

Abstract
Purpose
Effective cancer vaccines must both drive a strong CTL response and sustain long-term memory T cells capable of rapid, recall responses to tumor antigens. We sought to characterize the phenotype and function of gp100 peptide-specific memory CD8+ T cells in melanoma patients after primary gp100209-2M immunization, and to assess the anamnestic response to boosting immunization.
Experimental Design
8-color flow cytometry analysis of gp100-specific CD8+ T cells was performed on PBMCs collected shortly after the primary vaccine regimen, 12–24 months after primary vaccination, and following boosting immunization. The anamnestic response was assessed by comparing the frequency of circulating gp100-specific T cells before and after boosting. Gp100 peptide-induced in vitro functional avidity and proliferation responses, and melanoma-stimulated T-cell CD107 mobilization were compared for cells from all 3 time points for multiple patients.
Results
The frequency of circulating gp100-specific memory CD8+ T cells was comparable to CMV-and FLU-specific T cells in the same patients, and the cells exhibited anamnestic proliferation after boosting. Their phenotypes were not unique, and individual patients exhibited one of two distinct phenotype signatures that were homologous to either CMV-or FLU-specific memory T cells. Gp100-specific memory T cells demonstrated some properties of competent memory T cells, such as heightened in vitro peptide-stimulated proliferation and increase in central memory (TCM) differentiation when compared to T-cell responses measured after the primary vaccine regimen. However, they did not acquire enhanced functional avidity usually associated with competent memory T-cell maturation.
Conclusions
Although vaccination with class I – restricted melanoma peptides alone can break tolerance to self-tumor antigens it did not induce fully competent memory CD8+ T cells – even in disease-free patients. Data presented suggest other vaccine strategies will be required to induce functionally robust long-term memory T cells.
Keywords: Boosting Vaccine, Melanoma-Specific Memory T Cells, Memory T Cell Immunophenotyping, Melanoma Vaccine, Ploychromatic Flow Cytometry
Introduction
Previous vaccine trials performed primarily in patients with metastatic melanoma demonstrated that the administration of class I-restricted peptides from gp100 [1, 2], and MART-1 [3, 4] resulted in the in vivo expansion of functionally attenuated or senescent CD8+ T cells that were unable to induce long-lasting tumor regression (Reviewed in [5, 6]). These patients had potentially compromised immunity secondary to large tumor burdens, and there were few opportunities to study the long-term anti-melanoma memory T-cell response due to poor patient survival. Recent studies suggest that effective vaccine therapy must not only stimulate CTL activity, but must also sustain long-term memory T cells capable of mounting strong proliferative and functional responses to secondary tumor antigen challenge ([7, 8], Reviewed in [9]). The phenotype and functional characterization of tumor-specific T cells in similarly vaccinated melanoma patients with less advanced disease, and potentially more robust immunity, may demonstrate enhanced T-cell responses, and provide a better opportunity to study the activation and maintenance of long-term melanoma-specific memory T cells[10–12].
Given the “self” nature of many tumor-associated antigens and the fact that productive anti-tumor immunity requires some degree of abrogation of self-tolerance, it is reasonable to expect that self-tumor antigen-specific memory T cells may have phenotypes different from those described for T cells specific for common viral antigens such as CMV or FLU peptides. However, recent data also suggest that self-tumor antigen-specific memory CD8+ T cells exhibit phenotypes homologous to CMV peptide-specific T cells - characterized by low CCR7 expression and dominant expression of effector memory (TEM), and effector-like subpopulations ([10, 13, 14] Reviewed in [9, 15]). The characterization of memory and effector T-cell phenotypes in different tumor and infectious disease models has been limited by the use of 3- and 4-color flow cytometry analysis. The enhanced resolution of higher order (8–10 color) flow cytometry should produce a more comprehensive phenotype characterization of memory T cells, and more definitively determine whether or not tumor-specific memory T cells express phenotypes that differ significantly from competent memory T cells specific for common viral antigens.
We previously demonstrated that completely resected patients with stage I–III melanoma vaccinated with the HLA-A2-restricted (modified) melanoma-associated peptide, gp100209-2M, produced significant gp100-specific T-cell responses after six months of repetitive vaccination[11, 12]. Circulating gp100-specific CD8+ T cells collected 3–4 weeks after the last vaccine exhibited high levels of proliferation after in vitro stimulation (IVS) with cognate peptide. Native and modified gp100 peptide-loaded A2-restricted MHC class I dimers as well as A2+/gp100+ melanoma cell lines stimulated ex vivo production of IFN-γ by these T cells in the majority (>60%) of patients [16]. Long-term gp100-specific CD8+ memory T cells exhibiting high proliferative and functional responses after in vitro activation were detected in the peripheral blood ≥ 12 months after the last vaccination in the majority of patients. However, the limited numbers of PBMCs available prevented a detailed functional or phenotype analysis of these memory T cells. We were also interested in the anamnestic potential of these cells since, as with most infectious agents, long-term immunity to self-tumor antigens may require boosting immunization to maintain adequate numbers of tumor-specific T cells. We therefore sought to study the effect of boosting immunization on patients from our initial trial who were disease-free for at least one year after their last gp100 vaccine.
Sixteen patients from the initial trial were given multiple gp100209-2m boosting immunizations. The frequency of long-term memory gp100- specific CD8+ T cells, and the effect of boosting were assessed by ex vivo tetramer analysis. Herein we describe the 8-color phenotype of gp100 peptide-specific long-term memory and boosted CD8+ T cells, and compare the frequency of central memory (TCM), effector memory (TEM), and effector (TEFF) subpopulations at these two time points to frequencies observed 3 to 4 weeks after the initial vaccine regimen was completed. Additionally, these phenotypes were compared to the phenotypes of CMV (pp65) and FLU(M1)-specific memory CD8+ T cells. Correlated functional avidity, proliferation responses, and tumor-specific CD107 mobilization were also compared for gp100-specific CD8+ T cells from all three time points to determine the relative strength of long-term memory functional responses, and the effect of boosting immunization on these parameters.
Materials and Methods
Patient Samples
Thirty five HLA-A2+ patients with resected stage I–III melanoma were randomized to receive gp100209-2M peptide in Montanide ISA adjuvant every 2 or 3 weeks for 6 months [11]. The HLA-A2-restricted gp100209-2M peptide (NSC# 683472) was provided by the Cancer Therapy Evaluation Program (CTEP) under a National Cancer Institute Investigational New Drug Application (BB6123). A leukapharesis was performed before treatment and 1–2 weeks post completion of the initial vaccine regimen (PIVR). Subsequently, 16 of the patients who were disease-free for >12 months and willing to give informed consent were enrolled in a second booster vaccine trial. Both protocols were reviewed by CTEP-NCI and approved by the Providence Health System Institutional Review Board. All patients gave their written informed consent before screening for eligibility. Each patient underwent leukapharesis prior to boosting immunization with the gp100209-2M peptide in Montinide ISA 51 adjuvant to assess the frequency and functional properties of long-term memory (LTM) T cells. The first two boosting vaccines were given 30 days apart and a third leukopak was collected 3 weeks post the second boosting immunization (P2B). Thereafter boosting immunization and 50ml blood collections were performed every 6 months out to 3 years. All patients received subcutaneous injections at two separate sites that totaled 1 mg of peptide and 2ml of Montanide ISA 51 [11]. PIVR, LTM, and P2B cryopreserved PBMCs were used for all ex vivo polychromatic flow cytometry studies, and for in vitro stimulated (IVS) expansion cultures.
IL-15/Low-Dose IL-2 IVS
Cryopreserved PIVR, LTM, and P2B PBMCs were stimulated with gp100209-2M in 7 day IVS cultures in the presence of IL-15 and low-dose IL-2 as described previously [12]. Cells were thawed and washed twice in Dulbeccos PBS with 2% human AB serum (Irvine Scientific, Santa Ana, CA) before being cultured in X-vivo-15 medium (BioWhittaker, Walkersville, MD) with 5% human AB serum. Cells were plated at 12.5 × 105/ml in 200ul/well in a round bottom polystyrene 96-well plate in medium containing gp100209-2M peptide (0.01ug/ml), human IL-15 (50ng/ml – Peprotech, Rocky Hill, NJ) and cultured for 2 days at 37°C in a 5% CO2 humidified incubator. On day 2 rhIL-2 (PROLEUKIN®, Chiron Corporation, Emeryville, CA) was added to provide a final concentration of 60 IU/ml in each culture well. On day 7 of culture 2mM EDTA was added to each well for 5 minutes, cells were harvested and transferred to 5ml polystyrene tubes and washed 3x in Dulbeccos PBS prior to functional avidity or CD107 analysis.
8-color Flow Cytometry Analysis
Fluorescent antibodies were purchased or fluorescenated in-house with the use of commercially available QDot antibody conjugation kits (Invitrogen, Molecular Probes, Eugene, OR). The staining panel consisted of the following: CCR7-FITC (R&D Systems, Minneapolis, MN), HLA-A2-restricted gp100209-2M (IMDQVPFSV) and gp100209–217 (ITDQVPFSV) tetramer-PE (iTAg, Beckman Coulter, San Diego, CA), CD8β-PE-TR, CD28-PE-CY7 (Beckman Coulter, Fullerton, CA), CD14 PE-Cy5 (Beckman Coulter, Marseille, France), CD19-PE-Cy5 (eBioscience, San Diego, CA), CD45RA-APC, strepavidin-APC-CY7 (BD Bioscience, Pharmingen, San Diego, CA), CD57biotin (BD Bioscience, Immununocytometry Systems, San Jose, CA), and affinity-purified CD27 (BD Bioscience, Pharmingen, San Diego, CA) conjugated to Quantum Dot 605. HLA-A2-restricted FLU MI58–66 (GILGFVFTL) and CMVpp65495–503 (NLVPMVATV) peptide-specific, PE-conjugated tetramers (iTAg, Beckman Coulter) were used in the same 8-color panel to identify FLU and CMV-specific CD8+ T cells. The use of a “dump cocktail” of CD19&CD14-PE-Cy5 in combination with 5ug/ml of 7-aminoactinomycin D (7-AAD) (Invitrogen, Molecular Probes, Eugene Oregon) in 1X PBS was employed to stain cells with high cell surface Fc receptor(FcR)-mediated non-specific binding of antibodies, and to discriminate between live and dead cells [17]. All data was acquired on a 9-color Dako Cyan ADP Flow Cytometer equipped with three diode lasers (488nm, 635nm, and 407nm), and modified with optimal bandpass and dichroic filters (Dako, Fort Collins, CO). PIVR, LTM, and P2B PBMCs were examined by direct “ex vivo” interrogation of unmanipulated freshly thawed cells using the 8-color staining panel described above. Viable CD14−/CD19− lymphocytes were gated for positive CD8β and gp100 tetramer staining, and gp100-specific CD8β+ T-cells were further interrogated for expression of the remaining five cell-surface markers (CCR7, CD45RA, CD57, CD27, CD28) to determine their sub-phenotypes. All data were acquired in FCS format (Summit 4.2) and analyzed using Winlist™ 5.O Software (Verity House Software, Topsham, ME). Computer-assisted digital compensation was performed using single-color staining controls via the Hyperlog™ transform (Verity House Software) [18, 19]. FMO (“fluorescence minus one”) controls were used to set hinged-gating and define histogram regions that distinguished positive from negative events for each fluorescent parameter [17, 19, 20]. Fidelity controls were used to ensure that there was no loss of staining frequency and intensity between lower order panels and the corresponding fluorescence for each mAb in the 8-color panel.
CFSE Analysis
PBMCs were thawed, washed in Dulbecco’s PBS (DPBS) and pelleted by centrifugation. Cells were resuspended in pre-warmed (37°C) DPBS at 50×106 cells/ml and loaded with 7μl of a 0.05μM working stock of carboxyfluoroscein succinimidyl ester (CFSE) in DMSO(CellTrace™, Invitrogen) per 1ml of cell suspension. After CFSE incubation for 10 minutes at 37°C staining was quenched by the addition of 5 volumes of ice-cold culture medium containing 10% human AB serum and incubation on ice for 5 minutes. Cells were washed 3x in DPS, resuspended at 1×106/ml in X-vivo-15 medium supplemented with IL-15 (50ng/ml), IL-2 (60IU/ml) and 5% human AB serum, and plated at 2.5×105/well (250μl) in a 96 well plate. Cells were stimulated with 0.0025μg/well (0.01ug/ml) of gp100209-2M peptide for 2–7 days. Cells were collected at different time points and stained with anti-CD3 PE-Cy7 (BD Bioscience, San Jose, CA), CD8 APC-H7 (BD Bioscience, PharMingen), gp100209–217 tetramer APC (iTag, Beckman-Coulter), and Annexin PE (BD Bioscience, PharMingen). CFSE(FITC) staining of pre-gated viable, gp100 tetramer+ CD8+ T cells was analyzed on a DAKO Cyan ADP flow cytometer, and determination of the number of cell divisions from CFSE fluorescence decay data was calculated using ModFit™ software analysis (Verity House Software).
Data Reduction and Cluster Analysis
FCOM™ (which stands for “combination function”) is a parameter in Winlist™ that can be used to categorize and bin a fluorescent cellular event based on all the combinations of pre-defined (pre-gated) fluorescence regions that contain the event. This function uses gated positive regions of multiple fluorescence parameters to enumerate cells in all of the possible subphenotypes as defined by the number of gating parameters. The absolute number of gated cells of each defined subphenotype can be assessed and displayed as a discrete peak on the FCOM™ histogram array. The relative percentage of all cells in each of the subphenotypes are calculated in FCOM™ and displayed separately [19]. Since the number of subphenotypes for any given sample population is a power function (2n, where n = number of phenotype gates), FCOM™ delineated 32 subphenotypes with the five antibodies described in this study. The relative percentages of gp100 tetramer+ T cells with each subphenotype were used for unsupervised cluster analysis. Average-linkage hierarchical clustering was performed with The Institute for Genomic Research MultiExperiment Viewer (TIGR MeV 3.1) [19, 21]. Data files generated by FCOM™ were reformatted to mimic the Affymetrix data file format to accommodate loading into MeV software.
Functional Avidity Assay: IFN-γ Flow Cytometry Analysis
IFN-γ cytokine flow cytometry [CFC (IFN-γ)] functional avidity analysis was performed using freshly harvested IVS T Cells. 1×106 T cells/well were placed in a round-bottomed 96-well plate in 200 μl of X-VIVO 15 medium supplemented with 5% human AB serum (complete medium). Cells were restimulated with gp100209–2M or gp100209–217 peptide at various concentrations covering a 7-log range (0.00001 ug/mL to 10 ug/mL). The cells were incubated 1 h before brefeldin A (Sigma Chemical, St Louis, MO) was added at 1.25ug/well (5ug/ml). Cells were incubated an additional 14 h before they were collected and stained. Twenty-five μl of 20 mM EDTA was added to each well for 10 min at room temperature (RT) to stop the IFN-γ response before collection. Cells were washed twice with 3 ml of staining buffer and suspended in 50 –100 μl of staining buffer for subsequent fixation, permeabilization, and staining. Cells were fixed in 1 ml of 1x BD FACS-lysis solution (BD Biosciences) for 10 min at RT. All samples were washed in staining buffer and 500 μl of 1X BD FACS permeabilization solution (BD Biosciences) was added to each tube and incubated at RT for 15 min. Permeabilized cells were washed twice, suspended in 50–100 μl of staining buffer, and stained with CD3 (FITC)/anti-IFN-γ(PE)/CD8 (PerCP-Cy5.5) monoclonal antibodies (BD Biosciences) prior to incubation at RT for 30 min. After staining, cells were washed twice in staining buffer, fixed in 250 μl of 1% paraformaldehyde (Electron Microscopy Sciences), and stored at 4°C in the dark before analysis. Analysis was performed within 24 h of staining. Cells were acquired through a standard lymphocyte light scatter gate (90° versus forward angle), and 5 × 104 to 1 × 105 gated CD8+/CD3+ T- cells were analyzed for IFN-γ staining using a FACS Calibur flow cytometry system and CellQuest software (BD Biosciences). Net frequencies of IFN-γ+ cells were calculated by subtracting background “no antigen” (media only) values from the frequency of IFN-γ+ CD8+ T-cells after gp100209–2M peptide stimulation. Data was analyzed with Prism 4.0 (GraphPad Software, Inc., San Diego, CA). EC50 values were obtained from non-linear sigmoidal dose response curves (variable slope, 4-parameter) and the p-value was given when two curves were compared side by side.
CD107a/b Functional Assay
CD107a/b functional analysis was performed using freshly harvested IVS T-cells. 1×106 IVS cells were added to 1ml cultures in complete medium in a 6-well plate. They were stimulated with 0.25×106 Mel 118 (A2+/gp100+) or the negative control, Mel 103(A2+/gp100−), melanoma tumor cells per well in the presence of 2 uL of CD107 a/b Mab (PE) and 1 μl 1000X monensin (eBioscience) in each well. Cultures were incubated at 37°C for 6 hours after which cells were incubated with EDTA for 10 min at RT before they were transferred to staining tubes. Cells were washed 2x with 3 ml of staining buffer before staining with gp100209-2M tetramer (APC) for 1 hour at RT in the dark. Cells were again washed 2x with staining buffer and subsequently stained with CD3(FITC), and CD8 (PerCP-CY5.5) Mabs for 30 mins. at 4°C. Cells were washed 2x in staining buffer, fixed in 250μl of 1% paraformaldehyde and stored at 4°C prior to analysis within 24hrs. Cells were analyzed on a FACS Calibur using CellQuest software. Gating and acquisition were as described for the functional avidity assay – 1×104 to 2×104 CD8+/CD3+/tetramer+ T cells were analyzed for the frequency of CD107a/b expression. Background CD107a/b expression stimulated by the negative control cell line Mel 103 was subtracted from the response induced by Mel 118.
Statistical Analysis
Data in this study were evaluated using standard descriptive and graphical methods, nonlinear regression analyses, paired parametric and nonparametric tests, hierarchical clustering and heatmap construction techniques. Tests of significance, and four-parameter logistic nonlinear regression comparative analyses were performed using Prism 4.0 (GraphPad Software, Inc., San Diego, CA) and independently confirmed with the R Statistical Package, version 2.5.1, (R Development Core Team. R. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 20071. Hierarchical and alternative clustering algorithms were computed with and associated heatmaps were constructed with The Institute for Genomic Research MultiExperiment Viewer (TIGR MeV 3.1)2 [21] and independently confirmed with the R Statistical Package and Bioconductor, version 1.9.9 [22]. Two-sided probability values are reported for all tests of significance.
Results
Boosting immunization stimulates anamnestic proliferation of gp100-specific long-term memory T cells
Figure 1A shows the ex vivo frequency of gp100209-2M tetramer-specific CD8+ T cells for PIVR, LTM and P2B PBMCs from all 16 boosted patients. The frequency range of gp100-specific LTM CD8+ T cells in all patients (0.02% – 0.2%) was comparable to the frequency ranges of memory T cells specific for Flu (0.06% – 0.2%) and CMV (0.05% – 0.5%) antigens in the same patients. Boosted PBMCs showed evidence of a recall response with gp100-specific T-cell frequencies 1.5 to ≥ 20-fold higher than corresponding LTM values in 12/16 patients (mean = 6.2 fold increase). However, boosted responses equaled or exceeded PIVR values in only 5/16 patients. Comparable frequencies were observed in most patients when native peptide (gp100209–217) tetramers were used (Fig. 1B). Paired T test analysis showed tetramer binding was not statistically different for the modified and the native peptide at each time point (Fig. 1C). Mean cumulative frequencies of boosted T cells for both peptides were significantly higher than the mean frequencies of long-term memory cells (p=0.0019, native peptide), but were significantly lower than the mean PIVR frequencies at the end of the priming immunization (p=0.0407, native peptide) (Fig. 1D). The frequency of circulating gp100-specific CD8+ T cells did not increase beyond P2B levels for the majority of patients (15/16) after 6 more boosting immunizations over three years (data not shown). The absence of a more robust secondary response in the peripheral blood could reflect the trafficking of cells to lymph nodes and peripheral tissues as described in infectious disease models [23].
Figure 1.

Boosted P2B PBMCs exhibited an anamnestic increase in the frequency of circulating gp100-specific CD8+ T cells compared to LTM values. Both modified (A) and native (B) peptide-specific tetramer+ CD8+ T-cell frequencies were > LTM values for 12/16 patients, and there was no difference between the mean modified vs. native tetramer staining frequencies at each time point (C). The mean ex vivo gp100209–217 tetramer+ CD8+ T-cell frequency for P2B cells from all patients was significantly higher (p=0.0019) than the mean for LTM cells, but was lower (p=0.0407) than the mean PIVR response for all patients (D). Five patients (EA07, EA15, EA22, EA23, EA25) also exhibited boosted P2B tetramer frequencies ≥ PIVR values.
Gp100-specific LTM and P2B T cells do not exhibit enhanced functional avidity
Maturation of memory T cells, characterized by increased functional avidity and cytokine secretion following boosting immunization has been described ([24–29] Reviewed in [30]). To determine if long-term memory or boosted T cells exhibited increased functional avidity, PIVR, LTM and P2B cells were stimulated with low-dose gp100209-2M peptide (0.01μg/ml) and cultured in IL-15 + low-dose IL-2 for 7 days. Functional avidity was determined by titrating the modified and native gp100 peptide against IVS cells in a standard CFC (IFN-γ) assay. Non-linear regression analysis was performed, and EC50 values were calculated to compare functional avidity. Figure 2(A, B) shows the response curves and EC50 values for PIVR vs. LTM, PIVR vs. P2B, and LTM vs. P2B IVS cells from 2 of 7 patients studied. EA02 (Fig. 2A) LTM cells had a higher functional avidity than PIVR cells, and boosting sustained the higher avidity (i.e. LTM = P2B > PIVR). By comparison, the functional avidity of T cells from EA12 was the same at all three time points, suggesting that functional enhancement of LTM and P2B cells had not occurred (Fig. 2B). Overall, there was no statistical difference between the mean cumulative (i.e. for all patients studied) native peptide-induced EC50 values for PIVR vs. LTM (p= 0.76) or between LTM and P2B (P=0.52) by paired T test analysis (Fig. 2C). Similarly, the mean gp100209-2M stimulated EC50 values for all patients at all 3 time points were not statistically different, even though the modified peptide stimulated average EC50 values that were 1–1.5 logs lower than those stimulated by the native peptide, indicating higher functional avidity for the modified peptide (data not shown). Thus, on average, the functional avidity of LTM CD8+ T cells from seven patients did not exhibit functional enhancement compared to PIVR T cells, and boosting immunization did not improve T-cell functional avidity. In order to compare the relative strength of the functional avidity of native peptide-induced responses at all time points to that induced by a common infectious disease antigen we measured the response of CMV (pp65495–503) peptide-induced IVS cells to restimulation with the cognate pp65 peptide. As shown in Fig. 2C, the log of the mean EC50 peptide concentration for gp100 was 3 logs higher than the mean EC50 value for CMV pp65, indicating a significantly lower avidity for the self-tumor antigen-specific memory T cells.
Figure 2.
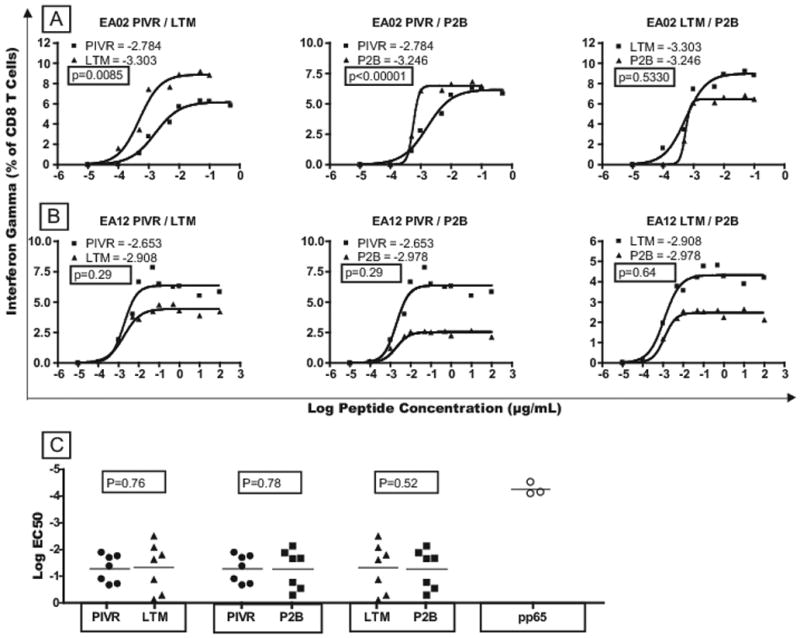
Comparison of functional avidity for PIVR, LTM, and P2B gp100-specific CD8+ T cells. Cytokine flow cytometry (CFC) analysis of IFN-γ expression by IVS CD8+ T cells after restimulation with gp100209-2M peptide. EC50 values were calculated as an index of functional avidity. CFC(IFN-γ) response curves and EC50 values for 2 patients demonstrate that LTM and P2B T cells exhibited significantly higher functional avidity than PIVR cells in some patients (A), but not in others(B). The cumulative mean log EC50 value for all LTM responses was not significantly higher than the mean PIVR value (p=0.76), nor did boosting increase the mean P2B log EC50 above the LTM value (p=0.52) – indicating no significant average increase in the functional avidity of LTM or boosted T cells (C).
Melanoma cells stimulate CD107 mobilization by gp100-specific T cells
CD107 cell surface expression has been correlated with perforin and granzyme B degranulation following CTL activation [31]. PIVR, LTM, and P2B IVS cells from 7 patients were stimulated in vitro with an A2+/gp100+ melanoma cell line, Mel 118, and an A2+/gp100− negative control melanoma cell line, Mel 103. CD107 expression was measured in a 6 hour assay, and the net CD107 response was calculated after subtracting the background Mel103-induced CD107 fluorescence. A no antigen control culture was also run to determine if melanoma cell activation of T cells induced concomitant down-regulation of TCR-specific tetramer staining. There was no statistical difference in the frequency of tetramer+ CD8+ T cells between the no antigen control and Mel 118 stimulated T cells (P=0.1557). The mean frequency of CD107+ tetramer+ CD8+ LTM T cells for all 7 patients was not statistically different from the mean frequency measured in PIVR samples – indicating no increase in the frequency of degranulating LTM compared to PIVR tumor-specific T cells as shown by paired T test analysis (P=0.42, Fig. 3). The same statistical analysis also showed that the mean frequency of P2B CD107+ T cells was not significantly different from the mean LTM value – indicating that boosting immunization did not increase the fraction of gp100 specific CD8+ T cells that responded to tumor cells by degranulation (P=0.78, Fig. 3).
Figure 3.

Comparative CD107 mobilization analysis of PIVR, LTM, and P2B-derived gp100-specific CD8+ T cells from 7 patients after in vitro challenge with an A2+/gp100+ melanoma tumor cell line, MEL 118. The mean frequency of LTM tetramer+ CD8+ IVS T cells expressing CD107 was not significantly higher than the mean PIVR CD107+ staining frequency (p=0.42); and boosting immunization did not increase the mean P2B CD107+ frequency above LTM levels (p=0.78). Background CD107 staining induced by the A2+/gp100− negative control melanoma cell line, MEL 103, was subtracted from MEL118 stimulated values.
Gp100 peptide stimulates rapid in vitro proliferation of LTM CD8+ T cells
ModFit™ analysis of the loss of CFSE fluorescence by labeled cells from all 3 time points for patient EA07 stimulated in a gp100 IVS culture illustrates the rapid proliferation kinetics of LTM gp100-specific T cells observed in many patients. As shown in Figure 4A, by 48hrs twice as many LTM gp100 specific T cells (43.9%) had divided ≥ 6 times compared to peptide-specific PIVR (20.9%) or P2B T cells (15.1%). PIVR, LTM, and P2B cells from all 16 patients were similarly stimulated in vitro with low-dose (0.01μg/ml) gp100209-2M for 7 days in the presence of IL-15 and low-dose IL-2, and proliferation was assessed by measuring the frequency of gp100209-2M tetramer+ CD8+ T cells and calculating the proliferative fold-increase over the ex vivo tetramer frequency at each time point. There was minimal antigen-induced apoptosis and cell death of CD8+ T cells during the 7 day culture (data not shown). Several patients (EA15, EA17, EA18, EA25) had low ex vivo tetramer+ frequencies (Fig. 1A) and their CD8+ T cells did not proliferate in vitro as shown by their low fold-increase values (Fig. 4). EA02 T cells also had a low proliferation response even though they were moderately gp100209-2M tetramer positive (range = 0.2% – 0.55%; Fig. 1A) at all time points. All other patients showed moderate (8–25 fold increase), to high (≥ 50 fold increase) proliferation of tetramer+ CD8+ T cells at all time points. LTM cells from 8/16 patients had very high in vitro proliferation responses (50–200 fold increases), which exceeded those observed for their respective PIVR and P2B cells, and equaled or exceeded the IVS fold increase values for CMV-specific (54.86± 9.5) and FLU-specific (60.5± 15.1) memory T cells. This suggested they had heightened proliferative capabilities characteristic of competent memory T cells. Similar results were obtained when native peptide was used (data not shown).
Figure 4.

In vitro proliferation of PIVR, LTM and P2B-derived gp100-specific CD8+ T cells after cognate peptide stimulation. CFSE (FITC) fluorescence decay of gp100 tetramer+ CD8+ T cells from a representative patient after 48hr. in vitro stimulation with gp100209-2M shows that LTM cells exhibited a very rapid proliferation response resulting in twice the frequency of T cells which doubled ≥ 6 times (43.9%) compared to PIVR (20.9%) and P2B (15.1%) proliferation responses (A). Seven day in vitro stimulation of PBMCs with gp100209-2M induced very high fold increases of LTM gp100-tetramer+ CD8+ T cells compared to the fold increase values for corresponding PIVR and P2B cells from 8/16 patients (B). EA35 exhibited increased proliferation of both LTM and P2B gp100-specific CD8+ T cells compared to PIVR cells.
Eight-color flow cytometry delineates multiple memory and effector T-cell subpopulations
Eight-color flow cytometry produces a more comprehensive phenotype characterization of memory and effector T cells than standard 3–4 color analysis. Figure 5 shows the gating and analysis strategy employed to resolve tumor antigen-specific T cell subphenotypes [19]. We identified both early TEM (CCR7−/CD45RA−/CD57−/CD27+/CD28+ ) and late TEM (CCR7−/CD45RA−/CD57−/CD27+/CD28−) subpopulations (Fig. 5 – CCR7− panel), as well as TCM (CCR7+/CD45RA−/CD57−/CD27+/CD28+) phenotypes (Fig. 5 – CCR7+ panel) predicted by consensus models based on 3–7 color analysis [9, 32–36]. We also delineated previously undescribed memory T-cell subpopulations. Using 4-color analysis Sallusto and Lanzavecchia first used the term “TEMRA” to designate CCR7−/CD45RA+ effector-memory CD8+ T cells [37]. Figure 5 shows that gp100-specific CCR7−/CD45RA+ T cells can be further divided into multiple subphenotypes based on staining with CD57, CD27, and CD28 mAbs (CCR7− panel; dot plots 1 and 2). Herein the “TEMRA” designation will refer only to the CCR7−/CD45RA+/CD57−/CD27+/CD28+/− subphenotypes since it describes the re-expression (or retention) of CD45RA on TEM CD8+ T cells prior to the acquisition of CD57 and concomitant loss of both CD27 and CD28 which characterizes the transition to terminal effector T cells [35, 36]. A similar, previously undescribed, subphenotype, which expressed CD45RA on the TCM background (CCR7+/CD45RA+/CD57−/CD27+/CD28+) was also detected (CCR7+ panel; dot plot 1), and will be referred to as “TCMRA”. Consensus models define this phenotype as unique to naïve T cells[9, 32]. However, we detected gp100-specific T cells with this phenotype signature only after repetitive gp100 peptide vaccination – suggesting “TCMRA” T cells are a true antigen-educated memory subpopulation.
Figure 5.

Sequential 2 parameter dot-plot gating strategy used for 8-color flow cytometry analysis of gp100-specific CD8+ memory T cells from a representative patient. The staining panel consisted of reagents specific for: CD8β/gp100 tetramer/CCR7/CD45RA/CD57/CD27/CD28/CD14, CD19, 7AAD cocktail. Only viable, gp100209-217 tetramer+ CD8+ T cells were selected for analysis and further divided into CCR7− (upper panel-dot plot A), or CCR7+ (lower panel-dot plot A) subpopulations. By example, CCR7−/tetramer+/CD8β+ T cells were selected (CCR7− panel; dot plot A) and further analyzed for CD45RA vs. CD57 expression (CCR7− panel; dot plot B). Cells from each of the CD45RA vs. CD57 dot plot quadrants were then analyzed for CD27 vs. CD28 staining (CCR7− panel; dot plots 1–4). 16 different subphenotypes were delineated in the CCR7− compartment, and, by similar analysis, another 16 subpopulations in the CCR7+ compartment.
Consensus models suggest that “terminal effector” CD8+ T cells express CD57 [38], and recapitulate CD45RA expression [32, 39]. Accordingly, fully differentiated effector T cells should have the following phenotype: CCR7−/CD45RA+/CD57+/CD27+/−/CD28−. T cells with a similar effector-like phenotype (i.e. CCR7−/CD45RA+/CD27+/−/CD28−) persist long-term in the blood of healthy CMV-seropositive individuals, and may be a result of chronic exposure to viral antigens [15, 33, 40, 41]. These cells have recently been called “resting vigilant effector” (TRVE) CD8+ T cells since they are believed to control latent virus expression [15]. Effector-like CD8+ T cells with similar TRVE phenotypes were found in the peripheral blood of some gp100 vaccinated melanoma patients, and may similarly reflect the sustained exposure to normal melanocyte or micrometastatic melanoma antigens (CCR7−panel; dot plot 2).
Low frequencies of previously undescribed CD8+ T cells expressing phenotypic traits of both effector cells (i.e. CCR7−/CD45RA+/−/CD57+), and memory cells (i.e. CD27+/CD28+) were also identified among LTM PBMCs (CCR7−;dot plots 2 and 4). Although these T cells may comprise TEM cells in transition to effector or TRVE -like cells they persisted in disease-free melanoma patients 12–24 months after any exogeneous exposure to gp100 peptide. A low frequency of T cells with a similar phenotype (CCR7+/CD45RA+/−/CD57+/CD27+/CD28+) was also seen in the CCR7+ compartment (CCR7+ panel; dot plots 2 and 4). Thus, the more comprehensive phenotype signatures produced using 8-color analysis confirmed the TEM and TCM phenotypes predicted from consensus models, expanded the phenotype characterization of previously described TEMRA and TRVE CD8+ T cells, and delineated previously undescribed subphenotypes such as TCMRA and putative “transitional” subpopulations with phenotype traits of both effector and memory T cells.
FCOM™ analysis generated detailed subphenotype signatures of gp100-specific CD8+ T cells
Efficient analysis of potentially different T-cell subpopulations between individual test samples was complicated by the fact that 8-color flow cytometry generated a large amount of data (32 subphenotypes) for each sample. Data reduction and analysis methods such as FCOM™ [19] were used to facilitate comparison of phenotypes between PIVR, LTM, and P2B samples within and between patients. Figure 6 shows bar graphs of the frequencies of all 32 FCOM™ subphenotypes after ex vivo analysis of PIVR/LTM/P2B cells from three patients using the aforementioned 8-color-staining panel. There were significant inter-patient differences, both qualitatively and quantitatively, in the subphenotype frequency distribution pattern (signature) of gp100 tetramer+ CD8+ T cells. While the frequency of tetramer+ CD8+ T cells in each subphenotype varied over the three time points for an individual patient, the qualitative subphenotype signature of antigen-specific cells remained uniform for each patient at all three time points – suggesting it was imprinted at the time of initial antigen exposure. The signatures of all three patients were distinct because of significant differences in the frequency of TEM, TCM, and TRVE/effector subpopulations. Most of the gp100-specific CD8+ T cells from patient EA07 were distributed in three major subsets, TEMRA (CCR7−/CD45RA+/CD57−/CD27+/CD28+), and two dominant TRVE subpopulations - CCR7−/CD45RA+/CD57+/CD27−/CD28− and CCR7−/CD45RA+/CD57+/CD27+/CD28−, respectively. The LTM CD27−/CD28− TRVE subpopulation was almost 2-fold higher compared to the PIVR value, and boosting increased its frequency almost 4-fold above PIVR. There were very low percentages of TEM and TCM at all three time points; TCMRA frequencies were low at PIVR and P2B but increased to 10% at LTM. The EA07 gp100-specific T-cell signature was composed of predominantly TRVE -like cells similar to the memory CD8+ T cells found in CMV+ patients [15]. By comparison, the signature for EA02 was characterized by significantly higher TEM frequencies at all 3 time points. However, similar to EA07, there was a significant TEMRA subpopulation (5%–12%), and the second largest subpopulation was the TRVE phenotype (11% –15%).
Figure 6.

FCOM™ generated gp100-specific CD8+ T cell subphenotype signatures for PIVR, LTM, and P2B PBMCs from 3 representative patients. The 32 possible subphenotypes of tetramer+ CD8+ T cells are displayed on the X axis and are divided into 16 CCR7− (left) and 16 CCR7+ (right) subpopulations. Subphenotype signatures are based on the combined positive and negative staining patterns for CD45RA/CD57/CD27/CD28 mAbs on PIVR, LTM, and P2B T cells within each CCR7 compartment. Each CCR7 staining compartment is subdivided into CD45RA− and CD45RA+ subsets, and each CD45RA subpopulation is then further divided into CD57− and CD57+ T cells. Final subphenotype delineation is defined by positive or negative expression of CD27 and CD28 on T cells in each CD57 subpopulation. Key dominant subphenotypes are designated for clarity of interpretation.
T cells from EA35 were very different than those of EA02 and EA07. The two dominant subpopulations were TEM and TCM cells; there were significant increases in the frequencies of these subpopulations among LTM and P2B gp100-specific T cells compared to PIVR. The EA35 LTM and P2B TEM, and TCM frequencies were also significantly elevated compared to those of EA02 and EA07. The EA35 T-cell signature indicated that the higher LTM frequencies of TCM and TEM CD8+ T cells were sustained after boosting immunization. Notably, EA35 long-term memory and boosted gp100-specific T cells did not include TRVE memory subpopulations. Thus, the three gp100-specific T-cell signatures shown in Fig. 6 suggested one patient had an immune response dominated by TRVE memory cells similar to the memory T cells specific for CMV (EA07), while a second patient’s immune response was characterized by high frequencies of TCM and TEM CD8+ T cells, devoid of TRVE expression (EA35). T cells from a third patient (EA02) were characterized by a variation of the TRVE phenotype, and exhibited the combined expression of TRVE as well as TEM memory cells. In order to determine if other patients expressed these or other memory/effector CD8+ T-cell signatures, direct ex vivo analysis of cryopreserved PIVR, LTM, and P2B PBMCs from 7 other patients was performed. Patients were selected because they had enough gp100 specific T cells to ensure the successful acquisition of at least 5×103 – 10×103 gated gp100 tetramer+ CD8+ T cells required for statistical accuracy of subphenotype delineation. FCOM signatures from all 7 patients were compared using unsupervised cluster analysis (Supplemental Data – Figure 1). Two patients (EA07, EA29) segregated together in a major cluster characterized by dominant TRVE subphenotype expression and low frequencies of TEM and TCM gp100-specific T cells. EA02 was closely associated in a second cluster characterized by high TRVE and TEM subphenotype expression. Four other patients (EA19, EA12, EA28, EA35) segregated together in a third major cluster characterized by cells with dominant TCM, TEM, TEMRA, and TCMRA subphenotypes, but devoid of TRVE subpopulations. Details show that long-term memory and boosted T cells from these four patients had significantly higher frequencies of gp100-specific TCM cells than were detected in PIVR PBMCs (Supplemental Data – Figure 1). Increased TCM differentiation following antigen depletion represents a memory T-cell maturation trait reported for competent memory T cells in infectious disease models [42–44]. Increased TCM expression was only observed in LTM, and P2B PBMCs from these four patients – all of whom had memory T-cell signatures similar to those described for memory T cells stimulated by acute viral infection such as influenza [45, 46].
Gp100-specific T-cell signatures are homologous to those of CMV- or FLU-specific memory T cells
In order to determine if the gp100 peptide-specific T-cell signatures resembled those specific for acute and chronic viral antigens we used A2-restricted FLU(M1) peptide, and CMV (pp65) peptide-specific tetramers in the same 8-color staining panel to phenotype memory T cells from 3 patients in the trial exposed to CMV and influenza respectively. The CMV-specific signature was similar for all 3 patients (Fig. 7). The memory T cells comprised very low frequencies of TEM and TEMRA, and very low total frequencies (≤ 7%) of CCR7+ CD8+ T cells. The two dominant CMV subpopulations were CD27− and CD27+ effector or TRVE-like phenotypes. By contrast, the FLU-specific memory CD8+ T cells in all 3 patients shared a different signature; the majority of FLU-specific T cells were in four subpopulations (TEM, TEMRA, TCM, TCMRA), although the frequency of each subpopulation varied between patients. In contrast to CMV, FLU-specific T cells were highly CCR7+ - EA12 (46%), EA18 (75.7%), EA19 (76.5%), and did not include high frequencies of TRVE-like subpopulations (≤ 5%). The contrast between the signatures of CMV and FLU-specific memory T cells was especially notable in patients EA18 and EA19 since they had memory CD8+ T-cell responses to both viral antigens. The difference between FLU and CMV memory signatures was further confirmed by cluster analysis of a larger number of samples – the signatures for 8 CMV samples were very homologous, and partitioned separately from the tightly clustered FLU-specific T-cell subpopulations from 7 patients (data not shown). Consensus models based on 3–4 color flow cytometry have suggested memory CD8+ T cells specific for acute viruses such as influenza would have a dominant CD45RA−/CCR7+/CD28+/CD27+ phenotype [42, 47], whereas CD8+ memory T cells specific for chronic viruses, such as CMV, would be characterized by a dominant CD45RA+/CD57+/CCR7−/CD27−/CD28− phenotype [15, 33, 40, 41]. Eight color analysis confirmed these predictions of the consensus models. Additional cluster analysis studies were also performed to determine whether gp100-specific CD8+ T-cell signatures for individual melanoma patients segregated with either the FLU-or CMV-specific memory T-cell phenotype signatures (Supplemental Data - Figure 2). The TRVE-rich T-cell signatures of patients EA02, EA07, and EA29 clustered exclusively with the 8 CMV-specific CD8+ T-cell signatures (Supplemental Fig. 2A). Conversely, the four patients with very low TRVE subphenotype expression, but high dominant TCM, TEM, TEMRA, TCMRA subpopulations (EA12, EA19, EA28, EA35) clustered exclusively with the 7 FLU-specific CD8+ T-cell signatures (Supplemental Data Fig. 2B). Cluster analysis thus confirmed that gp100-specific memory/effector CD8+ T cells had phenotype signatures which were not unique, but were closely homologous to the distinct signatures of either CMV- or FLU-specific T cells.
Figure 7.
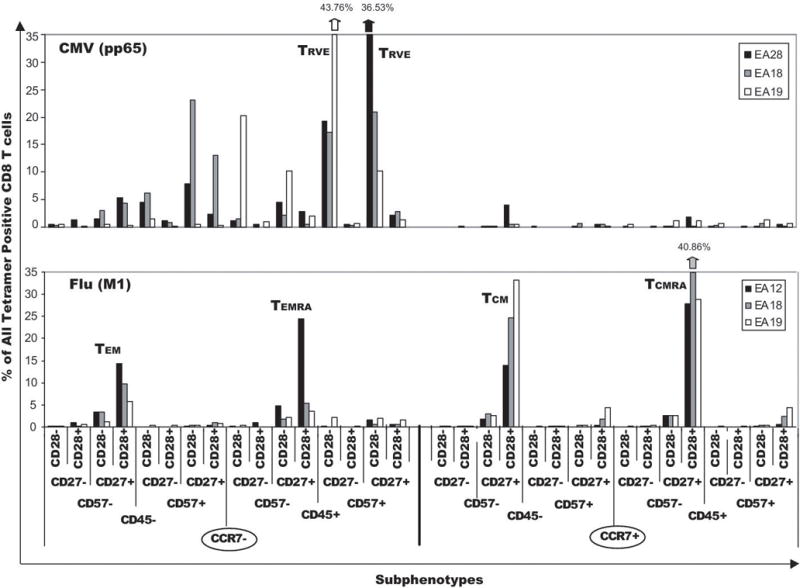
Eight-color flow cytometry subphenotype signatures of CMV- and FLU-specific CD8+ memory T cells. Subphenotypes of CMV (pp65) tetramer+ CD8+ T cells from 3 patients were all similar, and expressed dominant TRVE subpopulations (top panel) characterized by either CD27+ or CD27− staining on the CCR7−/CD45RA+/CD57+/CD28− background. FLU(M1) tetramer+ CD8+ T cells from 3 patients were also homologous, but expressed a distinctly different signature characterized by dominant TEM, TEMRA, TCM, and TCMRA subpopulations and negligible expression of TRVE T cells.
Discussion
Data presented herein show that the frequencies of long-term memory CD8+ T cells specific for the self-tumor antigen, gp100, in 16 disease-free patients previously vaccinated with gp100209-2M, were comparable to the frequencies of memory CD8+ T cells specific for the viral antigens CMV (pp65) and FLU (MI) detected in the same patients - suggesting homeostasis-driven proliferation of gp100-specific memory T cells. This conclusion is supported by the detection of high levels of CD127 on gp100-specific long-term memory and boosted T cells in some patients (data not shown). Boosted patients exhibited an anamnestic response shown by the significantly increased mean frequency of circulating gp100-specific T cells over corresponding long-term memory values for 12/16 patients after two boosting immunizations. By comparison, 4–6 repetitive vaccines given at 2–3 week intervals were required in the initial priming vaccine regimen to stimulate even marginal frequencies of gp100-specific T cells. However, the mean frequency of peptide-specific CD8+ T cells after boosting was statistically lower than the mean frequency measured after the initial series of immunizations, and did not improve despite 4–6 more boosting vaccinations over three years (data not shown). This indicates that the boosted response was not robust enough to equal or exceed the response measured following primary immunization in most patients. However, data from infectious disease prime-boost immunization studies indicate circulating memory CD8+ T cells are cleared from the blood within 3–4 weeks of immunization and rapidly traffic to peripheral organs [23, 48]. Repetitive boosting also increases the rate of trafficking to non-lymphoid organs [23]. After the second boosting immunization the subsequent follow up boosting vaccines and correlated blood draws in our study were six months apart. Thus, the failure to detect an increased frequency of circulating gp100-specific T cells may not reflect the true extent of the anamnestic response since trafficking of antigen-specific T cells to peripheral organs during the six-months between immunizations could significantly reduce circulating levels of memory T cells.
Even in the absence of high numbers of circulating memory T cells, competent memory T-cell differentiation could still be reflected by enhanced functional responses since memory T cells have increased functional avidity and proliferation potential as well as heightened cytokine responses after secondary antigen stimulation (Reviewed in [30]). Our patients exhibited no improvement in the functional avidity of long-term memory or boosted IVS T cells compared to the T-cell response measured after the initial vaccine regimen. In vitro stimulation of gp100209-2M IVS cells from all 3 time points with an A2+/gp100+ tumor cell line (Mel 118) demonstrated there was no increase in the mean frequency of degranulating CD107+ gp100-specific CD8+ T cells in the long-term memory or boosted cells compared to T cells at the completion of the initial adjuvant trial - suggesting that neither long-term memory nor boosted cells exhibited increased gp100+ tumor cell-specific CTL function. Additional statistical sub-analysis demonstrated there was no significant difference between patients with the FLU-like vs CMV-like phenotypes with regard to the functional avidity of LTM (p=0.66) or P2B (0.49) T cells. Similarly, there was no statistical difference in the LTM (p=0.79) or P2B (p=0.68) CD107 responses of T cells with the two phenotypes. By comparison, in vitro proliferation responses to cognate peptide at all time points for most patients were ≥10 fold higher than their respective ex vivo values. This was particularly true for long-term memory T cells which achieved increases (range = 50 – 242 fold) that were higher than those of either post adjuvant vaccine or boosted T cells in 8/16 patients. Notably, sub-analysis using an unpaired T test indicated that gp100-specific LTM T cells with a FLU-like signature exhibited a significant (p=0.0211) mean increase in in vitro proliferation compared to memory cells with a CMV-like signature. Memory T cells have a much reduced lag time from activation to cell cycle entry compared to naïve cells since they contain preformed cytoplasmic cyclin D3/CDK6 complexes that maintain the cells in a late G1 phase [30, 49]. This “ready-to-go” status facilitates rapid S-phase transition after re-activation [30, 49]. Memory T cells not only divide more rapidly but they also have a largely unexplained ability to survive the massive cell death that frequently occurs in vivo during the expansion phase of the immune response and also after IVS [30]. The average 6-fold increase in the in vivo frequencies of boosted circulating gp100-specific T cells over long-term memory T-cell frequencies, and the high in vitro proliferation responses of long-term memory CD8+ T cells compared to the absence of heightened functional responses in these cells may indicate that the proliferation of memory T cells is regulated by different mechanisms than those governing effector functions, and represents a separate control point for memory CD8+ T-cell differentiation [50]. Thus, repetitive class I peptide immunization in the absence of exogenous Th antigens appeared to result in circulating long-term memory T cells with increased potential for in vivo and in vitro proliferation after peptide re-challenge, but without the enhanced functional properties of fully competent memory T cells.
The inability to boost the frequency of gp100+/CD8+ T-cells to levels achieved following the initial series of vaccines could also be attributable to tolerogenic mechanisms mediated by increased regulatory T cells (Tregs) [51], myeloid suppressor cells [52], or increased expression of PD-1+/CD8+ T cells [53]. Although we have not studied myeloid suppressor cells, the frequency of PD-1+ gp100-specific T cells was comparable to those of CMV and FLU-specific memory T cells in the same patients (data not shown). Treg frequencies in individual patients varied from 2.5% – 9%, and were not higher among long-term memory or boosted T cells than in pre-vaccine and PIVR PBMCs (data not shown). Although not conclusive, this data suggested the limited anamnestic responses in some patients were not strictly due to immune suppression, but might also reflect alteration of intrinsic CD8+ memory T-cell function – possibly due to inadequate CD4+ Th cells at priming. Effective priming of CD8+ T cells in the presence of Th cells results in DNA changes that improve the transcription of certain genes [54]. Examples include the permanent and heritable demethylation of the IFN-γ locus in memory CD8+ T cells [55], and the demethylation of the IL-4 gene in Th2 CD4+ memory T cells [56]. These permanent modifications are thought to be regulated by the expression of T-box transcription factors such as T-bet and eomesodermin in memory CD8+ cells, and may be altered in the absence of CD4+ helper T cells [57, 58]. Memory CD8+ T cells generated in the absence of CD4+ Th cells (so-called “lethargic” cells), die upon re-activation with cognate antigen due to the upregulated expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which is not expressed in competent memory T cells [59]. Analysis of T-box transcription factor mRNA synthesis and TRAIL expression by long-term gp100-specific memory T cells has recently been initiated at our institute to determine if they are quantitatively different from those of competent memory T cells specific for CMV or FLU in the same patients.
Higher order polychromatic flow cytometry analysis of normal human CD8+ T cells has recently been performed using simultaneous staining with multiple antibodies specific for key cell surface markers and for perforin (Per), granzyme A (GraA) and granzyme B (GraB) [35, 36]. These studies suggested the following CD8+ T-cell differentiation pathway: early TEM (CCR7−/CD45RA−/CD27+/CCR5+/CD28+/Per−/low/GraA+/GraB−) → late TEM (CCR7−/CD45RA−/CD27low/CCR5low/CD28−/Perlow/GraA+/GraB+)→ effector (CCR7−/D45RA+/−/CD27−/CCR5low/−/CD28−/Per+/GraA+/GraB+). The sequential expression of the cytolytic molecules correlated with the concomitant loss of CD28, CD27, and CCR5 expression, and was the basis for the definitions of “early” and “late” TEM and effector CD8+ T-cell phenotypes. In a similar analysis Romero et al [60] described five major sub-phenotypes of normal human CD8+ T cells: CM (CD45RA−/CCR7+/CD27+/CD28+), EM1 (CD45RA−/CCR7−/CD27+/CD28+), EM2 (CD45RA−/CCR7−/CD27+/CD28−), EM3 (CD45RA−/CCR7−/CD27−/CD28−) and EM4 (CD45RA−/CCR7−/CD27−/CD28+). EM1 and EM4 subphenotypes were very similar, and PCR analysis of sorted cells demonstrated that, like CM T cells, they exhibited low levels of granzyme B and perforin, high levels of CD127/IL-7Rα and long telomeres suggesting a short replication history - all characteristics of memory T cells. By comparison, PCR analysis of sorted EM2 and EM3 T cells revealed elevated levels of granzyme B and perforin, minimal CD127 expression and shorter telomeres indicating more cycles of cell division - all characteristic traits of effector CD8+ T cells. EM3 cells also displayed CTL function comparable to fully differentiated effector T cells [60]. The CM and EM1/EM4 subpopulations are very similar to the gp100-specific TCM and early TEM subphenotypes respectively, and the terminal effector and EM2/EM3 T-cell subphenotypes appear very homologous to the TRVE CD8+ T-cell subpopulations described herein. Romero and coworkers further suggested that the up-regulation of cytolytic mediators, and the apparent step-wise loss of CCR7, CD27, and CD28 indicated a progressive differentiation from memory cells (EM1/EM4) to effector cells (EM2/EM3/terminal effectors) [60]. The similarity of the gp100-specific CD8+ T cells described in our study to the putative memory and effector subpopulations recently described in antigen non-specific normal human T cells, and the close homology of the gp100-specific T-cell phenotypes to those of CMV- and FLU-specific CD8+ T cells indicates that self-tumor antigen-specific memory CD8+ T cells exhibit conventional phenotypes.
It is tempting to assume that the TRVE-rich, gp100-specific CD8+ T-cell signatures of patients EA02, EA07, and EA29 indicate they may be responding to chronic antigen activation, as might occur with micrometastatic disease or from autoimmune stimulation. TCM/TEM-rich, FLU-like gp100-specific signatures of patients EA19, EA12, EA28, and EA35 might therefore conversely suggest the maintenance of competent resting memory T cells following the complete clearance of melanoma tumor antigen. However, this interpretation is not supported by other published data. CMV-specific, TRVE-like CD8+ T cells are maintained long-term in seropositive individuals and have long telomeres and increased telomerase activity, which may explain their memory-like maintenance [15, 47, 61, 62]. Analysis of CMV-specific CD8+ T cells in rhesus macaques strongly suggests that CD28− effector-memory T cells retain replication potential and are sustained by independent homeostasis-driven mechanisms comparable to those that maintain central memory CD8+ T-cell levels [63]. Homeostasis of CMV-specific effector-memory or TRVE-like T cells may not depend on IL-7, since chronic viral infection down regulates IL-7Rα (CD127) on antigen-specific T cells [15, 64]. Other data suggest the dominant TCM and TEM subpopulations that characterize some gp100-specific CD8+ T-cell memory responses may not be unique to the clearance of an acute antigen challenge since they are also detected at high frequencies in the memory T-cell response to chronic infections with EBV and HCV [65]. Moreover, increased tumor burden in melanoma patients has been correlated with the increased presence of tumor-specific CD8+ T cells exhibiting a very early resting memory phenotype (CCR7+/CD45RA+/CD27+/CD28+/perforin−) in tumor-invaded lymph nodes. This was also correlated with the absence of tumor regression in metastatic lesions [66]. Thus, the expression of memory (TCM/TEM) subpopulations of circulating CD8+ T cells without concomitant effector T cells may reflect the influence of tolerogenic/suppressive down-regulation of effector T-cell differentiation rather than the potential for fully competent memory T-cell proliferative and functional activation. This notion is also supported by a recent study that showed that the dominant CEA, MAGE and HER-2/neu-specific CD8+ T cells detected in non-immunized metastatic breast cancer patients exhibited early TEM-like phenotypes (i.e. CD45RA−/CD27+/−/CD28+), and exhibited high levels of IL-2 expression as compared to CMV- or FLU-specific T cells in the same patients [67]. While IL-2 expression is characteristic of memory T cells, the highly restricted TAA-specific memory phenotype, and the correlated absence of IFN-γ+ or TNF-α+ T cells compared to CMV- or FLU- specific T cells suggested impaired TAA-specific protective immunity in these patients [67]. The authors argued that the heightened IL-2 expression by TAA-specific memory T cells may actually contribute to immunological quiescence through the upregulation of T regulatory cells. Taken together these data suggest that a phenotype signature rich in terminal - effector or TRVE subphenotypes, similar to that commonly found in CMV+ donors, could indicate the homeostasis-driven maintenance of a memory T-cell component capable of self-replication. The data similarly support the concept that a TCM/TEM-rich resting memory T-cell signature like that found after the clearance of an acute viral challenge could reflect a state of immunological anergy rather than anamnestic potential if tolerogenic forces are in play. Notably, our study indicated there was no statistical difference in functional avidity, or CD107 mobilization exhibited by gp100-specific IVS CD8+ T cells from patients expressing the two major phenotypes. By contrast, there was a significantly higher peptide-stimulated in vitro proliferation response of memory T cells with the FLU-like signature. However, the clinical significance of this observation is unclear at present since there was no clinical correlation with either subphenotype signature, and there was no evidence of vitiligo in any patient. Moreover, both the CMV-like and FLU-like phenotype signatures were observed with equal frequency in the four booster patients who have subsequently developed progressive disease (data not shown). At present the biological/clinical significance of these phenotype differences in our study remains unresolved. Future clinical trials with much larger patient enrollment may provide the statistical power to determine if there is diagnostic/prognostic relevance to the two distinct self-tumor antigen-specific memory T-cell phenotype signatures.
In conclusion, these data demonstrate that, even in otherwise healthy disease-free melanoma patients, the therapeutic vaccine strategy employed did not induce fully competent gp100-specific memory CD8+ T cells. The data presented suggest that while repetitive class I-restricted melanoma peptide vaccination by itself can break tolerance to self-tumor antigens, other vaccine strategies will be required to induce functionally competent long-term memory T cells capable of robust anamnestic response to tumor challenge.
Supplementary Material
Acknowledgments
We thank Dr. Andrew Weinberg and Dr. Nora Disis for reviewing the manuscript. We also thank Tanisha Meeuwsen and Iliana Gonzalez for their technical assistance and Molly Torgeson for her excellent assistance in preparation of the paper. We additionally thank the Cancer Therapy Evaluation Program (CTEP) for providing the gp100209-2M peptide (NSC#683472) and Montanide ISA 51 for our study.
Grant Support
Supported by NIH IR21-CS82614-01 (W.J.U.), NIH RA21-CA099265-02 (W.J.U), NIH R01-CA119123 (B.A.F.) the Murdock Charitable Trust and the Chiles Foundation.
Footnotes
References
- 1.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–7. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 3.Ayyoub M, Zippelius A, Pittet MJ, et al. Activation of human melanoma reactive CD8+ T cells by vaccination with an immunogenic peptide analog derived from Melan-A/melanoma antigen recognized by T cells-1. Clin Cancer Res. 2003;9:669–77. [PubMed] [Google Scholar]
- 4.Speiser DE, Romero P. Toward improved immunocompetence of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1467–9. doi: 10.1172/JCI25427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–6. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts AD, Ely KH, Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med. 2005;202:123–33. doi: 10.1084/jem.20050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–24. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powell DJ, Jr, Rosenberg SA. Phenotypic and functional maturation of tumor antigen-reactive CD8+ T lymphocytes in patients undergoing multiple course peptide vaccination. J Immunother (1997) 2004;27:36–47. doi: 10.1097/00002371-200401000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JW, 2nd, Walker EB, Fox BA, et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol. 2003;21:1562–73. doi: 10.1200/JCO.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Walker EB, Haley D, Miller W, et al. gp100(209-2M) peptide immunization of human lymphocyte antigen-A2+ stage I-III melanoma patients induces significant increase in antigen-specific effector and long-term memory CD8+ T cells. Clin Cancer Res. 2004;10:668–80. doi: 10.1158/1078-0432.ccr-0095-03. [DOI] [PubMed] [Google Scholar]
- 13.Champagne P, Ogg GS, King AS, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–11. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 14.Kuijpers TW, Vossen MT, Gent MR, et al. Frequencies of circulating cytolytic, CD45RA+CD27−, CD8+ T lymphocytes depend on infection with CMV. J Immunol. 2003;170:4342–8. doi: 10.4049/jimmunol.170.8.4342. [DOI] [PubMed] [Google Scholar]
- 15.van Leeuwen EM, de Bree GJ, ten Berge IJ, van Lier RA. Human virus-specific CD8+ T cells: diversity specialists. Immunol Rev. 2006;211:225–35. doi: 10.1111/j.0105-2896.2006.00379.x. [DOI] [PubMed] [Google Scholar]
- 16.Meijer SL, Dols A, Jensen SM, Hu HM, et al. Induction of circulating tumor-reactive CD8+ T cells after vaccination of melanoma patients with the gp100 209-2M peptide. J Immunother (1997) 2007;30:533–43. doi: 10.1097/CJI.0b013e3180335b5e. [DOI] [PubMed] [Google Scholar]
- 17.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–55. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 18.Bagwell CB. Hyperlog-a flexible log-like transform for negative, zero, and positive valued data. Cytometry A. 2005;64:34–42. doi: 10.1002/cyto.a.20114. [DOI] [PubMed] [Google Scholar]
- 19.Petrausch U, Haley D, Miller W, et al. Polychromatic flow cytometry: a rapid method for the reduction and analysis of complex multiparameter data. Cytometry A. 2006;69:1162–73. doi: 10.1002/cyto.a.20342. [DOI] [PubMed] [Google Scholar]
- 20.Tung JW, Parks DR, Moore WA, Herzenberg LA. New approaches to fluorescence compensation and visualization of FACS data. Clin Immunol. 2004;110:277–83. doi: 10.1016/j.clim.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–8. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 22.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–9. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- 24.Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med. 1999;189:701–10. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kedl RM, Schaefer BC, Kappler JW, Marrack P. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat Immunol. 2002;3:27–32. doi: 10.1038/ni742. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez F, Harkins S, Slifka MK, Whitton JL. Immunodominance in virus-induced CD8(+) T-cell responses is dramatically modified by DNA immunization and is regulated by gamma interferon. J Virol. 2002;76:4251–9. doi: 10.1128/JVI.76.9.4251-4259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2:711–7. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 28.Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169:3492–8. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- 29.Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol. 2000;1:47–53. doi: 10.1038/76907. [DOI] [PubMed] [Google Scholar]
- 30.Rocha B, Tanchot C. The Tower of Babel of CD8+ T-cell memory: known facts, deserted roads, muddy waters, and possible dead ends. Immunol Rev. 2006;211:182–96. doi: 10.1111/j.0105-2896.2006.00378.x. [DOI] [PubMed] [Google Scholar]
- 31.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 32.van Baarle D, Kostense S, van Oers MH, Hamann D, Miedema F. Failing immune control as a result of impaired CD8+ T-cell maturation: CD27 might provide a clue. Trends Immunol. 2002;23:586–91. doi: 10.1016/s1471-4906(02)02326-8. [DOI] [PubMed] [Google Scholar]
- 33.Kern F, Khatamzas E, Surel I, et al. Distribution of human CMV-specific memory T cells among the CD8pos. subsets defined by CD57, CD27, and CD45 isoforms. Eur J Immunol. 1999;29:2908–15. doi: 10.1002/(SICI)1521-4141(199909)29:09<2908::AID-IMMU2908>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 34.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–6. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 35.Tomiyama H, Matsuda T, Takiguchi M. Differentiation of human CD8(+) T cells from a memory to memory/effector phenotype. J Immunol. 2002;168:5538–50. doi: 10.4049/jimmunol.168.11.5538. [DOI] [PubMed] [Google Scholar]
- 36.Takata H, Takiguchi M. Three memory subsets of human CD8+ T cells differently expressing three cytolytic effector molecules. J Immunol. 2006;177:4330–40. doi: 10.4049/jimmunol.177.7.4330. [DOI] [PubMed] [Google Scholar]
- 37.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 38.Brenchley JM, Karandikar NJ, Betts MR, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–20. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 39.Faint JM, Annels NE, Curnow SJ, et al. Memory T cells constitute a subset of the human CD8+CD45RA+ pool with distinct phenotypic and migratory characteristics. J Immunol. 2001;167:212–20. doi: 10.4049/jimmunol.167.1.212. [DOI] [PubMed] [Google Scholar]
- 40.Khan N, Shariff N, Cobbold M, et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169:1984–92. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- 41.Chen G, Shankar P, Lange C, et al. CD8 T cells specific for human immunodeficiency virus, Epstein-Barr virus, and cytomegalovirus lack molecules for homing to lymphoid sites of infection. Blood. 2001;98:156–64. doi: 10.1182/blood.v98.1.156. [DOI] [PubMed] [Google Scholar]
- 42.Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–34. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 43.Marzo AL, Klonowski KD, Le Bon A, et al. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–9. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Faassen H, Saldanha M, Gilbertson D, et al. Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J Immunol. 2005;174:5341–50. doi: 10.4049/jimmunol.174.9.5341. [DOI] [PubMed] [Google Scholar]
- 45.de Bree GJ, Heidema J, van Leeuwen EM, et al. Respiratory syncytial virus-specific CD8+ memory T cell responses in elderly persons. J Infect Dis. 2005;191:1710–8. doi: 10.1086/429695. [DOI] [PubMed] [Google Scholar]
- 46.He XS, Mahmood K, Maecker HT, et al. Analysis of the frequencies and of the memory T cell phenotypes of human CD8+ T cells specific for influenza A viruses. J Infect Dis. 2003;187:1075–84. doi: 10.1086/368218. [DOI] [PubMed] [Google Scholar]
- 47.Wallace DL, Zhang Y, Ghattas H, Worth A, Irvine A, Bennett AR, Griffin GE, Beverley PC, Tough DF, Macallan DC. Direct measurement of T cell subset kinetics in vivo in elderly men and women. J Immunol. 2004;173:1787–94. doi: 10.4049/jimmunol.173.3.1787. [DOI] [PubMed] [Google Scholar]
- 48.Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J Immunol. 2005;175:4686–96. doi: 10.4049/jimmunol.175.7.4686. [DOI] [PubMed] [Google Scholar]
- 49.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol. 2004;5:31–7. doi: 10.1038/ni1015. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez J, Aung S, Marquardt K, Sherman LA. Uncoupling of proliferative potential and gain of effector function by CD8(+) T cells responding to self-antigens. J Exp Med. 2002;196:323–33. doi: 10.1084/jem.20011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 52.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 53.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 54.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–75. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- 55.Fitzpatrick DR, Shirley KM, Kelso A. Cutting edge: stable epigenetic inheritance of regional IFN-gamma promoter demethylation in CD44highCD8+ T lymphocytes. J Immunol. 1999;162:5053–7. [PubMed] [Google Scholar]
- 56.Ouyang W, Lohning M, Gao Z, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 57.Pearce EL, Mullen AC, Martins GA, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–3. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 58.Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–44. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 59.Janssen EM, Droin NM, Lemmens EE, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 60.Romero P, Zippelius A, Kurth I, et al. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J Immunol. 2007;178:4112–9. doi: 10.4049/jimmunol.178.7.4112. [DOI] [PubMed] [Google Scholar]
- 61.Hathcock KS, Kaech SM, Ahmed R, Hodes RJ. Induction of telomerase activity and maintenance of telomere length in virus-specific effector and memory CD8+ T cells. J Immunol. 2003;170:147–52. doi: 10.4049/jimmunol.170.1.147. [DOI] [PubMed] [Google Scholar]
- 62.Hamann D, Kostense S, Wolthers KC, et al. Evidence that human CD8+CD45RA+CD27− cells are induced by antigen and evolve through extensive rounds of division. Int Immunol. 1999;11:1027–33. doi: 10.1093/intimm/11.7.1027. [DOI] [PubMed] [Google Scholar]
- 63.Pitcher CJ, Hagen SI, Walker JM, et al. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 64.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004;101:16004–9. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Appay V, Dunbar PR, Callan M, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–85. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 66.Mortarini R, Piris A, Maurichi A, et al. Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res. 2003;63:2535–45. [PubMed] [Google Scholar]
- 67.Inokuma M, dela Rosa C, Schmitt C, et al. Functional T cell responses to tumor antigens in breast cancer patients have a distinct phenotype and cytokine signature. J Immunol. 2007;179:2627–33. doi: 10.4049/jimmunol.179.4.2627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.