

Abstract
General base catalysis supplied by the histidine-12 (H-12) residue of ribonuclease (RNase) A has long been appreciated as a major component of the catalytic power of the enzyme. In an attempt to harness the catalytic power of a general base into antibody catalysis of phosphodiester bond hydrolysis, the quaternary ammonium phosphate 1 was used as a bait and switch hapten. Based on precedence, it was rationalized that this positively charged hapten could induce a counter-charged residue in the antibody binding site at a locus suitable for it to deprotonate the 2′-hydroxyl group of the anhydroribitol phosphodiester substrate 2. After murine immunization with hapten 1, mAb production yielded a library of 35 antibodies that bound to a BSA-1 conjugate. From this panel, two were found to catalyze the cyclization-cleavage of phosphodiester 2. Kinetic studies at pH 7.49 (Hepes, 20 mM) and 25°C showed that the most active antibody, MATT.F-1, obeyed classical Michaelis–Menten kinetics with a Km = 104 μM, a kcat = 0.44 min−1, and a kcat/kuncat = 1.7 × 103. Hapten 1 stoichiometrically inhibits the catalytic activity of the antibody. MATT.F-1 is the most proficient antibody–catalyst (1.6 × 107 M−1) yet generated for the function of phosphodiester hydrolysis and emphasizes the utility of the bait and switch hapten paradigm when generating antibody catalysts for processes for which general-base catalysis can be exploited.
The hydrolysis of phosphodiester bonds, such as those found in DNA and RNA, is a reaction of fundamental importance in living systems. Consequently, there are intense efforts centered around the development of novel phosphodiesterases for use in biochemistry and medicine (1–5). Our approach to this area has been to exploit the diversity of the murine immune system (6, 7) to generate antibodies possessing phosphodiesterase activity. Preliminary success in this area was based on our work with oxotechnetium(v) and oxorhenium(V) complexes that inhibit ribonuclease (RNase) U2 (EC 3.1.27.4) activity (8–10). The oxorhenium(v) hapten 3, a putative transition state analog for the cyclization–cleavage of oligonucleotides, generated a catalytic antibody, 2G12, that catalyzes the hydrolysis of uridine 3′-(p-nitrophenyl) phosphate (UpOC6H4-p-NO2) 4 (11) (Fig. 1).
Figure 1.

The bait and switch hapten 1 used to generate antibodies for the catalysis of the cyclization–cleavage of anhydroribitol phosphosphodiester 2. Below are juxtaposed the oxorhenium(v) metallochelate 3 used as a transition state analog (TSA) hapten to elicit antibodies that catalyzed the same cyclization cleavage of phosphodiester 4 (11).
The most studied of the natural ribonucleases is bovine pancreatic RNase A (EC 3.1.27.5) (12–14). X-ray diffraction (15), chemical modification (16), and pH-rate studies (17) are all consistent with an enzymatic reaction mechanism in which the rate-limiting transition state for RNA cleavage is similar to that shown in Fig. 2.
Figure 2.

The classical structure assigned to the transition state for RNase A-catalyzed hydrolysis of RNA.
Although this mechanism has stimulated much speculation in the fields of bioorganic chemistry and enzymology (18–22), it does seem clear that the imidazole group of histidine 12 (H-12) acts as a general base by deprotonating the 2′ oxygen and that the imidazolium group of histidine 119 (H-119) acts as a general acid by either protonating the 5′′ oxygen of the leaving group (classical mechanism) or a nonbridging phosphoryl oxygen (triester-like mechanism). Furthermore, Raines (23) has shown by mutagenesis studies that the imidazolium group of H-119 is not required as a general acid if the leaving alcohol is activated, i.e., with a pKa much lower than for its native natural substrate. In fact, it was noted that the ratio of kcat/Km for an H119A mutant and wild-type RNase A was only 0.63 for the substrate UpOC6H4-p-NO2 4 emphasizing this point. Therefore, it seemed possible that, to produce a de novo catalyst for cleavage of the activated phosphodiester substrate 2 as an alternative strategy to our transition state analog approach (11) vide supra, the incorporation of a general base at a locus in the binding pocket proximate to the 2′-hydroxyl group may be sufficient.
The bait and switch paradigm is a well established strategy for the elicitation of antibody catalysts (24–27). The methodology involves the use of a charged hapten that elicits a complementary charged residue in the antibody combining site during the evolutionary time scale of the immunization process. This counter-charged residue then may be able to act in a catalytic manner when the hapten is “switched” to the substrate in question. Significant success has been realized when the hapten is positively charged, yielding negatively charged catalytic residues in the antibody combining sites capable of acting as general bases (24, 26, 27). In an attempt to mimic the key features of general base catalysis used by RNase A, herein we describe the implementation of a quaternary ammonium, bait and switch hapten 1 for the elicitation of antibody catalysts for phosphodiester hydrolysis of substrate 2 (Fig. 1).
MATERIALS AND METHODS
Synthesis of Hapten 1.
The quaternary ammonium hapten 1 was synthesized in 14 steps from the anhydro d-lyxo acetate 5 (Fig. 3), which was prepared by our previously described method from l-xylose (9, 10). Removal of the acetate ester of 5 was achieved by stirring in a solution of potassium cyanide (Aldrich) in methanol. After addition of a mixed bed ion-exchange resin (Bio-Rad) and filtration and evaporation of solvent, the azido alcohol 6 was obtained in 96% yield. C-3 epimerization of alcohol 6 was achieved by an initial activation to the trifluoromethylsulfonate ester 7 with trifluoromethanesulfonic anhydride (Aldrich) in a pyridine/dichloromethane (DCM) mixture followed by displacement with cesium acetate (Sigma) and 18-crown-6 (Aldrich). This gave the anhydro d-ribo acetate 8 in good yield (68% from 6). The acetate group was removed by the same method as described for 5, vide supra. The alcohol product 9 was protected as a tert-butyldimethylsilyl (TBDMS) ether 10 with TBDMS triflate (Aldrich) and 2,6-lutidine (Aldrich) in DCM (0°C, 20 min) in excellent yield (97%). The diethylacetal group was removed by treatment in a DCM/H2O/trifluoroacetic acid (TFA) mixture (1:0.2:1) to give the aldehyde derivative 11. Aldehyde 11 was oxidized smoothly with a 5% aqueous potassium permanganate solution to the carboxylic acid 12, which, without further purification, was acylated with tert-butyl 6-aminohexanoate in the presence of triethylamine (Aldrich) and O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (Aldrich). The crude reaction mixture was purified by silica gel chromatography to give the amide product 13 in 57% yield from 7. The TBDMS ether group was removed by tetrabutylammonium fluoride (Aldrich) in tetrahydrofuran (THF). The azide group of alcohol 14 was reduced by catalytic hydrogenation, and the amine product 15 was methylated reductively with a 37% aqueous formaldehyde (Aldrich), sodium cyanoborohydride (NaCNBH3, Aldrich), and zinc chloride (ZnCl2, Aldrich) mixture to give the dimethylamino product 16 in 97% yield from 14. The 4-nitrophenylphosphate group then was added by treatment of 16 with 1.1 equivalents of lithium diisopropylamide (Aldrich) in THF followed by 4-nitrophenylphosphorodichloridate (Aldrich) in THF to yield the anhydro ribo aminophosphate 17 in 43% yield after preparative scale silica gel chromatography. Methylation of the tertiary amine 17 with an excess of iodomethane followed by acid (TFA) hydrolysis of the tert-butyl ester gave 1 in 33% yield from 17. Rf = 0.25 [acetonitrile:H2O (4:1)]; 1H NMR (500 MHz, CD3OD) δ 8.21 (2H, d, J = 9.0 Hz, Ar-H), 7.35 (2H, d, J = 9.0 Hz, Ar-H), 5.30 (1H, dd, J = 7.8 Hz, J = 4.4 Hz, C-H), 4.67 (1H, s, C-H), 4.44 (1H, t, J = 8.1 Hz, C-H), 4.33 (1H, t, J = 8.7 Hz, C-H), 4.08 (1H. m. C-H), 3.34 (9H, s, 3 x N-CH3), 3.14–3.21 (2H, m, CH2), 2.25 (2H, t, J = 7.2 Hz, CH2), 1.45–1.61 (6H, m, 3 x CH2); ESI+ MS calculated for C20H30N3PO10 504 (M+H)+, observed 504 (M+H)+; ESI−MS calculated for C20H30N3PO10 502 (M-H)−, observed 502 (M-H)−.
Figure 3.

Synthetic route to the quaternary ammonium hapten 1. Reagents and conditions: (a) potassium cyanide (KCN) and methanol; (b) trifluoromethanesulfonic anhydride, pyridine, and dichloromethane (DCM); (c) cesium acetate, 18-crown-6, and toluene, under reflux; (d) KCN and methanol; (e) tert-butyldimethylsilyl (TBDMS) triflate, 2,6-lutidine, and DCM, 0°C; (f) DCM, trifluoroacetic acid (TFA), and H2O (1:1:0.2); (g) potassium permanganate (5% aqueous solution); (h) tert-butyl-6-aminohexanoate, triethylamine, and O-benzotriazol-1-yl-N, N,N′,N′-tetramethyluronium hexafluorophosphate; (i) tetrabutylammonium fluoride and tetrahydrofuran (THF); (j) H2 [40 psi (280 kPa)], Pd/C, and methanol; (k) aqueous formaldehyde (37%), sodium cyanoborohydride (NaCNBH3), and zinc (II) chloride; (l) lithium diisopropylamide (LDA), −78°C, 4-nitrophenylphosphorodichloridate, and THF; (m) methyliodide; and (n) TFA and DCM (1:1).
Synthesis of Substrate 2.
The poor stability of phosphodiester 2 precluded its direct synthesis and isolation. However, the 5′- dimethoxytrityl (DMT)-2′-tetrahydropyranyl (THP) derivative 18 was sufficiently stable that it could be stored as a 10-mM stock solution in acetonitrile/H2O (1:1) with 1% Tween-20 for up to 6 months essentially unchanged. Substrate 2 is generated from 18 when treated with Amberlyst H-15 (strongly acidic) resin for 45 min in acetonitrile/H2O (1:1) with 1% Tween-20. The route to 18 involves five steps from anhydroribitol 19, which was synthesized via the methyl glycoside of d-ribose (Aldrich) by the method of Bennek and Gray (28). Triol 19 was reacted with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPS-Cl) (Aldrich) and imidazole (Aldrich) to give the TIPS-protected derivative 20 in excellent yield (87%). The 2′ hydroxyl then was protected selectively as a tetrahydropyranyl (THP) ether 21 by using the method of Cardillo et al. (29). The TIPS group was removed by treatment with tetrabutylammonium fluoride in THF to give the diol 22 in excellent yields (95%) after silica gel chromatography. The 5′-hydroxyl group of 22 then was protected as a dimethoxytrityl ether by using DMT–chloride (Aldrich) and triethylamine with pyridine as the solvent. After silica gel chromatography, the 5′-DMT ether 23 was isolated in good yield (76%). Phosphorylation of the 3′ position of 23 was achieved by using 4-nitrophenylphosphorodichloridate, triazole, triethylamine, and N-methylimidazole. The product 18 was isolated as its triethylammonium salt after an aqueous extraction (4 M triethylammonium bicarbonate) and preparative scale thin layer chromatography in good yield (53%). Rf 0.4 [DCM/methanol (95:5), 1% triethylamine]; 1H NMR (400 MHz, CDCl3) δ 8.02 (2H, d, J = 9.0 Hz, Ar-H), 7.39 (2H, d, J = 9.0 Hz, Ar-H), 7.27–7.21 (9H, m, Ar-H), 6.76 (2H, d, J = 7.5 Hz, Ar-H), 6.75 (2H, d, J = 7.5 Hz, Ar-H), 4.82 (1H, dd, = 5.7 Hz, J = 4.2 Hz, OCHO), 4.67 (1H, m, C-H), 4.53 (1H, dd, J = 7.2 Hz, J = 4.7 Hz, C-H), 4.27 (1H, dd, C-H), 4.17 (1H, dd, J = 8.7 Hz, C-H), 3.92 (1H, dd, J = 6.7 Hz, C-H), 3.90, (1H, m, C-H), 3.76 (6H, s, OCH3), 3.46 (1H, m, C-H), 3.28 (1H, dd, J = 6.7 Hz), 2.97 (6H, m, 3 x CH2), 1.61–1.48 (8H, m, THP-ring protons), 1.23 (9H, t, J = 7.8 Hz, 3 x CH3); 31P NMR (400 MHz, CDCl3) δ −5.47 (s); ESI− MS calculated for C37H39NO12P 720 (M-H)−, observed 720 (M-H)−.
mAb Generation.
Hapten 1 was activated to its N-hydroxysuccinimide ester by treatment with N-hydroxysuccinimide (Aldrich) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (Aldrich) in dimethylformamide. The hapten then was conjugated to the ɛ-amino groups of the lysine groups of carrier proteins, keyhole limpet hemocyanin (Pierce), and BSA (Sigma) 5 mg⋅ml−1 in aqueous sodium phosphate (20 mM, pH 7.4) buffer. Female 129GIX mice were hyperimmunized with the keyhole limpet hemocyanin 1 conjugate, and mAb production occurred by standard techniques (30–32). Hybridoma cell lines that secreted BSA-1-specific antibodies were grown in tissue culture or ascites. Antibody purification involved a three-stage process including precipitation from ammonium sulfate and then affinity, protein-G, column chromatography followed by ion-exchange chromatography on a DEAE column. At this stage, all antibody preparations were >99% pure based on silver staining by using SDS/PAGE gels. Immediately before kinetic evaluation, all antibody preparations were dialyzed into the reaction buffer.
Kinetic Studies.
Buffers used for kinetic studies were prepared with deionized water. The pH of all aqueous solutions was measured with an Orion model 920A pH electrode at room temperature. Hepes buffer (10–400 mM; Sigma, ultragrade, >99.5% pure) was used between pH 7.0–8.0.
Aqueous preparations of 2 were generated fresh for each set of kinetic studies by treatment of a 5′-DMT-2′-THP-protected derivative 18 solution in acetonitrile/H2O (1:1, with 1% Tween-20) (Aldrich) (1 ml, 10 mM) with water-swollen Amberlyst-H15 (Aldrich) strongly acidic ion exchange resin (30 mg) for 45 min. The resulting solution was pipetted from the resin and added immediately to a buffer solution such that the final concentration of acetonitrile = 2.5%.
Analysis of the time course of the acid-catalyzed hydrolysis of the DMT and THP protecting groups of 18 occurred by high performance liquid chromatography. Aliquots (5 μl) of the stock substrate solution (10–50 mM) in acetonitrile/H2O (1:1 with 1% Tween-20) were removed from the resin at times from 0 to 120 min and quenched into Hepes (20 mM, pH 7.49, 95 μl) containing 4-methyl-3-nitroanisole (100 μM, Aldrich), as an external standard, and immediately injected onto an high performance liquid chromatography column. The products were separated on an Alltech Adsorbosphere HS C-18 reversed-phase column at 254 nm by using an isocratic elution system of 54% acetonitrile and 46% water (0.1% TFA) at a flow rate of 1 ml⋅min−1. The first step involves a rapid loss of the DMT ether from 18 to form the hydroxyphosphate 24 and dimethoxytrityl alcohol 25 (Fig. 5). This reaction is complete after 15 min. The loss of the THP ether is, however, much slower, taking >1 h to reach completion. Unfortunately, there is a competing acid-catalyzed cyclization-cleavage of 2 occurring during this process, so it is difficult to generate substrate 2 independently of the 2′,3′-cyclic phosphate 26 and p-nitrophenol. Throughout these studies, the acid-catalyzed deprotection of 18 was stopped at 45 min, where the ratio of 2 to 24 is ≈95:5 and only a very small proportion (<1%) of 26 and p-nitrophenol are present.
Figure 5.

Intermediates in the acid-catalyzed deprotection of 18. All intermediates containing a UV chromophore were separated by using high performance liquid chromatography and their rates of formation were determined by peak area ratio with an external standard.
Kinetic analysis of the base-catalyzed hydrolysis of 2, initial screening of antibody clones, and in depth kinetic analyses of catalytic antibodies were performed by monitoring p-nitrophenol release by using a 96-well microtitre plate and ELISA plate reader (Spectramax 250) and repetitively scanning at 405 nm and 25°C. In a typical assay, a stock solution of substrate 2 (10–50 mM, 1 ml) in acetonitrile/H2O (1:1 with 1% Tween-20) was deprotected for 45 min and then diluted serially in acetonitrile/H2O (1:1 with 1% Tween-20) before being added (5 μl) to a buffer solution (Hepes, 20 mM, pH 7.49, 95 μl) with or without antibody present in a 96-well microtitre plate. In all cases, the reported rate constants were calculated by the method of initial slopes (2–5% conversion, >20 points). The rate of cyclization-cleavage of 2 was followed at 6 substrate concentrations and four buffer concentrations at pH 7.49; all points were at least duplicate determinations. The background or noncatalyzed rate of hydrolysis of 2 kuncat is calculated by plotting rate vs. buffer concentration and extrapolating to zero buffer by using the cricket graph computer program (Computer Associates International, Islandia, NY).
MATT.F-1 steady state kinetic parameters were determined by using the assay conditions as described above. Substrate concentration varied from 20 to 500 μM and antibody concentrations, determined by UV spectroscopy, were 0.14–0.28 mg⋅ml−1. The reaction was followed for only the linear part of the slope (<5%, correlation coefficient >0.985). The data were analyzed by using nonlinear regression analysis by using the grafit computer program (Erithacus and Microsoft) on a Compaq PC.
RESULTS AND DISCUSSION
The problems of mimicking the penta-coordinate anionic phosphorane transition state for phosphodiester hydrolysis have kept antibody-catalysis of the phosphodiester bond from being reported until only recently. The successful strategy used a penta-coordinate metallochelate as a transition state analog hapten (11). In an attempt to expand and improve the scope of this original approach, we have designed hapten 1 with the sole purpose of incorporating a general base in an antibody binding site proximate to the 2′ hydroxyl of substrate 2 such that it facilitates nucleophilic attack of this hydroxyl group on the adjacent phosphoryl center. For this hapten design transition, state mimicry is sacrificed and is replaced by a point charge with the expectation of yielding a counter-charged residue in the antibody binding site.
Of key import in the hapten and substrate design was to minimize the risk of contaminating phosphodiesterases that could contribute to the observed catalysis of the antibody preparations. For this reason, substitution at the 1′-locus of the d-ribose ring of 2, with a heterocyclic base, was omitted. Indeed, kinetic studies revealed that phosphodiester 2 is not a substrate for RNase A, U2, and H under the assay conditions outlined vide supra, and only in the presence of high concentrations (micromolar) of phosphodiesterase I was catalytic activity detected. Based on these results, it was believed that any contaminating phosphodiesterases would have to be present in high concentration to generate any observable catalysis and thus would be easily detectable during the purification stages described above.
After immunization protocols with the keyhole limpet hemocyanin 1 conjugate and mAb production, 35 hapten-specific hybridoma cell lines were isolated. All of these antibodies then were screened for their ability to catalyze the cyclization-cleavage of 2.
The base-catalyzed cleavage of 2 was found to be first order with respect to hydroxide ion, and the observed rate constant (kobs) was found to have a small linear dependence on the concentration of total Hepes buffer. The apparent second-order rate constant (k2 = dkobs/d[Hepestot] for 2 is proportional to the fraction of Hepes as the free base, so the rate law is given by Eq. 1 (33).
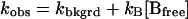 |
1 |
The true background rate kbkgrd, at pH 7.49, corrected for buffer effects, was determined from a plot of kobs vs. [Bfree]. The gradient, kB = 6.0 × 10−3 M−1 min−1 and extrapolating to zero buffer, yielded the background rate as kbkgrd = 2.66 × 10−4 min−1, a value in complete accord with previous examples of aryl phosphodiester bond hydrolysis (33, 34).
Two antibodies from the library vide supra were found to catalyze phosphodiester hydrolysis of substrate 2. The most active clone, MATT.F-1, was studied in depth and was found to obey classical Michaelis–Menten kinetics. Steady-state kinetic parameters were measured at pH 7.49 (Hepes, 20 mM), yielding a kcat = 0.44 min−1, a Km = 104 ± 11 μM, and a kcat/kuncat = 1.65 × 103. The reaction progress curves remained linear when followed for more than one turnover, suggestive of no product inhibition.
When studying any catalyst for processes in which natural enzymes catalyze the same reaction, it is essential to exclude the possibility of enzyme contamination. In addition to the inherent stability of substrate 2 to naturally occurring phosphodiesterases, vide supra, several experiments also have been performed that help rule out the potential contribution of enzymatic contamination. First, hapten 2 was found to be a stoichiometric inhibitor of MATT.F-1.† Second, antibody activity was destroyed upon boiling for 3 min, mitigating against contamination by thermostable RNases. Finally, MATT.F-1 preparations purified in three steps, precipitation, affinity chromatography (Protein-G column), and ion-exchange chromatography (DEAE column), from different batches of ascites retain the same catalytic activity.
Attempts to dissect the mechanism of action of MATT.F-1 have been limited severely by the proclivity of the antibody to precipitate from buffer solutions at pHs below ≈7.3 and at temperatures at or below 25°C. Isotyping of MATT.F-1 by ELISA revealed that it is an IgG with a κ light chain and a γ3 heavy chain. Standard texts report that γ3 subtypes are susceptible to precipitation under ambient conditions (35). In our hands, all attempts to improve the solubility of MATT.F-1 by modifications of salt concentration and temperature were unsuccessful. Therefore, pH-rate studies had to be confined to the narrow range of 7.49–8.0. In this range, the kcat of MATT.F-1 remains constant, offering little information about the contribution of binding site residues, except that any ionizable group present in the binding site does not have a pKa in that range.
Wolfenden (36) has defined the catalytic proficiency of a biocatalyst by division of its second order specificity constant (kcat/Km) with the background rate (kbkgd) of substrate hydrolysis. This analysis allows a direct comparison of different biocatalysts catalyzing the hydrolysis of different substrates. By this rationale, MATT.F-1 has a proficiency of 1.6 × 107 M−1, which is higher than that reported for our phosphodiesterase antibody, 2G12, with its substrate 4 (1.3 × 106 M−1) (11) elicited by the transition state analog hapten 3 and is only 3 orders of magnitude lower than the proficiency of the naturally occurring enzyme RNase A for substrate 4.
CONCLUSIONS
In this report, we have shown the powerful utility of a bait and switch hapten paradigm for the generation of antibody catalysts. By producing antibodies to the quaternary ammonium hapten 1, we have elicited the most catalytically proficient antibody, MATT.F-1, for phosphodiester hydrolysis yet reported. It is now feasible to envisage a hapten design that incorporates the salient features of both a transition state analog and bait and switch approach and that may lead to the elicitation of antibodies with RNase-like catalytic activity.
Figure 4.

Synthetic route to phosphodiester substrate 2. Reagents and conditions: (a) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane, imidazole, and dimethylformamide (DMF); (b) dihydropyran, Amberlyst H-15 resin, and hexane; (c) TBAF and THF; (d) 4,4′-dimethoxytrityl (DMT) chloride, triethylamine, and pyridine; (e) 4-nitrophenylphosphorodichloridate, triazole, N-methylimidazole, triethylamine, and THF; (f) Amberlyst H-15 resin, CH3CN/H2O (1:1), and 1% Tween-20.
Acknowledgments
This work was supported in part by the National Institutes of Health (Grant GM 43858 K.D.J.) and The Skaggs Institute for Chemical Biology.
ABBREVIATIONS
- DCM
dichloromethane
- TCA
trifluoroacetic acid
- THF
tetrahydrofuran
- THP
tetrahydropyranyl
- DMT
dimethoxytrityl
Footnotes
Reaction conditions: 10 μM MATT.F-1 active sites, 1–100 μM inhibitor 2, and 200 μM substrate in Hepes (20 mM, pH 7.49).
References
- 1.Chin J. Acc Chem Res. 1991;24:145–152. [Google Scholar]
- 2.Magda D, Miller R A, Sessler J L, Iverson B L. J Am Chem Soc. 1994;116:7439–7440. [Google Scholar]
- 3.Lorsch J R, Szostak J W. Acc Chem Res. 1996;29:103–110. doi: 10.1021/ar9501378. [DOI] [PubMed] [Google Scholar]
- 4.Anslyn E, Breslow R. J Am Chem Soc. 1989;111:5972–5973. [Google Scholar]
- 5.Breslow R, Schmuck C. J Am Chem Soc. 1996;118:6601–6605. [Google Scholar]
- 6.Wentworth P, Jr, Janda K D. Curr Opin Chem Biol. 1998;2:138–144. doi: 10.1016/s1367-5931(98)80046-x. [DOI] [PubMed] [Google Scholar]
- 7.Schultz P G, Lerner R A. Science. 1995;269:1835–1842. doi: 10.1126/science.7569920. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y C J, Janda K D. J Am Chem Soc. 1992;114:1488–1489. [Google Scholar]
- 9.Wentworth P, Jr, Wiemann T, Janda K D. J Am Chem Soc. 1996;118:12521–12527. [Google Scholar]
- 10.Wentworth, P., Jr., & Janda, K. D. (1997) SYNLETT 537–543.
- 11.Weiner D P, Wiemann T, Wolfe M M, Wentworth P, Jr, Janda K D. J Am Chem Soc. 1997;119:4088–4089. [Google Scholar]
- 12.Richards F M, Wyckoff H W. Enzymes. 1971;4:647–806. [Google Scholar]
- 13.Blackburn P, Moore S. Enzymes. 1982;15:317–433. [Google Scholar]
- 14.Eftink M R, Biltonen R L. In: Hydrolytic Enzymes. Neuberger A, Brocklehurst K, editors. New York: Elsevier; 1987. pp. 333–376. [Google Scholar]
- 15.Kartha G, Bello J, Harker D. Nature (London) 1967;213:862–865. doi: 10.1038/213862a0. [DOI] [PubMed] [Google Scholar]
- 16.Lennette E P, Plapp B V. Biochemistry. 1979;18:3938–3946. doi: 10.1021/bi00585a015. [DOI] [PubMed] [Google Scholar]
- 17.Herries D G, Mathias A P, Rabin B R. Biochem J. 1962;85:127–134. doi: 10.1042/bj0850127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breslow R, Labelle M. J Am Chem Soc. 1986;108:2655–2659. [Google Scholar]
- 19.Anslyn E, Breslow R. J Am Chem Soc. 1989;111:4473–4482. [Google Scholar]
- 20.Herschlag D. J Am Chem Soc. 1994;116:11631–11635. [Google Scholar]
- 21.Breslow R, Dong S D, Webb Y, Xu R. J Am Chem Soc. 1996;118:6588–6600. [Google Scholar]
- 22.Menger F M. J Org Chem. 1991;56:6251–6252. [Google Scholar]
- 23.Thompson J E, Raines R T. J Am Chem Soc. 1994;116:5467–5468. doi: 10.1021/ja00091a060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shokat K M, Leumann C J, Sugasawara R, Schultz P G. Nature (London) 1989;338:269–271. doi: 10.1038/338269a0. [DOI] [PubMed] [Google Scholar]
- 25.Shokat K M, Schultz P G. In: The Generation of Antibody Combining Sites Containing Catalytic Residues. Chadwick D J, Marsh J, editors. New York: Wiley; 1991. pp. 118–134. [DOI] [PubMed] [Google Scholar]
- 26.Janda K D, Weinhouse M I, Schloeder D M, Lerner R A, Benkovic S J. J Am Chem Soc. 1990;112:1274–1275. [Google Scholar]
- 27.Janda K D, Weinhouse M I, Danon T, Pacelli K A, Schloeder D M. J Am Chem Soc. 1991;113:5427–5434. [Google Scholar]
- 28.Bennek J A, Gray G R. J Org Chem. 1987;52:892–897. [Google Scholar]
- 29.Bongini A, Cardillo G, Orena M, Sandri S. Synthesis. 1979;11:618–620. [Google Scholar]
- 30.Köhler G, Milstein C. Nature (London) 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 31.Köhler G, Milstein C. Eur J Immunol. 1976;6:511–519. doi: 10.1002/eji.1830060713. [DOI] [PubMed] [Google Scholar]
- 32.Galfré G, Milstein C. Methods Enzymol. 1981;73:1–46. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- 33.Davis A M, Hall A D, Williams A. J Am Chem Soc. 1988;110:5105–5108. [Google Scholar]
- 34.Usher D A, Richardson D I, Oakenfull D G. J Am Chem Soc. 1970;92:4699–4700. doi: 10.1021/ja00718a037. [DOI] [PubMed] [Google Scholar]
- 35.Goding J W. Monoclonal Antibodies: Principles and Practice. New York: Academic; 1986. pp. 14–15. [Google Scholar]
- 36.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]