

Abstract
The importance of cation->aromatic polarization effects on cation-π interactions has been explored. Theoretical calculations demonstrate that polarization is a large contribution to cation-aromatic interactions, and particularly to cation-π interactions. For a series of compounds with a similar aromatic core, polarization is constant and makes small influence in the relative cation-binding energies. However, when the aromatic core changes polarization contributions might be very different. We found that the generalized molecular interaction potential with polarization is a very fast and powerful tool for the prediction of cation binding of aromatic compounds.
Cation-π is a strong, and quite specific, interaction, which plays a key role in molecular recognition (for an excellent review see ref. 1). Thus, cation-π interactions are relevant in host-guest complexes (1–5). In addition, they are common in protein structure (6–8), where these interactions seem to be related with general mechanisms of substrate-enzyme binding (8–12) and probably with some catalytic processes (13–16). Finally, Dougherty and coworkers (17, 18) recently have suggested that these interactions are essential for the recognition and action of ion channels.
Cation-π complexes involve aromatic molecules having large and well-defined π-electron distributions, and the cations lie perpendicular to the aromatic plane. Dougherty and coworkers (1, 7, 18, 19) have shown that, in the case of nonpolarizable cations such as Na+, the preferential binding to different aromatic compounds can be explained from electrostatic considerations. Particularly, for a series of 11 derivatives of benzene, they found an excellent correlation (r = 0.991, slope = 1.01, intercept = 11.6 kcal/mol) between the self-consistent field (SCF) binding energies and the molecular electrostatic potential (MEP; Eq. 1) at the SCF-optimized position of the Na+ in the complex (19). More interesting, when the MEP was computed at a common position for all of the molecules (2.47 Å above the aromatic ring), the correlation with the SCF binding energy was also excellent (r = 0.992, slope = 1.04, intercept = 12.3 kcal/mol). This finding suggests that a simple MEP calculation can provide very accurate estimates of the relative values of cation binding energy for a given series of aromatic compounds (18, 19).
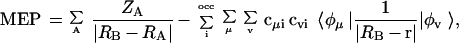 |
1 |
where RB and RA stands for the positions of the cation and nuclei, and c stands for the coefficients of atomic orbitals in the molecular orbital-linear combination atomic orbitals (MO-LCAO) approximation.
The pure electrostatic model seems very useful for a qualitative description of cation-π interactions. However, the electrostatic component of the cation-π interaction energy is always larger (in absolute terms) than the MEP value. Therefore, other interactions are involved in the formation of cation-π complexes. For the particular case of Na+, the charge transfer is expected to be small. Indeed, the polarization of the π-electron system by the cation will have a significant contribution, whereas the reverse effect is expected to be sensibly lower. In fact, the relevance of polarization effects in cation-π interactions has been previously emphasized by Kollman and Caldwell (20) by using classical force-field calculations.
In this paper we present a systematic quantum mechanical study on the contribution of polarization effects to cation-π interactions. We also analyze the possibility to introduce these polarization effects into “reactivity indexes,” which are used for the prediction of the strength of cation-π interactions.
METHODS
The study is performed by using a perturbational approach to compute the polarization energy. Particularly, we analyze the suitability of the recently developed generalized molecular interaction potential with polarization (GMIPp; refs. 21–23) as an improved computational generalization of the MEP for the a priori description of chemical reactivity. Three terms contribute to the interaction energy in the GMIPp: (i) an electrostatic term identical to the MEP, (ii) a classical dispersion-repulsion term (refs. 22 and 23; §), and (iii) a polarization term derived from perturbational theory (21–23). Let us note that the polarization effects are estimated at a reduced computational effort and that the perturbational and SCF values of the polarization energy are very similar (ref. 23; ¶). For the case of the interaction with Na+, the GMIPp is given by Eq. 2.
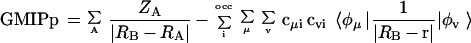 |
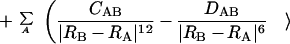 |
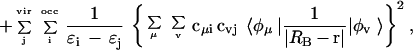 |
2 |
where CAB and DAB are the van der Waals interaction parameters§, and ɛi denotes the energy of molecular orbital i.
Calculations were performed by using the HF/6–31G(d,p) geometries and wavefunctions, because this level of theory provides (probably because of fortuitous error cancellation) very accurate values of the cation-π binding energy (refs. 1, 7, 18, and 19; ‖). Empirical van der Waals parameters were used§. In all calculations the Na+ was considered as a classical nonpolarizable particle.
Wavefunction calculations have been performed by using the gaussian 94 computer program (27); MEP and GMIPp calculations were performed with the mopete program (28). All calculations have been carried out on the IBM-SP2 computer of the Centre de Supercomputació de Catalunya and on workstations in our laboratory.
RESULTS AND DISCUSSION
We first explored the profiles for the electrostatic (Eele), polarization (Epol), and total (Etot) binding energies when a Na+ approaches a benzene molecule. Three orientations were considered (see Fig. 1): (i) along the middle of a C-C bond (x orientation), (ii) along the C-H bond (y orientation), and (iii) perpendicular to the center of the aromatic ring (z orientation). In all cases we determined the “hard particle radius,” which was defined as the point where dispersion and repulsion cancel each other. Then, we computed Eele, Epol, and Etot there and in points closer to and farther away from the molecule.
Figure 1.

Orientations of cation-aromatic interactions in the study. The z axis corresponds to the cation-π interaction.
The energy profiles (see Fig. 2) provide insight into the nature of cation-π interactions. At very short distances the repulsive term dominates the interaction, whereas the electrostatic component is the leading term at very large distances. At intermediate values (near the hard sphere radius) the magnitude of Eele and Epol is in general similar. As expected, the electrostatic term is positive for x and y orientations, whereas it is large and negative for the approach along the z axis. The polarization term is always negative and seems to follow a r−3 dependence, compared with well-known r−1 dependence of the electrostatic term.
Figure 2.

GMIPp energy profiles for the approach of Na+ to benzene (x axis: dotted line; y axis: dashed line; z-axis: solid line). (Top) Electrostatic energy. (Middle) Polarization energy. (Bottom) Total energy. All the values are in kcal/mol. Distances (in Å) are referred in all cases to the hard sphere radius (see text). Hard sphere radii are: 4.58 (x axis), 4.97 (y axis), and 3.24 Å (z axis).
When the Na+ approaches along the x and y axis, the polarization energy is larger (in absolute value) than the electrostatic term for distances shorter than the hard sphere radius plus 0.8 (y axis) or 1.2 (x axis) Å. This suggests that the energy profile for the cation approach to the benzene in the aromatic plane is not fully repulsive, as noted in the minima in the Etot profiles of −3.8 (at −0.6 Å; around 4 Å from the center of the ring) and −1.5 (at −0.4 Å; around 4.6 Å from the center of the ring) kcal/mol for the x and y orientations. The existence of these minima was verified by performing SCF calculations, which provided binding energies of −4.6 and −2.0 kcal/mol, respectively. It then is clear that the large magnitude of the aromatic polarization stabilizes the in-plane approach of cations in spite of the strong electrostatic repulsion.
The approach of the cation along the z axis is clearly favored by the electrostatic term. This component is greater (in absolute terms) than the polarization for distances larger than −1.3 Å. The polarization energy is 44% the magnitude of the electrostatic energy at the hard sphere radius, and 70% the magnitude of the electrostatic term at the optimum distance of 2.47 Å (around −0.8 Å). Note in Fig. 2 that the SCF and GMIPp optimum Na+-benzene distances are very similar (within 0.1 Å). It is then clear that polarization has a non-negligible contribution to the total cation-π binding energy.
Inspection of Epol profiles shows that at large distances Epol is very similar irrespectively of the cation orientation. At intermediate distances the x orientation leads to the largest polarization, whereas the smallest effects are found for the z orientation, but the differences are in any case small. At the hard sphere radius Epol is −5.2 (x axis), −4.3 (y axis), and −4.2 (z axis) kcal/mol. At inner distances the y orientation provides the largest polarization, as noted in the values of Epol 95 at a distance of −1.0 Å: −16.0 (x axis), −16.5 (y axis), and −13.2 kcal/mol (z axis). In summary, polarization favors the in-plane approach of cations, but the small difference with respect to the z-axis orientation cannot revert the electrostatic preference for the perpendicular orientation.
After examining the dependence of Eele, Epol, and Etot on orientation factors, we explored the polarization contribution to the interaction of Na+ for a series of 16 aromatic compounds (see Fig. 3). This also allowed us to explore the suitability of the GMIPp as an alternative to the MEP for the a priori description of cation-π interactions. Following Dougherty and coworkers (18, 19), we computed the MEP and the GMIPp at 2.47 Å above the aromatic ring and compared these values with the optimum SCF interaction energies**. The results in Table 1 point out the importance of the polarization component, which is in general similar to the electrostatic one, and for six of the 16 compounds it is even larger than the electrostatic term. The polarization energy is mainly related to the size of the aromatic system, with little dependence on the nature of the substituents. Thus, for benzene and their substituted derivatives Epol ranges from −10 to −11 kcal/mol, a value that is around 1 kcal/mol larger (in absolute terms) to those found for five-membered rings. Larger aromatic rings provide larger values of Epol. Thus, the Epol for indole is −12.4, and for the 10-membered rings of naphthalene and azulene amounts to −13.7 and −16.2 kcal/mol.
Figure 3.

Aromatic structures considered in this study.
Table 1.
Electrostatic (MEP) energy, polarization energy, and total energy (GMIPp and SCF) for the interaction of Na+ with several aromatic systems
| Molecule | Eele(MEP) | Epol | GMIPp | E(SCF) |
|---|---|---|---|---|
| 1 | 15.0 | 9.9 | 21.9 | 27.1 |
| 2 | 14.2 | 13.7 | 25.1 | 28.7 |
| 3 | 18.8 | 12.4 | 28.2 | 32.6 |
| 4 | 17.1 | 16.2 | 29.5 | 34.1 |
| 5 | 9.8 | 10.0 | 16.8 | 22.0 |
| 6 | 14.0 | 10.3 | 21.3 | 26.9 |
| 7 | 19.3 | 10.7 | 27.0 | 31.8 |
| 8 | 11.1 | 11.1 | 19.2 | 24.4 |
| 9 | 9.3 | 10.9 | 17.1 | 21.5 |
| 10 | 3.3 | 11.4 | 11.7 | 15.7 |
| 11 | 4.6 | 10.1 | 11.6 | 16.8 |
| 12 | −0.3 | 10.3 | 6.9 | 12.4 |
| 13 | 18.1 | 9.7 | 24.4 | 29.6 |
| 14 | 11.2 | 9.2 | 16.7 | 21.0 |
| 15 | 11.2 | 9.4 | 17.3 | 21.0 |
| 16 | 8.4 | 9.6 | 15.2 | 20.0 |
MEP, Epol, and GMIPp were computed by placing the Na+ at 2.47 Å above the aromatic ring. The SCF values were computed by optimizing the complex geometry. All the values are in kcal/mol. (See Fig. 3.)
Inspection of the benzene series shows that the introduction of electron withdrawing groups leads to a dramatic reduction of the electrostatic energy, as noted by Dougherty and coworkers (18, 19), whereas Epol shows little variation (see Table 1). Accordingly, the nature of the cation-π interaction changes from mostly electrostatic to be dominated by polarization effects. Thus, Epol is 55% of Eele for aniline, 102% for the fluoro derivative, 345% for the cyano derivative, and more than 3,000% for the tri-fluoro derivative. Therefore, even though a pure electrostatic model is not able to reproduce accurately the cation-π interaction, it can provide excellent relative values for a series of aromatic compounds of similar size, in agreement with previous studies (18).
To analyze the reliability of the electrostatic model on the size of the aromatic system, we examined the correlation between SCF and MEP values. The results (see Fig. 4A) show that the electrostatic model performs well, but obviously worse than on application to the series of benzene derivatives. Thus, the model explains 93% of the variance of the results, the intercept is −11.6 kcal/mol, and the slope is 1.08. However, a detailed inspection of the results in Table 1 allows us to detect some of the most clear shortcomings of the electrostatic model. For instance, it predicts benzene as better cation binder than naphthalene, which is in disagreement with SCF results. Similarly, the boron derivative of benzene and furan show the same electrostatic energy, but the first binds Na+ better according to the SCF values. Another example is furnished by the cation binding to indole and azulene, which is predicted to be better for indole by the MEP, whereas the opposite trend is found at the SCF level. In all of these cases the polarization varies sensibly and modulates the total interaction energy.
Figure 4.

Correlations between SCF and MEP (A) and GMIPp (B) values.
The performance of the GMIPp model can be examined from the correlation analysis given in Fig. 4B, which shows the superiority of this computational tool. Thus, 99% of the variance in the SCF results is reproduced by the model, the slope is 0.984 and the intercept amounts to −5.0 kcal/mol. The value of the intercept reflects the basis set superposition error (BSSE), which was not corrected in the SCF values following Dougherty’s suggestions (18, 19) (for instance, the BSSE for benzene is −1.8 kcal/mol, and for naphthalene is −1.6 kcal/mol), and the remaining 60% of the intercept might arise from uncertainties in the classical evaluation of the repulsion contribution (the van der Waals component is around 3 kcal/mol), to Na+ polarization, and obviously to charge transfer effects. In fact, Bader’s analysis (29) for the benzene-Na+ system shows that around 0.05 electrons are transferred from benzene to Na+, which can account for a few kcal/mol of stabilization in the complex formation‡‡.
Overall, the improvement of the GMIPp with respect to the MEP is clear, as shown by the increase of ≈6% in the descriptive ability of the model, a reduction of ≈7% in the deviation of the slope from the ideal value, and the notable reduction of ≈6.5 kcal/mol in the systematic deviation noted by the intercept. Furthermore, the GMIPp reproduces the differences in Na+ binding to benzene/naphthalene, furan/Phe-BH2, and indole/azulene. Computationally, the GMIPp requires a limited increase in computer time compared with the MEP, because the recomputation of the wavefunction is not necessary, and Epol is evaluated by using the same monoelectron integrals involved in the calculation of the MEP. Furthermore, because the GMIPp is based on perturbational treatment, it provides a natural partitioning of the interaction energy into intuitive components, without the uncertainties derived from basis set superposition error. All of these results suggest the GMIPp as a more precise alternative to the MEP for the quantitative analysis of cation-π interactions, specially in cases where aromatic systems of different sizes are to be studied.
Acknowledgments
This work has been supported by the Spanish Dirección General de Investigación Cientifica y Técnica (PB96-1005), and by the Centre de Supercomputació de Catalunya (CESCA, Molecular Recognition Project).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: SCF, self-consistent field; MEP, molecular electrostatic potential; GMIPp, generalized molecular interaction potential with polarization; Eele, electrostatic binding energy; Epol, polarization binding energy; Etot, total binding energy.
C and D were computed with geometrical combinatorial rules: CAB = 4ɛABσAB12 and DAB = 4ɛABσAB6, where ɛAB = (ɛA ɛB)0.5 and σAB = (σA σB)0.5. van der Waals parameters (ɛ, σ) for C, N, and H were taken from an in-house quantum mechanical–molecular mechanical (QM-MM) parametrization (H: ɛ = 0.0116, σ = 2.272; C: ɛ = 0.0488, σ = 3.77, N: ɛ = 0.0521, σ = 3.225), and the rest of van der Waals parameters for aromatic compounds were taken from the optimized potentials for liquid simulations (OPLS) force-field (24). Parameters for Na+ were taken from Aqvist’s parameters (25). Parameters for boron were taken equal to those of carbon.
As an example, the SCF estimates (kcal/mol) of cation-π interactions at the optimum SCF distances are −9.3 for benzene and −8.9 for furan, which compare with the GMIPp values of −9.9 and −9.1 kcal/mol, respectively.
Single point calculations (HF/6–31G(d,p) geometries) were performed for the Na+… Phe dimer at the MP2/6–311++G(2d,p) level. The interaction energy was (after basis set superposition error correction) −22.0 kcal/mol (−21.2 if the Na+ is fixed at 2.47 Å above the aromatic ring). These values compare well with the HF/6–31G(d) values (−27.1 kcal/mol). Experimental enthalpy of interaction determined by Guo et al. (26) for this system is −28 kcal/mol.
Geometry optimization leads to optimum distances that can be different from the “average” value of 2.47 Å, as noted previously by Dougherty and coworkers (18, 19). However, the difference between the optimized distance and 2.47 Å is less than 0.05 Å for all molecules, except for compounds 10, 11, and 12 (optimum distances of 2.53 Å, 2.54 Å, and 2.59 Å, respectively). In addition to the situation for compounds 15 and 16 (see text), geometry optimization can lead to slight deviations of the Na+ with respect to the center of the aromatic ring. The largest deviations are found for compounds 3 (0.20 Å), 14 (0.20 Å), and 4 (0.19 Å). Finally, as noted in ref. 18, molecules containing heteroatoms might have an additional minima corresponding to a cation-σ interaction.
Correlations were performed from the GMIPp at 2.47 Å above the aromatic ring instead of the SCF or GMIPp optimized geometry. Calculations performed by using these optimized geometries provided almost identical results to those obtained for the frozen geometry, as previously reported for MEP calculations by Dougherty and coworkers (18, 19).
References
- 1.Ma J C, Dougherty D A. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 2.Shepodd T J, Petti M A, Dougherty D A. J Am Chem Soc. 1986;108:6085–6087. doi: 10.1021/ja00279a093. [DOI] [PubMed] [Google Scholar]
- 3.Shepodd T J, Petti M A, Dougherty D A. J Am Chem Soc. 1988;110:1983–1985. doi: 10.1021/ja00279a093. [DOI] [PubMed] [Google Scholar]
- 4.Petti M A, Shepodd T J, Dougherty D A. J Am Chem Soc. 1988;110:6825–6840. doi: 10.1021/ja00279a093. [DOI] [PubMed] [Google Scholar]
- 5.Arnecke R, Böhmer V, Cacciapaglia R, Cort A D, Mandolini L. Tetrahedron. 1997;53:4901–4908. [Google Scholar]
- 6.Burley S K, Petsko G A. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 7.Dougherty D A. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 8.Scrutton N A, Raine A R C. Biochem J. 1996;319:1–8. doi: 10.1042/bj3190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sussman J L, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman L. Science. 1991;256:7781–7785. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 10.Ortiz A R, Pisabarro T, Gallego J, Gago F. Biochemistry. 1992;31:2887–2896. doi: 10.1021/bi00126a007. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz A R, Pisabarro T, Gallego J, Gago F. J Med Chem. 1993;36:1866–1879. doi: 10.1021/jm00065a010. [DOI] [PubMed] [Google Scholar]
- 12.Galzi J L, Revah F, Bessis A, Changeux J P. Annu Rev Pharmacol. 1992;31:37–72. doi: 10.1146/annurev.pa.31.040191.000345. [DOI] [PubMed] [Google Scholar]
- 13.Stauffer D A, Barrans J R E, Dougherty D A. Angew Chem Int Ed Engl. 1990;29:915–918. [Google Scholar]
- 14.McCurdy A, Jiménez L, Stauffer D A, Dougherty D A. J Am Chem Soc. 1992;114:10314–10321. [Google Scholar]
- 15.Shi Z, Butel C J, Griffin J H. Proc Natl Acad Sci USA. 1994;91:7370–7374. doi: 10.1073/pnas.91.15.7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heginbotham L, MacKinnon R. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- 17.Kumpf R A, Dougherty D A. Science. 1993;261:1708–1710. doi: 10.1126/science.8378771. [DOI] [PubMed] [Google Scholar]
- 18.Mecozzi S, West A P, Dougherty D A. Proc Natl Acad Sci USA. 1996;93:10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mecozzi S, West A P, Dougherty D A. J Am Chem Soc. 1996;118:2307–2308. [Google Scholar]
- 20.Caldwell J W, Kollman P A. J Am Chem Soc. 1995;117:4177–4178. [Google Scholar]
- 21.Francl M M. J Phys Chem. 1985;89:428–434. [Google Scholar]
- 22.Orozco M, Luque F J. In: Molecular Electrostatic Potentials: Concepts and Applications. Murray J S, Sen K, editors. Vol. 3. Amsterdam: Elseiver; 1996. pp. 181–218. [Google Scholar]
- 23.Luque, F. J. & Orozco, M. (1998) J. Comp. Chem., in press.
- 24.Jorgensen W L, Tirado-Rives J. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 25.Aqvist J. J Phys Chem. 1990;94:8021–8024. [Google Scholar]
- 26.Guo B C, Purnell J W, Castleman A W. Chem Phys Lett. 1990;168:155–160. [Google Scholar]
- 27.Frisch M J, Trucks G W, Schlegel H B, Gill P M W, Johnson B G, Robb M A, Cheeseman J R, Keith T A, Petersson G A, Montgomery J A, et al. gaussian 94, Rev. A.1. Pittsburgh: Gaussian; 1995. [Google Scholar]
- 28.Luque F J, Orozco M. mopete. Spain: University of Barcelona; 1997. [Google Scholar]
- 29.Bader R F W, Carroll M T, Cheeseman J R, Chang C. J Am Chem Soc. 1987;109:7968–7979. [Google Scholar]