotted (100 μg protein applied), or DS-epi1 was enriched by Red-Sepharose, starting with 5 mg of initial lysates. (Center gel) Twenty micrograms each of lysates from control and DS-epi1-overexpressing 293HEK cells. (Right gel) Twenty micrograms each of lysates from adult skin primary fibroblast from Dse+/+ and Dse−/− mutant mice. Membranes were probed with anti-DS-epi1 antibody. (B) The same amount of protein from each organ, irrespective of the genotype of origin, was assayed for epimerase activity. Values (± 2 standard deviations of triplicates) are reported as percent epimerase specific activity referred to that of the wild-type organ. Empty bars, Dse+/−; black bars, Dse−/−. As an example, the wild-type skin homogenates released 3,067 dpm of tritium from the labeled substrate during 20 h of incubation.
Whole-body-derived Dse−/− mutant DS lacks iduronic acid blocks and has a reduced amount of iduronic acid-2-sulfated-GalNAc-4-sulfated structures.
To obtain a general picture of the alterations in CS/DS, GAG chains were 35S labeled in vivo and extracted from entire mice. Similar amounts of 35S radioactivity were recovered in the CS/DS chains from mice of all three genotypes. Moreover, the size distribution of the 35S-labeled CS/DS chains, as seen on gel permeation chromatography, was similar in the three genotypes (data not shown). Purified CS/DS chains were analyzed for the amount and distribution of iduronic acid by chondroitinases B and AC-I, which cleave GalNAc∼IdoA and GalNAc∼GlcA linkages, respectively. Digestion of wild-type chains by chondroitinase B yielded a disaccharide peak, derived from at least three contiguous IdoA-GalNac sequences in the native chain, amounting to 7.3% of the total radioactivity of the cleavage products (Fig. 4A). Wild-type tetrasaccharides, derived from ∼IdoA-GalNAc-GlcA-GalNAc∼IdoA sequences in the native chains, amounted to 11.5%. The corresponding sample from a Dse−/− mutant mouse showed 1% of the disaccharides (an approximately sevenfold reduction compared to the wild type) and 2.3% of the tetrasaccharides (an approximately fivefold reduction). The sum of hexa-, octa-, and decasaccharide fractions, coming from ∼IdoA-(GalNAc-GlcA)n 2-4-GalNAc∼IdoA sequences in the native chains, were a minor component of both the wild-type and Dse−/− mutant samples and were very similar, amounting to 7.2% and 6.9%, respectively. The cleavage product size distributions coming from wild-type and Dse+/− samples were almost superimposable. Disregarding the possible occurrence of nonsulfated disaccharide units, lacking radiolabel, calculations from the disaccharide until decasaccharide fractions resulted in total IdoA contents of 14.9% in wild-type chains, 16.8% in Dse+/− chains, and 3.4% in Dse−/− chains, which therefore have 23% remaining IdoA compared to wild-type chains (Fig. 4B). Digestion by chondroitinase AC-I leaves unaffected structures constituted by adjacent (IdoA-GalNAc)n units. Wild-type mice produced such structures with more than four adjacent units, eluting most of them in the void volume of the column (Fig. 4C). These structures, called iduronic acid blocks, amounted to 19% of the total radioactivity in Dse+/+ chains, decreased to 15.1% in Dse+/− chains, and were missing in Dse−/− chains.
FIG. 4.

Analysis of Dse−/− mutant CS/DS present in the whole body. Ten-day-old pups were 35S labeled in vivo, and CS/DS was purified from their whole bodies. Chains were cleaved with chondroitinase B (A) or chondroitinase AC-I (C). Cleavage products were separated by size permeation chromatography on a Superdex peptide column. Elution positions of di-, tetra-, hexa-, octa-, and decasaccharides are indicated by arrows. AC-I-resistant iduronic acid blocks are shown. Squares, Dse+/−; triangles, Dse+/+; circles, Dse−/−. (B) Proportions of iduronic acid residues in the CS/DS chains, as calculated from panel A.
Dse+/+ and Dse−/− CS/DS chains were also cleaved by chondroitinase ABC, and the resulting disaccharides were separated by high-performance liquid chromatography. The iduronic-2-sulfated-GalNAc-4-sulfated structures, recovered as ΔB after the analysis, were reduced by 85% in Dse−/− chains (Table 1) .
TABLE 1.
Disaccharide composition of Dse+/+ and Dse−/− CS/DS chainsa
| Unit | Whole-body total CS/DSb
|
Skin decorin/ biglycan CS/DSc
|
||
|---|---|---|---|---|
| Dse+/+ | Dse−/− | Dse+/+ | Dse−/− | |
| ΔO, ΔHexA-GalNAc | NAd | NA | 1.0 | 1.3 |
| ΔA, ΔHexA-GalNAc4S | 86.6 | 86.6 | 89.5 | 95.1 |
| ΔC, ΔHexA-GalNAc6S | 7.6 | 10.1 | 0.6 | 0.8 |
| ΔB, ΔHexA2S-GalNAc4S | 3.4 | 0.5 | 9.0 | 2.7 |
| ΔE, ΔHexA-GalNAc4S6S | 1.7 | 1.7 | NDe | ND |
| ΔD, ΔHexA2S-GalNAc6S | 0.7 | 1.1 | ND | ND |
CS/DS preparations were digested with chondroitinase ABC, and the resulting disaccharides were analyzed by anion-exchange high-pressure liquid chromatography. Values are in moles percent.
In vivo-labeled CS/DS chains were prepared from whole mice as described in Materials and Methods.
Unlabeled CS/DS chains were prepared from skin decorin/biglycan from 4-month-old mice. The disaccharides obtained after chondroitinase ABC digestion were labeled with the reducing reagent NaB3H4.
NA, not applicable.
ND, none detected (less than 0.1%).
Skin decorin/biglycan- and versican-CS/DS Dse−/− chains contain less iduronic acid blocks and iduronic acid-2-sulfated-GalNAc-4-sulfated structures.
35S-sulfate in vivo-labeled versican and a mixture of decorin and biglycan were purified, and their CS/DS chains were recovered. Versican-derived and decorin/biglycan-derived chains were comparable in size, as shown by gel filtration, irrespective of the different genotypes (Fig. 5A). Chondroitinase B or AC-I digestion made it possible to determine the amount and distribution of iduronic acid along the chain, as outlined above. In skin, the decorin/biglycan-CS/DS iduronic acid content decreased from 67.1% in the wild type to 17.9% in the Dse−/− mutant, as calculated from the chondroitinase B cleavage product distribution (Fig. 5B). The entire decrease in iduronic acid content could be attributed to the 90% decrease in the iduronic acid blocks, which accounted for 58.2% of the total chain in the wild type and 5.6% in the Dse−/− mutant (Fig. 5C). Skin versican CS/DS wild-type chains contained 13.3% iduronic acid, which decreased to 1.4% in Dse−/− mutant chains (Fig. 5D). Iduronic acid blocks were 7.5% of the total wild-type chain and 0.6% of the Dse−/− chains (Fig. 5E).
FIG. 5.

Analysis of Dse−/− mutant skin decorin/biglycan- and versican-CS/DS chains. Ten-day-old pups were 35S labeled in vivo. (A) Skin decorin/biglycan and versican were isolated, and their CS/DS chains were analyzed by size permeation chromatography on a Superose 6 column run in 0.2 M NH4HCO3. Heparins of different molecular weights were used as markers. Filled triangles, wild-type decorin/biglycan chains; empty triangles, Dse−/− mutant decorin/biglycan chains; filled squares, wild-type versican chains; empty squares, Dse−/− mutant versican chains. (B to E) CS/DS chains were cleaved by chondroitinases B and AC-I as indicated. Cleavage products were separated as described in the legend to Fig. 4. Squares, Dse+/+; triangles, Dse+/−; circles, Dse−/−. (F) Wild-type decorin/biglycan-derived iduronic acid blocks, as obtained after chondroitinase AC-I cleavage and pooled as indicated in panel C, were analyzed by size permeation chromatography on a Superose 6 column run in 1 M NaCl.
Size distribution of decorin/biglycan-derived Dse+/+ iduronic acid blocks, obtained after chondroitinase AC-I digestion, was analyzed, and the modal molecular mass was 15 kDa, corresponding to approximately 30 disaccharide residues (Fig. 5F), as long as the intact, i.e., uncleaved, chain (Fig. 5A).
Furthermore, the iduronic-2-sulfated-GalNAc-4-sulfated structures were reduced by 70% in Dse−/− decorin/biglycan chains (Table 1).
DS-epi1 deficiency alters skin morphology, collagen fibril ultrastructure, and skin tensile strength.
Skin samples obtained from five Dse−/− mutant mice contained sparser loose connective tissue in the hypodermal layer (Fig. 6A to D). These structural changes did not alter the morphology or the numbers of hair follicles (wild type, 33 ± 10 hair follicles/tail cross section [n = 7]; Dse−/− mutant, 29.5 ± 4.5 [n = 8]). The collagen content, as determined by hydroxyproline, was not significantly different in the skin of wild-type (20.8 ng hydroxyproline/mg [wet weight], n = 4, range = 16.8 to 25.3) and Dse-deficient mice (18.6 ng hydroxyproline/mg [wet weight], n = 4, range = 17.3 to 20.8). Immunostaining of decorin showed a similar staining pattern and indicated that the distribution and amount of decorin are similar in wild-type and Dse-deficient skin (Fig. 6E to G). Similarly, staining with an antibiglycan antiserum showed no change in the distribution or amount of biglycan (data not shown).
FIG. 6.

Skin morphology and immunohistochemistry. Shown is tail skin (A and B) stained with Chromotrope 2R and visualized with a light microscope or (C and D) stained with Sirius Red and visualized with polarized light. Sparser loose connective tissue appears in the hypodermis of Dse−/− mutant skin (arrow). Bars, 100 μm. D, dermis; E, epidermis; H, hair follicles; T, tendons. (E and F) Histoimmunological detection of decorin. (G) Preimmune serum control.
TEM pictures were taken at different levels of the dermis and hypodermis (Fig. 7A). Analysis of the skin collagen fibril ultrastructure by TEM showed a shift toward thicker fibrils in Dse−/− mutant mouse skin. The mean fibril diameter shifted from 60 nm in wild-type skin to 85 nm in Dse−/− mutant skin. The differences in fibril diameter remain in the dermal and hypodermal areas. In some Dse−/− hypodermal areas, collagen fibrils were absent. This agrees with the observations by light microscopy (Fig. 6). On the other hand, Dse−/− tail tendons showed a minor shift toward thicker fibrils compared to wild-type tendons, and most of the fibrils had the same diameter in both genotypes (Fig. 7B). There were no differences in collagen fibril diameter or morphology in the Achilles tendon (Fig. 7C). Fibril density did not change between the two genotypes (Table 2).
FIG. 7.

Ultrastructural analysis of collagen fibrils in skin, tail tendon, and Achilles tendon. (Right side of panels A to C) Skin (A), tail tendon (B), and Achilles tendon (C) samples were examined by electron microscopy. (Left side of panels A to C) The diameters of 1,000 fibrils from each source were measured, and the results are presented in histograms. Solid bars, wild type; open bars, Dse−/− mutant. (D) qRT-PCR of Dse mRNAs from different wild-type tissues. Values (± 2 standard deviations of triplicates) are normalized to spleen mRNA.
TABLE 2.
Fibril density calculated from TEM images
| Tissue | Avg fibril densitya ± SE |
|---|---|
| Wild-type dermis | 72 ± 5 |
| Dse−/− mutant dermis | 68 ± 7 |
| Wild-type hypodermis | 62 ± 5 |
| Dse−/− mutant hypodermis | 60 ± 8 |
| Wild-type tail tendon | 75 ± 7 |
| Dse−/− mutant tail tendon | 88 ± 8 |
| Wild-type Achilles tendon | 74 ± 8 |
| Dse−/− mutant Achilles tendon | 77 ± 10 |
Percentage of area occupied by fibrils in TEM images.
We compared the relative amount of Dse mRNA in wild-type tissues by qRT-PCR. Dse mRNA was more abundant in skin than in the tendons, with spleen and lung having the greatest amounts (Fig. 7D).
The altered collagen fibrils in Dse−/− mutant skin were accompanied by a decrease in mechanical strength (Fig. 8). The stress at failure was 5.86 ± 1.88 MPa (n = 5) in wild-type skin and 3.48 ± 1.12 MPa (n = 5) in Dse−/− mutant skin, which is a 41% reduction.
FIG. 8.
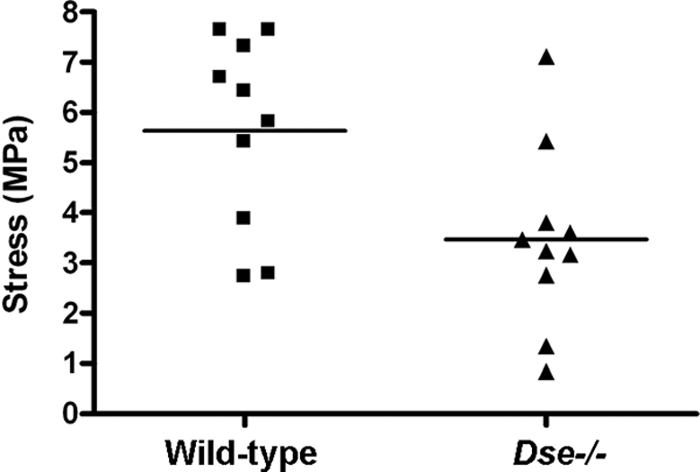
Biomechanics of skin. Stress at failure was measured by applying force to skin samples and measuring the stress at which the skin ruptured. Duplicate samples from five mice of each genotype were measured. Bars indicate mean values; P = 0.018.
Skin decorin and biglycan from wild-type mice and Dse−/− mutant mice have differential effects on collagen fibril formation in vitro.
To compare the biophysical properties of skin collagen from wild-type and Dse−/− mutant mice, collagen denaturation was analyzed in a differential scanning calorimeter. Skin from Dse−/− mutant mice denatures more heterogeneously, having two denaturation peaks at 60 and 62°C, compared to wild-type skin which has only the 60°C peak (Fig. 9A).
Decorin and biglycan are prominent CS/DS PGs in skin and have been shown to participate in collagen fibril formation (1). Decorin and biglycan were extracted from the skin of wild-type and Dse−/− mutant adult mice with similar yields and obtained as a mixture containing 90 to 95% decorin. Both PGs migrated in a similar manner on SDS-PAGE (Fig. 9B), irrespective of the genotype. Similar amounts of decorin (Fig. 9C) and biglycan (data not shown) were present in the skin of the two genotypes. Acid-solubilized collagen was incubated in the presence of extracted decorin/biglycan preparations from the different genotypes. Differential scanning calorimeter analysis of the resulting collagen fibrils, which melted at around 50°C, showed a two-peak fibril denaturation pattern in the presence of Dse−/− mutant decorin/biglycan and a single peak after the addition of wild-type decorin/biglycan (Fig. 9D), similar to the results obtained with intact skin. The denaturation of collagen monomers, occurring at 42°C, was unaffected. We also investigated the kinetics of in vitro collagen fibril formation after addition of the decorin/biglycan preparations, by turbidimetry. In this assay, decorins isolated from the skin of three wild-type mice all inhibited the formation of collagen fibrils (Fig. 9E). In contrast, decorins and biglycans isolated from the skin of three Dse−/− mutant mice stimulated fibril formation. After removal of the CS/DS chains by chondroitinase ABC, all of the decorin/biglycan preparations inhibited fibril formation, regardless of the genotype. These results demonstrate that the iduronic acid blocks in CS/DS chains carried by decorin/biglycan have a direct effect on collagen fibril formation.
DISCUSSION
Biosynthesis of CS/DS is a multistep process requiring at least 22 enzymes directly involved in the assembly and modification of the chain, starting from the activated monosaccharides and the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate. Multienzyme complexes are thought to accomplish these coordinated and fast reactions. The final structure of CS/DS is subject to different levels of regulation in vivo, which is still poorly understood despite all of the enzymes having been cloned and studied in vitro. DS-epi1 is one of the two DS epimerases that convert glucuronic acid to iduronic acid (23, 34). An in vivo model with ablated DS-epi1 can provide new insights into the biological roles of specific CS/DS structures and the regulation of the biosynthetic machinery.
DS-epi1 mRNA and protein were not detectable in Dse−/− mutant mice. In adult animals, DS-epi1 is the enzyme responsible for the majority of the epimerase activity in the spleen, kidneys, lungs, and skin. The remaining activity in these tissues can most likely be attributed to DS-epi2. Adult brain, where the activity of DS-epi2 is predominant, is an exception. This is in agreement with data on mRNA expression in brain tissue showing the highest expression of DS-epi2 and the lowest expression of DS-epi1 (13, 27). DS-epi1 deficiency results in a 77% reduction of iduronic acid in newly synthesized CS/DS by Dse−/− mutant mice. Skin decorin/biglycan and versican contain 67% and 13% iduronic acid in their wild-type chains, respectively; they were similarly affected by ablation of DS-epi1, which led to 73% and 89% reductions in iduronic acid, respectively. Clearly, DS-epi1 plays a role in the modification of both PGs, at least in skin. In wild-type skin decorin/biglycan, the iduronic acid blocks, with a modal molecular mass of 15 kDa, can make up almost the entire chain. In Dse−/− mutant mouse skin, only 10% of the iduronic acid blocks remain in decorin/biglycan. On the other hand, isolated iduronic acid residues, i.e., surrounded by glucuronic acid, are not affected in skin decorin/biglycan. On the contrary, in skin versican and whole-body CS/DS, the latter sample probably being predominantly composed of versican-type chains, both isolated iduronic acid and iduronic acid blocks are downregulated. Our report is the first showing that versican contains iduronic acid blocks, at least in skin.
Interestingly, Dse+/− mice have approximately half the amount of the enzyme but still produce chains which are almost identical to the wild-type chains. Our data also show that decreased epimerization does not affect the length of the CS/DS chains, in contrast to the effect of CS4ST1 ablation (50). Disaccharide fingerprinting of CS/DS chains showed that, as a consequence of the reduction of the iduronic acid blocks, the disulfated structures in the native chains (IdoA-2OS-GalNAC-4OS)n are also greatly reduced, both in whole-body CS/DS and in skin decorin/biglycan. Our results support previous findings that locate the position of these disulfated structures within the iduronic acid blocks (17, 24). In summary, DS-epi1 is the main, but not the only, enzyme that synthesizes iduronic acid blocks in vivo. The data presented here confirm the results obtained with lung fibroblasts (34) and further suggest that the difference in the levels of expression of the two epimerases in the different tissues, together with the expression of dermatan 4-O-sulfotransferase 1 (35), is a primary determinant of the amount and distribution of iduronic acid.
Dse−/− mutant mice in a mixed 129/Sv-C57BL/6 genetic background were vital, without any gross organ alteration. This is different from the perinatal mortality of HS epimerase-deficient mice, which lack kidneys and have altered lungs (21). This probably reflects a different role for iduronic acid in HS and CS/DS in embryo development and the fact that only one epimerase is present in HS biosynthesis. Dse−/− mutant mice were smaller at birth but grew normally. We can speculate that the maternal/fetal placenta—an organ rich in DS and decorin (12)—might have a heparin cofactor II-mediated thrombotic imbalance and/or an altered extracellular matrix and therefore could play a role in the retardation of intrauterine growth. An indication of a combined maternal and fetal contribution to the severity of the intrauterine phenotype is the reduced litter size generated by knockout parents.
Many organs might be affected under close examination by the deficiency of DS-epi1, which is ubiquitously expressed, with the possible exception of the brain, where DS-epi2 is the predominant epimerase. Therefore, it will be interesting to study if organs rich in DS-epi1, like the spleen and kidney, despite a normal morphological appearance, present some functional defects. We initially analyzed the skin of Dse-deficient mice since this tissue showed histological changes under light microscopy and has a high content of DS. Dse−/− mutant skin has a histological appearance different from that of wild-type skin, and the hypodermal region contains sparse connective tissue. This resembles the skin from decorin-deficient mice (see Fig. 3C and D in reference 7). The skin morphology did not lead to any alterations in the amount of collagen, as determined by hydroxyproline content. Light microscopic investigation of Dse-deficient tail and Achilles tendons revealed no morphological abnormalities. Collagen fibrils in the skin of Dse−/− mutant mice are thicker than those in wild-type mouse skin. Decorin is an abundant CS/DS PG in skin, binds to collagen, and affects collagen fibril formation in vitro (38). Decorin (Dcn)- and Dse-deficient mice both have changes in skin collagen fibril structure (7). In Dcn-deficient skin, the fibrils show irregular outlines and a larger variation in diameter than wild-type skin fibrils. The mean fibril diameters in Dcn-deficient skin and wild-type skin are similar, whereas Dse-deficient skin has collagen fibrils with an increased mean diameter of 85 nm, compared to 60 nm in the wild type. Dcn-deficient tail tendons have abnormal collagen fibrils, some larger than 600 nm, with multiple lateral fusions. In contrast, Dse-deficient tail and Achilles tendons were largely unaffected. The lack of a tendon phenotype is likely due to the measured lower level of DS-epi1 mRNA in tendons compared to that in skin and to a lower content of iduronic acid in tendons (4). The skin of Dcn- and Dse-deficient mice has reduced tensile strength, as a possible result of altered collagen fibril assembly. The stress at failure was reduced by 40% and 70%, compared to the wild type, in the skin of Dse- and Dcn-deficient mice (7), respectively. In summary, Dcn−/− skin and Dse−/− skin have overlapping but not identical alterations in collagen structure. This can be explained by the total ablation of the PG in Dcn−/− mutant mice, as opposed to the ablation of some of its functional components in Dse−/− mutant mice. The Dse-deficient skin phenotype was not the result of an altered level of decorin or biglycan expression. In fact, the amount of decorin and biglycan in skin, as determined by Western blotting, immunohistochemistry, and chemical quantification after extraction, indicated no change in the amount due to Dse ablation. This is different from the functionally defective enzyme galactosyltransferase I that is responsible for a variant of the Ehlers-Danlos syndrome, which leads to an altered amount of decorin, both as a PG and as a nonglycanated core protein (42). Our results therefore suggest that the iduronic acid blocks carried by decorin chain have a role in determining the structure and mechanical properties of collagen. We obtained further support by using preparations containing predominantly decorin and minor amount of biglycan, isolated from wild-type and Dse-deficient mouse skin. The denaturation of collagen fibrils preformed in the presence of Dse−/− PGs showed two closely spaced melting temperatures in differential scanning calorimetry, while wild-type PGs had a single melting temperature. These patterns of denaturation were reproduced with intact skin from wild-type and Dse−/− mutant mice and indicate that the iduronic acid blocks of DS affect the biophysical property of collagen fibrils. We also used decorin/biglycan from the two genotypes in turbidimetric measurements of collagen fibril formation. PGs from wild-type skin inhibited fibril formation, whereas Dse−/− PGs promoted fibrillogenesis. These results suggest that IdoA blocks have a role in the fibril formation process and agree with previously reported roles of the DS chain (39).
The interaction between antiparallel CS/DS chains of two decorin molecules has been proposed to contribute to the lateral growth of collagen fibrils (40). In a skin healing model and in skin development from the embryonic stage to adulthood, longer decorin chains correlate with wider interfibrillar spacing (19, 30). In the mentioned skin healing model, the longer chains contained fewer (IdoA-2OS-GalNAC-4OS)n structures and therefore presumably fewer iduronic acid blocks (20), and thinner fibrils were present. Therefore, the structural changes described in our study in skin decorin/biglycan CS/DS chains, which contain less iduronic acid but have similar length, are different from the changes described during development and healing.
Understanding the role of the CD/DS chains is complicated by their multiple potential interactions. In Dse−/− mutant mice, an altered amount and/or distribution of iduronic acid-containing structures, thought to be important for CS/DS-CS/DS interaction (10), might lead to a modified affinity of these CS/DS self-interactions. This, in turn, could explain the changes in the fibril structures. CS/DS chains have been shown to contribute to the binding of decorin to collagen I (29). Iduronic acid affects CS/DS chain flexibility, which often favors protein binding (8). Dse−/− chains might have an altered conformation affecting collagen I/decorin binding and ultimately the growth of the fibrils. Alternatively, fibril surface-associated proteins like FACIT-collagen XIV (2, 9) might bind not optimally via decorin CS/DS to the growing fibril, affecting its structure. Finally, the behavior of skin cells, for instance, fibroblasts, could be modified by altered DS chains.
In conclusion, our results indicate that decorin/biglycan iduronic acid blocks have a role in skin collagen fibril formation and in the biophysical characteristics and mechanical properties of skin.
Acknowledgments
This work was supported by grants from the Swedish Science Research Council, the Medical Faculty of Lund University, the Albert Österlund Foundation, the Greta and Johan Kock Foundation, Polysackaridforskning AB, and the Tissue in Motion Medical Faculty Program.
We are grateful to Ragnar Matsson (Lund Transgenic Core Facility) for help in generating the knockout mice and for precious advice and to Ricardo Feinstein (National Veterinary Institute, Uppsala, Sweden) for help in histological examinations.
Footnotes
Published ahead of print on 17 August 2009.
REFERENCES
- 1.Ameye, L., and M. F. Young. 2002. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology 12:107R-116R. [DOI] [PubMed] [Google Scholar]
- 2.Ansorge, H. L., X. Meng, G. Zhang, G. Veit, M. Sun, J. F. Klement, D. P. Beason, L. J. Soslowsky, M. Koch, and D. E. Birk. 2009. Type XIV collagen regulates fibrillogenesis: premature collagen fibril growth and tissue dysfunction in null mice. J. Biol. Chem. 284:8427-8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao, X., T. Muramatsu, and K. Sugahara. 2005. Demonstration of the pleiotrophin-binding oligosaccharide sequences isolated from chondroitin sulfate/dermatan sulfate hybrid chains of embryonic pig brains. J. Biol. Chem. 280:35318-35328. [DOI] [PubMed] [Google Scholar]
- 4.Cheng, F., D. Heinegard, A. Malmström, A. Schmidtchen, K. Yoshida, and L.-Å. Fransson. 1994. Patterns of uronosyl epimerization and 4-/6-O-sulphation in chondroitin/dermatan sulphate from decorin and biglycan of various bovine tissues. Glycobiology 4:685-696. [DOI] [PubMed] [Google Scholar]
- 5.Choi, H. U., T. L. Johnson, S. Pal, L. H. Tang, L. Rosenberg, and P. J. Neame. 1989. Characterization of the dermatan sulfate proteoglycans, DS-PGI and DS-PGII, from bovine articular cartilage and skin isolated by octyl-Sepharose chromatography. J. Biol. Chem. 264:2876-2884. [PubMed] [Google Scholar]
- 6.Cöster, L., L.-Å. Fransson, J. Sheehan, I. A. Nieduszynski, and C. F. Phelps. 1981. Self-association of dermatan sulphate proteoglycans from bovine sclera. Biochem. J. 197:483-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danielson, K. G., H. Baribault, D. F. Holmes, H. Graham, K. E. Kadler, and R. V. Iozzo. 1997. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 136:729-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferro, D. R., A. Provasoli, M. Ragazzi, B. Casu, G. Torri, V. Bossennec, B. Perly, P. Sinay, M. Petitou, and J. Choay. 1990. Conformer populations of l-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 195:157-167. [DOI] [PubMed] [Google Scholar]
- 9.Font, B., E. Aubert-Foucher, D. Goldschmidt, D. Eichenberger, and M. van der Rest. 1993. Binding of collagen XIV with the dermatan sulfate side chain of decorin. J. Biol. Chem. 268:25015-25018. [PubMed] [Google Scholar]
- 10.Fransson, L.-Å., L. Cöster, A. Malmström, and J. K. Sheehan. 1982. Self-association of scleral proteodermatan sulfate. Evidence for interaction via the dermatan sulfate side chains. J. Biol. Chem. 257:6333-6338. [PubMed] [Google Scholar]
- 11.Fransson, L.-Å., and L. Roden. 1967. Structure of dermatan sulfate. II. Characterization of products obtained by hyaluronidase digestion of dermatan sulfate. J. Biol. Chem. 242:4170-4175. [PubMed] [Google Scholar]
- 12.Giri, T. K., and D. M. Tollefsen. 2006. Placental dermatan sulfate: isolation, anticoagulant activity, and association with heparin cofactor II. Blood 107:2753-2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goossens, D., S. Van Gestel, S. Claes, P. De Rijk, D. Souery, I. Massat, D. Van den Bossche, H. Backhovens, J. Mendlewicz, C. Van Broeckhoven, and J. Del Favero. 2003. A novel CpG-associated brain-expressed candidate gene for chromosome 18q-linked bipolar disorder. Mol. Psychiatry 8:83-89. [DOI] [PubMed] [Google Scholar]
- 14.Hannesson, H. H., A. Hagner-McWhirter, K. Tiedemann, U. Lindahl, and A. Malmström. 1996. Biosynthesis of dermatan sulphate. Defructosylated Escherichia coli K4 capsular polysaccharide as a substrate for the d-glucuronyl C-5 epimerase, and an indication of a two-base reaction mechanism. Biochem. J. 313(Pt. 2):589-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iozzo, R. V. 1999. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J. Biol. Chem. 274:18843-18846. [DOI] [PubMed] [Google Scholar]
- 16.Kalamajski, S., A. Aspberg, and A. Oldberg. 2007. The decorin sequence SYIRIADTNIT binds collagen type I. J. Biol. Chem. 282:16062-16067. [DOI] [PubMed] [Google Scholar]
- 17.Karamanos, N. K., P. Vanky, A. Syrokou, and A. Hjerpe. 1995. Identity of dermatan and chondroitin sequences in dermatan sulfate chains determined by using fragmentation with chondroitinases and ion-pair high-performance liquid chromatography. Anal. Biochem. 225:220-230. [DOI] [PubMed] [Google Scholar]
- 18.Kusche-Gullberg, M., and L. Kjellen. 2003. Sulfotransferases in glycosaminoglycan biosynthesis. Curr. Opin. Struct. Biol. 13:605-611. [DOI] [PubMed] [Google Scholar]
- 19.Kuwaba, K., M. Kobayashi, Y. Nomura, S. Irie, and Y. Koyama. 2001. Elongated dermatan sulphate in post-inflammatory healing skin distributes among collagen fibrils separated by enlarged interfibrillar gaps. Biochem. J. 358:157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwaba, K., Y. Nomura, S. Irie, and Y. Koyama. 1999. Temporal changes in disaccharide composition of dermatan sulfate in the skin after epicutaneous application of hapten. J. Dermatol. Sci. 19:23-30. [DOI] [PubMed] [Google Scholar]
- 21.Li, J. P., F. Gong, A. Hagner-McWhirter, E. Forsberg, M. Abrink, R. Kisilevsky, X. Zhang, and U. Lindahl. 2003. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking l-iduronic acid and in neonatal lethality. J. Biol. Chem. 278:28363-28366. [DOI] [PubMed] [Google Scholar]
- 22.Liu, X., M. L. Yeh, J. L. Lewis, and Z. P. Luo. 2005. Direct measurement of the rupture force of single pair of decorin interactions. Biochem. Biophys. Res. Commun. 338:1342-1345. [DOI] [PubMed] [Google Scholar]
- 23.Maccarana, M., B. Olander, J. Malmström, K. Tiedemann, R. Aebersold, U. Lindahl, J. P. Li, and A. Malmström. 2006. Biosynthesis of dermatan sulfate: chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 281:11560-11568. [DOI] [PubMed] [Google Scholar]
- 24.Maimone, M. M., and D. M. Tollefsen. 1991. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J. Biol. Chem. 266:14830. [PubMed] [Google Scholar]
- 25.Malmström, A., and L.-Å. Fransson. 1975. Biosynthesis of dermatan sulfate. I. Formation of l-iduronic acid residues. J. Biol. Chem. 250:3419-3425. [PubMed] [Google Scholar]
- 26.McBurney, M. W., L. C. Sutherland, C. N. Adra, B. Leclair, M. A. Rudnicki, and K. Jardine. 1991. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 19:5755-5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakao, M., S. Shichijo, T. Imaizumi, Y. Inoue, K. Matsunaga, A. Yamada, M. Kikuchi, N. Tsuda, K. Ohta, S. Takamori, H. Yamana, H. Fujita, and K. Itoh. 2000. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J. Immunol. 164:2565-2574. [DOI] [PubMed] [Google Scholar]
- 28.Nandini, C. D., and K. Sugahara. 2006. Role of the sulfation pattern of chondroitin sulfate in its biological activities and in the binding of growth factors. Adv. Pharmacol. 53:253-279. [DOI] [PubMed] [Google Scholar]
- 29.Nareyeck, G., D. G. Seidler, D. Troyer, J. Rauterberg, H. Kresse, and E. Schonherr. 2004. Differential interactions of decorin and decorin mutants with type I and type VI collagens. Eur. J. Biochem. 271:3389-3398. [DOI] [PubMed] [Google Scholar]
- 30.Nomura, Y. 2006. Structural change in decorin with skin aging. Connect. Tissue Res. 47:249-255. [DOI] [PubMed] [Google Scholar]
- 31.Obrink, B. 1973. A study of the interactions between monomeric tropocollagen and glycosaminoglycans. Eur. J. Biochem. 33:387-400. [DOI] [PubMed] [Google Scholar]
- 32.Oldberg, A., S. Kalamajski, A. V. Salnikov, L. Stuhr, M. Morgelin, R. K. Reed, N. E. Heldin, and K. Rubin. 2007. Collagen-binding proteoglycan fibromodulin can determine stroma matrix structure and fluid balance in experimental carcinoma. Proc. Natl. Acad. Sci. USA 104:13966-13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osoegawa, K., M. Tateno, P. Y. Woon, E. Frengen, A. G. Mammoser, J. J. Catanese, Y. Hayashizaki, and P. J. de Jong. 2000. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 10:116-128. [PMC free article] [PubMed] [Google Scholar]
- 34.Pacheco, B., A. Malmström, and M. Maccarana. 2009. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 284:9788-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pacheco, B., A. Malmström, and M. Maccarana. 6 August 2009, posting date. Dermatan 4-O-sulfotransferase 1 is pivotal in the formation of iduronic acid blocks in dermatan sulfate. Glycobiology doi: 10.1093/glycob/cwp110. [DOI] [PubMed]
- 36.Prabhakar, V., and R. Sasisekharan. 2006. The biosynthesis and catabolism of galactosaminoglycans. Adv. Pharmacol. 53:69-115. [DOI] [PubMed] [Google Scholar]
- 37.Rauch, U., and J. Kappler. 2006. Chondroitin/dermatan sulfates in the central nervous system: their structures and functions in health and disease. Adv. Pharmacol. 53:337-356. [DOI] [PubMed] [Google Scholar]
- 38.Reed, C. C., and R. V. Iozzo. 2002. The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj. J. 19:249-255. [DOI] [PubMed] [Google Scholar]
- 39.Rühland, C., E. Schonherr, H. Robenek, U. Hansen, R. V. Iozzo, P. Bruckner, and D. G. Seidler. 2007. The glycosaminoglycan chain of decorin plays an important role in collagen fibril formation at the early stages of fibrillogenesis. FEBS J. 274:4246-4255. [DOI] [PubMed] [Google Scholar]
- 40.Scott, J. E., and A. M. Thomlinson. 1998. The structure of interfibrillar proteoglycan bridges (′shape modules') in extracellular matrix of fibrous connective tissues and their stability in various chemical environments. J. Anat. 192(Pt. 3):391-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seidler, D. G., E. Breuer, K. J. Grande-Allen, V. C. Hascall, and H. Kresse. 2002. Core protein dependence of epimerization of glucuronosyl residues in galactosaminoglycans. J. Biol. Chem. 277:42409-42416. [DOI] [PubMed] [Google Scholar]
- 42.Seidler, D. G., M. Faiyaz-Ul-Haque, U. Hansen, G. W. Yip, S. H. Zaidi, A. S. Teebi, L. Kiesel, and M. Gotte. 2006. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (β4GalT-7). J. Mol. Med. 84:583-594. [DOI] [PubMed] [Google Scholar]
- 43.Shively, J. E., and H. E. Conrad. 1976. Formation of anhydrosugars in the chemical depolymerization of heparin. Biochemistry 15:3932-3942. [DOI] [PubMed] [Google Scholar]
- 44.Sugahara, K., and T. Mikami. 2007. Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 17:536-545. [DOI] [PubMed] [Google Scholar]
- 45.Svensson, L., A. Aszodi, F. P. Reinholt, R. Fassler, D. Heinegard, and A. Oldberg. 1999. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J. Biol. Chem. 274:9636-9647. [DOI] [PubMed] [Google Scholar]
- 46.Taylor, K. R., J. A. Rudisill, and R. L. Gallo. 2005. Structural and sequence motifs in dermatan sulfate for promoting fibroblast growth factor-2 (FGF-2) and FGF-7 activity. J. Biol. Chem. 280:5300-5306. [DOI] [PubMed] [Google Scholar]
- 47.Tiedemann, K., T. Larsson, D. Heinegard, and A. Malmström. 2001. The glucuronyl C5-epimerase activity is the limiting factor in the dermatan sulfate biosynthesis. Arch. Biochem. Biophys. 391:65-71. [DOI] [PubMed] [Google Scholar]
- 48.Tollefsen, D. M. 2007. Heparin cofactor II modulates the response to vascular injury. Arterioscler. Thromb. Vasc. Biol. 27:454-460. [DOI] [PubMed] [Google Scholar]
- 49.Trowbridge, J. M., and R. L. Gallo. 2002. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology 12:117R-125R. [DOI] [PubMed] [Google Scholar]
- 50.Uyama, T., M. Ishida, T. Izumikawa, E. Trybala, F. Tufaro, T. Bergstrom, K. Sugahara, and H. Kitagawa. 2006. Chondroitin 4-O-sulfotransferase-1 regulates E disaccharide expression of chondroitin sulfate required for herpes simplex virus infectivity. J. Biol. Chem. 281:38668-38674. [DOI] [PubMed] [Google Scholar]