

Abstract
Background & Aims
Leptin has pro-fibrogenic effects in liver, although the mechanisms of this process are unclear. We sought to elucidate the direct and indirect effects of leptin on hepatic stellate cells (HSCs).
Methods
HSCs isolated from Sprague-Dawley rats were exposed to leptin; expression of collagen-I, tissue inhibitor of matrix metalloproteinases-1 (TIMP1), transforming growth factor β1 (TGFβ1) and connective tissue growth factor (CTGF/CCN2) was assessed by quantitative PCR. The effects of medium from Kupffer cells (KCs) and sinusoidal endothelial cells (SECs) following incubation with leptin were evaluated in HSCs; α-smooth muscle actin (αSMA) production and KC signaling were analyzed.
Results
HSCs were not activated by incubation with leptin. However, HSCs cultured with medium taken from KCs that had been incubated with leptin increased expression of genes that encode the pro-fibrogenic factors collagen I, TIMP1, TGFβ1and CTGF/CCN2, as well as αSMA protein levels and proliferation. These effects were leptin-receptor dependent, because conditioned medium from KCs that were isolated from leptin receptor-deficient Zucker (fa/fa) rats did not activate HSCs. In KCs incubated with leptin, mRNA and protein expression of TGFβ1 and CTGF/CCN2 increased. Leptin potentiated STAT3, AKT and ERK1/2 phosphorylation in KCs and increased AP-1 and NF-κB DNA binding. Finally, addition of anti-TGFβ to KC-conditioned medium inhibited HSC expression of collagen I, TIMP1 and CTGF/CCN2, whereas a STAT3 inhibitor attenuated TGFβ1 production by KC.
Conclusions
Leptin mediates HSC activation and liver fibrosis through indirect effects on KC; these effects are partly mediated by TGFβ1.
Introduction
Leptin, an adipocyte-derived hormone has important effects in regulating body weight, metabolism and reproductive function. Circulating levels of leptin are known to be increased in overweight and obese persons, in individuals with nonalcoholic steatohepatitis,1,2 and in those with alcoholic liver disease and chronic viral hepatitis.3–5 More recently, leptin has been shown to possess direct profibrogenic activity in the liver,6–8 while the absence of leptin is associated with a marked attenuation of the hepatic response to a diverse range of fibrotic stimuli.6,7 We previously demonstrated that leptin-deficient ob/ob mice failed to develop hepatic fibrosis in a rodent nutritional model of steatohepatitis and in response to chronic CCl4-induced liver injury.6 Restitution of physiological levels of circulating leptin restored the liver’s ‘fibrogenic’ capacity.6 Similar results have been obtained in models of fibrosis associated with bile duct ligation9 and following the administration of thioacetamide.10 The cellular and molecular mechanisms for this effect, however, have not been fully elucidated.
Since HSCs are the main source of extracellular matrix (ECM) during the evolution of fibrosis, the effects of leptin on HSC behavior have been examined, but results are conflicting. One view holds that leptin acts directly on HSCs to trigger downstream response pathways that ultimately lead to ECM deposition.7,11 Others suggest that KCs and/or SECs contain a functional leptin receptor, which can stimulate the release of profibrogenic mediators such as TGFβ1 that in turn drives HSC activation.12 To date, there is no evidence to indicate that leptin-primed KCs or SECs exert direct stimulatory effects on HSCs. However, it is well known that KCs and SECs play important roles in modulating stellate cell behavior by releasing proinflammatory and profibrogenic factors such as TGFβ1 and reactive oxygen species (ROS) upon stimulation by various noxious stimuli. In addition, recent data indicate that KC dysfunction as evidenced by decreased TNFα production and down-regulation of TGFβ1 gene expression occurs in leptin receptor-deficient Zucker rats and may account for the attenuation of liver fibrosis in these rodents following the administration of pig serum.13
In this study, we undertook detailed in vitro experiments to clarify the cellular and molecular mechanisms whereby leptin exerts profibrogenic effects on the liver. Using primary cell culture models of hepatic non-parenchymal cells alone and in co-culture, we demonstrate that the principle profibrogenic effects of leptin are mediated via direct effects on KCs leading to the release of soluble mediators including TGFβ1 and CTGF/CCN2.
Materials and Methods
Animals
Male Sprague-Dawley rats were obtained from the Animal Resources Centre (Perth, WA, Australia). Zucker rats (fa/fa) and their lean littermates (Fa/Fa) were obtained from Professor Greg J. Barritt (Flinders University, Adelaide). All animals were maintained under 12 h light/dark cycles with food and water ad libitum. Experimental protocols were approved by the Sydney West Area Health Service Animal Research Ethics Committee.
Materials
Rat recombinant leptin was purchased from Sigma-Aldrich. Phospho-STAT3, p38, pERK1/2(44/42), pAKT and pJNK mouse monoclonal antibodies were purchased from Cell Signaling Tec. Recombinant TGFβ1, monoclonal TGFβ antibody, PDGF BB and PDGF ELISA Kit were purchased from R&D Systems. AP-1 and NF-κB consensus double stranded oligonucleotides were purchased from Promega. STAT3 inhibitor peptide (5730956), the MEK inhibitor PD98059 and the PI3-kinase inhibitor LY294002 were obtained from Calbiochem. Soluble TGFβ receptor (sTGFβR) fusion protein was a gift from Biogen Inc.
Non-parenchymal cell isolation and culture
HSCs were isolated by two-step (collagenase B and pronase E) perfusion.14 KCs and SECs were further obtained and purified by elutriation.15 HSCs were cultured in DMEM supplemented with 10% FCS and 1% penicillin-streptomycin and plated on 6 well plates at a density of 0.8×106 cells/well. Viability was routinely over 95% for all experiments. Purity was 95% as determined by morphology, vitamin A autofluorescence and desmin positivity.
KCs were identified by their ability to phagocytose latex beads; viability was >96% and purity >98%. The viability of SECs was >98% and purity at least 94% as determined by morphology (cobblestone appearance) and absence of latex bead phagocytosis. KCs were cultured in 10% FCS/DMEM/1% penicillin-streptomycin in 6-well plates. SECs were cultured in M199 with 20% FCS, 1% penicillin-streptomycin, insulin (20 mU/mL), heparin (10 U/mL), VEGF (5 ng/mL) and dexamethasone (10 μmol/L). SEC culture wells were pre-coated with type I collagen (Nalge Nunc International).
Immunoblot assays for protein expression
Culture media was removed, cells washed with PBS then lysed on ice in a buffer containing 20 mmol/L Tris, 0.5 mmol/L MgCl2, 1 mmol/L DTT, 3mmol/L NaN3, with protease- and phosphatase-inhibitors. Cell lysates were disrupted with a sonicator on ice. Immunoblots were performed as previously described.16 Immunoblotting was performed for TGFβ1 protein in culture medium after concentrating the media using Microcon YM-10 Centrifugal Filters (Millipore).
Real time reverse-transcription polymerase chain reaction
Total cellular RNA was prepared from HSCs, KCs and SECs using TRI REAGENT® (Molecular Research Center). Complementary DNA (cDNA) was synthesized from 1 μg RNA using SuperScript III reverse transcriptase and 0.5 nmol of random primers (Invitrogen). Real-time quantitative reverse-transcription polymerase chain reaction (qPCR) was performed using SYBR Green JumpStart Taq ReadyMix (Sigma). The relative amount of mRNA was calculated by reference to a calibration curve. Each sample was normalized to the respective 18S value.
Cell proliferation
HSC proliferation was assessed by using the Cell Proliferation Reagent WST-1 according to the manufacturer’s instructions (Roche Diagnostics).
Immunocytochemistry for CTGF/CCN2
CTGF/CCN2 protein expression in KCs was assessed as previously described.17
Nuclear protein extraction and Electrophoretic Mobility Shift Assay (EMSA)
KC nuclear protein preparation and EMSA were performed as previously described.18
Sirius red staining and quantification of collagen I in HSCs
These experiments were performed as previously described.19
ELISA for platelet-derived growth factor (PDGF)
PDGF concentration in KC media was determined according to the manufacturer’s directions (R&D systems).
H2O2 Generation by KCs
Intracellular H2O2 production in KCs was assessed as previously described.20
Statistical analysis
The results are expressed as mean ± SD. Comparisons between 2 groups was analyzed using the Student’s t-test. A 2-sided P value <0.05 was used to connote significance.
Results
Leptin has minimal direct effects on profibrotic gene expression in HSCs in vitro
As shown in Fig. 1A, there was no change in the expression of TIMP1, TGFβ1 and CTGF/CCN2 mRNA in HSCs after treatment with either 10 nmol/L or 100 nmol/L leptin. Similarly, leptin did not affect TGFβ1 protein expression in 2 day cultured HSCs (Fig. 1B). αSMA protein was unaltered following leptin treatment in 2 day (Fig. 1C) and 6 day HSC cultures (Fig. 1D). However, exposure to high dose leptin (100 nmol/L) was associated with a minimal (~30%) increase in collagen 1 gene expression in 2 day cultured HSCs (Fig. 1A), but not in HSCs cultured for 6 days (Fig. 1A).
Figure 1. Direct effects of leptin on profibrotic gene and αSMA protein expression in HSCs.

(A) Collagen I, TIMP1, TGFβ1 and CTGF/CCN2 mRNA expression in HSCs determined by qPCR. HSCs were cultured for 48 h and 6 days before leptin (10 or 100 nmol/L) was added to the medium for 24 h. (B) TGFβ1 protein expression in HSCs cultured for 48 h then treated with leptin (10 or 100 nmol/L) for 24 h, as determined by immunoblot on cell lysates. (C and D) αSMA protein expression in HSCs (48 h and 6 days in culture, respectively) treated with leptin (10 nmol/L or 100 nmol/L) and/or TGFβ1 (1 or 10 ng/mL). (E) mRNA expression of collagen I and TIMP1 in HSCs (cultured for 48 h) treated with leptin (10 or 100 nmol/L) and/or TGFβ1 (10 ng/mL) for 24 h. Mean ± SD for at least 3 experiments performed on 3 cell preparations, except for Fig. 1A (day 2 HSC) where results from 7 different cell isolations were pooled. * P<0.05; ** P<0.01 compared with control HSCs.
Leptin (10 nmol/L and 100 nmol/L) and TGFβ1 (1 ng/mL and 10 ng/mL) alone or the combination of leptin and TGFβ1 did not enhance αSMA protein expression in 2 day (Fig. 1C) or 6 day HSC cultures (Fig. 1D). TGFβ1 treatment was associated with upregulation of collagen I and TIMP1 mRNA expression (Fig. 1E). However, co-administration of TGFβ1 (10 ng/mL) with leptin (10 nmol/L or 100 nmol/L) in HSCs failed to have any synergistic effects on profibrotic gene expression (Fig. 1E).
Leptin enhances stellate cell proliferation
In contrast to its minimal direct profibrotic effects, leptin significantly and in a dose-dependent manner enhanced the proliferation of HSCs after both 2 (Fig. 2A) and 6 days in culture (Fig. 2C). As PDGF is known to be the most potent mitogen for HSCs, we assessed whether leptin facilitates PDGF-induced HSC proliferation. As shown in Figs. 2A and 2C, leptin did not have additional effects on HSC proliferation above that mediated by PDGF in cultured HSCs.
Figure 2. Direct and indirect effects of leptin on the proliferation of HSCs in culture.

(A) Effect of leptin on HSC proliferation. HSCs (48 h after isolation) were incubated with leptin (10 or 100 nmol/L) and/or PDGF BB (25 ng/mL) for 24 h. PDGF was used as a positive control. Proliferation was assessed by WST-1 assay. (B) HSC proliferation by KC- and SEC-conditioned medium with leptin treatment. Primary cultured KCs and SECs (cultured for 48 h) were incubated with or without leptin and/or LPS for 24 h and subsequently the KC- or SEC-conditioned medium was transferred onto 3 day HSCs. After 24 h, HSC proliferation was analyzed by WST-1 assay. ** P<0.01 compared to control HSC; # P<0.05, compared to HSCs treated with KC-conditioned medium only. (C) Day 6 HSC proliferation by leptin or leptin treated KC-conditioned medium. Methods were the same as described for Figs. 2A and 2B except that HSCs were cultured for 6 days before treatments. (D) PDGF concentration in KC-conditioned medium. (E). mRNA expression of PDGF α and β receptors in HSCs co-cultured with KC conditioned medium by leptin. (F). mRNA expression of OB-Rb in cultured HSCs (day 1, day 3 and day 7) and KCs (day 1 and day 3). ** P<0.01 compared to D1 HSCs; # P<0.05, compared to D3 HSCs. Results are means ± SD of at least three independent experiments performed at least in triplicate.
We next determined whether leptin indirectly affects HSC proliferation. KC-conditioned medium in the absence of leptin increased HSC proliferation by 2.3 fold compared to control medium, while pre-treatment of KC with leptin (100 nmol/L) enhanced HSC proliferation 3.1 fold (Fig. 2B). SEC-conditioned medium did not affect HSC proliferation (Fig. 2B). Similar results were obtained when KC-conditioned medium was applied on activated HSC, i.e. after 6 days in culture (Fig. 2C). High-dose LPS to activate KCs enhanced the proliferative effects of KC-conditioned medium on HSCs, but together with leptin had no additional effect on proliferation (Fig. 2B).
PDGF concentration and PDGF receptors were assessed. As shown, neither PDGF concentration in leptin treated KC-conditioned medium (Fig. 2D), nor PDGF receptor (α and β subtypes) mRNA expression in HSCs that had been exposed to leptin treated KC-conditioned medium (Fig. 2E) were altered. These data suggest that PDGF and its receptors are not the principal mediators of the HSC proliferative effects of KC conditioned medium.
HSCs and KCs express OB-Rb but demonstrate differential expression patterns in culture
Quiescent HSCs and KCs express OB-Rb (Fig. 2F), however, receptor expression was dramatically down-regulated during the process of HSC activation in vitro. Thus, OB-Rb expression was reduced to 10% by day 3 and to 4% at day 7 of culture, when compared to the levels in quiescent HSCs (day 1). In contrast, the expression of Ob-Rb remained stable in KC cultures.
Leptin promotes stellate cell activation by acting on Kupffer cells
Given the minimal direct effects of leptin on profibrotic gene expression in HSCs, we examined whether leptin might exert these actions indirectly, by modulating KC and/or SEC behavior. For these studies, 24 h leptin-treated KC- or SEC-conditioned medium was transferred onto 3-day old HSCs in culture. After 24 h incubation in conditioned medium, collagen I, TIMP1, TGFβ1 and CTGF/CCN2 mRNA expression in HSCs was examined. The expression of all profibrogenic genes was elevated at least 2 fold (P<0.05) in HSCs incubated with KC-, but not with SEC-conditioned medium (Fig. 3A). Consistent with these data, collagen I protein was also augmented (Fig. 3B).
Figure 3. Indirect profibrogenic effects of leptin on HSCs via soluble mediators released from KCs.
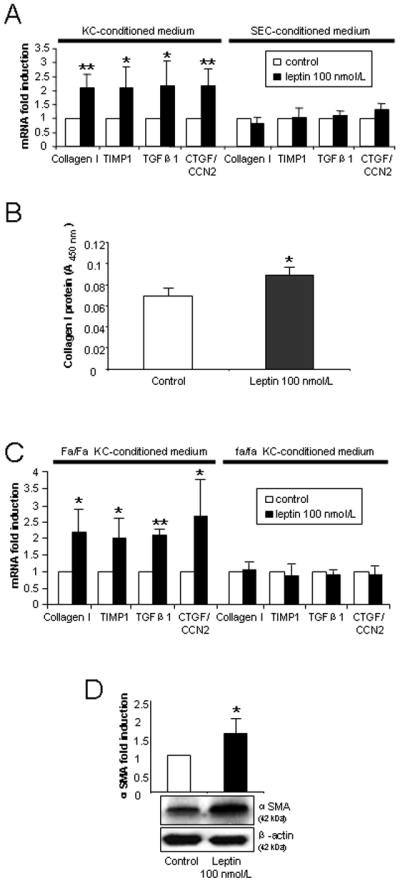
(A) KCs or SECs were cultured for 48 h before being treated with leptin 100 nmol/L for 24 h. Conditioned medium was then transferred onto 3 day HSCs and cultured for another 24 h. Collagen I, TIMP1, TGFβ1 and CTGF/CCN2 gene expression was determined by qPCR in HSCs. (B). Total collagen protein released by HSCs cocultured with KC conditioned medium. (C). Profibrogenic gene expression of HSCs incubated with leptin-treated lean (Fa/Fa) rat or fatty (fa/fa) Zucker KC-conditioned medium. (D). αSMA protein expression in HSCs incubated with control or leptin-treated KC-conditioned medium. Culture methods for 3B, 3C and 3D are the same as Fig. 3A. The results are means ± SD of at least three independent experiments. * P<0.05 and ** P<0.01 between control and leptin-treated conditioned medium.
To exclude the possibility that this effect was due to endotoxin contamination of the recombinant leptin protein, we treated KCs with 0.16 ng/mL LPS. LPS-treated KC-conditioned medium was then transferred onto HSCs. We observed no increase in collagen I, TIMP1, TGFβ1 or CTGF/CCN2 mRNA expression in HSCs (supplementary Fig. 1). Additional experiments showed that leptin-treated Zucker (fa/fa; Ob-Rb receptor deficiency) rat KC-conditioned medium failed to elicit any profibrogenic effects when transferred onto wild type (WT) HSCs (Fig. 3C). In contrast, KC-conditioned medium from leptin-treated lean littermates (Fa/Fa) reproduced the profibrogenic effects that were observed when using leptin-treated WT KC-conditioned medium (Fig. 3A and 3C). This confirmed that the observed effects were specifically due to leptin acting through its receptor.
We next determined whether leptin indirectly affects HSC αSMA protein expression. Leptin-treated WT KC-conditioned medium significantly increased αSMA protein expression in HSC (Fig. 3D). SEC-conditioned medium, however, did not result in increased αSMA expression (supplementary Fig. 2).
Collectively, these data suggest that leptin activates KCs to release soluble profibrogenic mediators that are transferred in the conditioned medium to HSCs. These factors promote the activation, proliferation and profibrogenic activities of HSCs.
Leptin up-regulates TGFβ1 and CTGF/CCN2 expression in Kupffer cells
TGFβ1 and CTGF/CCN2 are major profibrogenic cytokines in the development of liver fibrosis.21–23 Leptin (100 nmol/L) treatment for 24 h significantly up-regulated TGFβ1 and CTGF/CCN2 mRNA expression in KCs (Fig. 4A). As expected, TGFβ1 protein was elevated in culture medium of WT KCs treated with leptin (100 nmol/L) (Fig. 4B), while CTGF/CCN2 protein was also expressed at higher levels in leptin-treated KCs than in control (Fig. 4C). Considering that CTGF/CCN2 is induced via TGFβ1-dependent pathways, we analyzed the inhibitory effects of pan-TGFβ blockade on CTGF/CCN2 protein expression by using a sTGFβR fusion protein. Co-exposure of KCs to leptin and sTGFβR significantly attenuated CTGF/CCN2 protein expression induced by leptin, while, human IgG as a control, did not (Fig. 4C). In additional studies, we demonstrated that on leptin treatment, Zucker (fa/fa) rat KCs failed to augment TGFβ1 and CTGF/CCN2 mRNA expression (Fig. 4D), while the results in lean (Fa/Fa) rat KCs as expected, were similar to that in wild type rat KCs (Fig. 4D). These results further corroborate our data that the effects of leptin in inducing TGFβ1 and CTGF/CCN2 are indeed leptin receptor-dependent.
Figure 4. Gene and protein expression of TGFβ1 and CTGF/CCN2 mediated by leptin in cultured KCs.

All KCs were cultured for 48 h prior to treatment. Three independent experiments were performed. (A) mRNA expression of TGFβ1 and CTGF/CCN2 in wt rat KCs cultured for 24 h in the presence of leptin (10 or 100 nmol/L). (B) Immunoblot of TGFβ1 in KC-conditioned medium treated with leptin 100 nmol/L for 24 h. (C) Immunocytochemical determination of CTGF/CCN2 protein in KCs. KCs were cultured for 24 h with leptin 100 nmol/L, leptin 100 nmol/L + sTGFβR fusion protein (50 μg/mL) or leptin 100 nmol/L + human IgG (50 μg/mL). Scale bar: 20 μm. (D) mRNA expression of TGFβ1 and CTGF/CCN2 in Zucker (fa/fa) and lean (Fa/Fa) rat KCs cultured for 24 h in the presence of leptin (100 nmol/L). (E). KC intracellular H2O2 generation by leptin. Leptin 100 nmol/L was incubated with KC (48 h cultured) for 24 h. The excitation wavelength was 485 nm and emission wavelength 535nm. (F). Collagen I, TIMP1, TGFβ1 and CTGF/CCN2 mRNA expression in HSCs incubated with KC-conditioned medium in the presence or absence of TGFβ antibody (10 μg/mL).* P<0.05 and ** P<0.01 compared to control group. # p<0.05 compared to the leptin treatment group without TGFβ antibody.
Leptin does not induce intracellular hydrogen peroxide production in KCs
H2O2 is a well recognized profibrogenic factor,24 and leptin is reported to stimulate H2O2 production in HSC cell lines.11 However, we were unable to demonstrate significant increase in H2O2 production by leptin treated KCs (Fig. 4E). This data suggests that at least in vitro, leptin’s profibrogenic effects that are mediated via KCs, are not due to the production of H2O2.
TGFβ neutralization attenuates the profibrogenic effects of leptin on KCs
In order to further clarify whether TGFβ1 is indeed one of the soluble mediators of the profibrogenic effects of leptin on KC, we undertook TGFβ neutralization studies. As shown (Fig. 4F), after TGFβ antibody (10 μg/mL) treatment of leptin treated KC medium, collagen I mRNA was reduced by 67%, TIMP1 by 78%, TGFβ1 by 76% and CTGF by 102% in HSCs. These data confirm that TGFβ1 is likely to be the principal profibrogenic mediator that is released on leptin treatment of KCs.
Leptin activates the phosphorylation of STAT3, ERK1/2 and AKT and activates the transcription factors AP-1 and NF-κB in Kupffer cells
Leptin acts principally through the long form OB-Rb leptin receptor and downstream JAK/STAT pathways. Leptin also activates MAPK and PI-3K/AKT.25–27 Therefore, we determined whether leptin activates these pathways in KCs to target downstream components leading to profibrotic gene transcription. Exposure of KCs to leptin resulted in increased activation of ERK1/2 (particularly the 44 kDa isoform) and AKT after 5 and 10 minutes incubation respectively, and in a time dependent manner (Figs. 5A and 5B). A rapid and time-dependent increase in STAT3 phosphorylation was also observed upon leptin treatment of KCs (Figs. 5A and 5B). Leptin did not increase JNK or p38 phosphorylation (Fig. 5C).
Figure 5. Leptin activates JAK/STAT3, MAPK/ERK1/2 and PI-3K/Akt pathways as well as AP1 and NF-κB transcription factors in Kupffer cells.
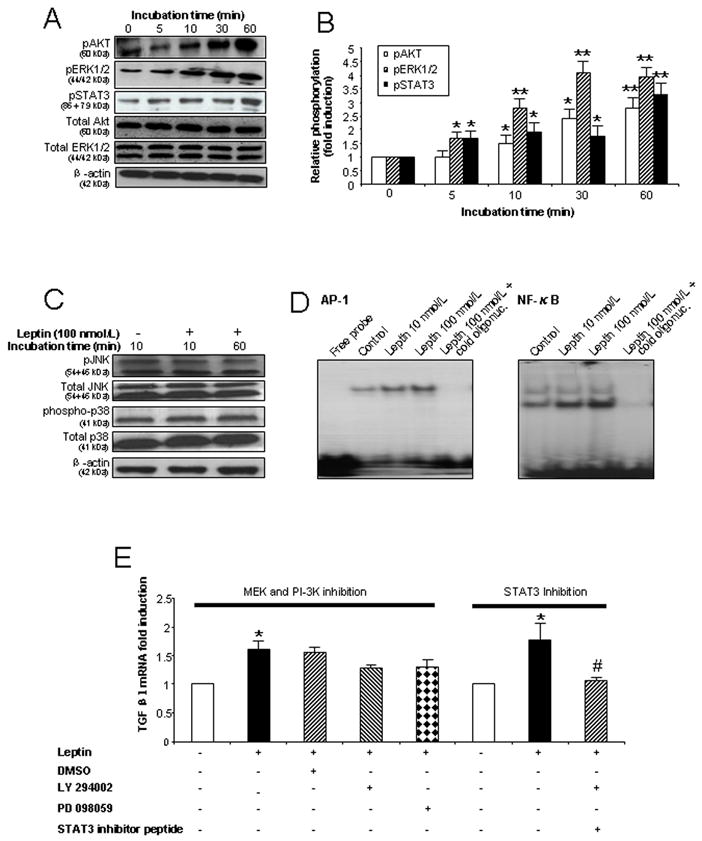
(A) Leptin enhances phosphorylation of STAT3, ERK1/2 and Akt in KCs in a time dependent manner. KCs were exposed to leptin 100 nmol/L for 0, 5, 10, 30 and 60 min and cell lysates used for immunoblot analysis. (B) Densitometry analysis of Fig. 5A as the ratio of p-protein to total protein (ERK1/2 or AKT) or the ratio of pSTAT3 to β-actin. * P<0.05; ** P<0.01 (C) Leptin did not influence phosphorylation of p38 or JNK in KCs. Cell lysates from KCs exposed to leptin 100 nmol/L for 10 or 60 min were used for immunoblotting. (D) Leptin activates AP-1 and NF-κB DNA binding in KCs. Nuclear protein from KCs cultured for 60 min with leptin (10 or 100 nmol/L) was used for EMSA. The disappearance of the shifted band in the presence of a molar excess of unlabeled (cold) oligonucleotide confirms the specificity of the binding. (E). TGFβ1 mRNA expression in KCs in the presence or absence of STAT3 inhibitor peptide (50 μmol/L), a MEK inhibitor (50 μmol/L) and a PI-3K inhibitor (25 μmol/L). RNA was extracted after 24 h treatment. * P < 0.05 and ** P <0.01 vs. control group. # P < 0.05 vs. leptin treatment group without STAT3 inhibitor peptide. Each experiment was conducted in at least 3 independent sets.
We speculated that leptin may also activate downstream transcription factors such as AP-1 and/or NF-κB since these transcription factors can be activated by MAPK/ERK1/2 and PI-3K/AKT signaling,28,29 and AP-1 and/or NF-κB binding motifs are known to be present in the promoter regions of TGFβ1 and CTGF/CCN2.30–32 Electrophoretic mobility shift assays (EMSAs) were therefore performed on nuclear protein extracted from leptin-treated and control KCs. As shown, (Fig. 5D), leptin, in a dose dependent fashion, increased AP-1 and NF-κB DNA binding abilities in KCs.
Activated STAT3 mediates TGFβ1 expression in KCs
To clarify which of the activated signaling pathways contributes to the observed increase in TGFβ1 gene expression, we undertook studies to inhibit the various signaling pathways and then measured TGFβ1 expression. As shown (Fig. 5E), the STAT3 inhibitor but not the MEK (PD98059) or PI-3K (LY294002) inhibitors, resulted in a significant reduction of TGFβ1 mRNA expression in KCs.
Discussion
Leptin not only regulates body weight and metabolism but also exerts pleiotropic effects on other organs such as the liver. Leptin has been shown to accelerate and enhance the process of liver fibrosis induced by various stimuli in vivo,6–9 whereas leptin-deficient animals are resistant to fibrosis.6,7,12 A number of studies have assessed whether leptin has direct profibrogenic effects on HSCs, but the results have been conflicting.7,9,11,12,33 Saxena and colleagues reported that leptin binds to the signaling form of the leptin receptor (OB-Rb) on primary rat HSCs and HSC-T6 cells, an immortalized rat HSC cell line, and that leptin activates STAT3 to enhance the expression of α2 (I) collagen mRNA.7 However, others have failed to confirm the expression of OB-Rb in activated primary rat HSCs or in the human HSC cell line LX-1,12,33 and have failed to demonstrate induction of collagen gene expression in HSCs by leptin.12 In contrast, expression of the short form leptin receptor (OB-Ra) in activated primary HSCs and LX-1 cell lines has been established.12,33 Therefore, whether leptin directly signals to HSCs via the OB-Rb remains unresolved.
Using leptin at pharmacological concentrations in a range similar to those of earlier studies,7,12,33 we have shown that only high leptin concentrations (100 nmol/L-1600 ng/mL) marginally increased collagen I mRNA expression in HSCs at day 2. This result is similar to the study by Tang et al. demonstrating that only high concentrations of leptin (1000 ng/mL) resulted in increases of collagen I expression in HSCs.33 In addition, we noted that expression of the other profibrotic genes (TIMP1, TGFβ1 and CTGF/CCN2) and the protein expression of αSMA was not altered by leptin treatment. Similarly, leptin neither enhanced TGFβ1 protein expression nor facilitated TGFβ1-induced collagen I or TIMP1 gene expression. Based on these composite data, the direct profibrogenic effects of leptin on HSCs, if present, appears modest.
As distinct from the above, leptin significantly and directly increased the proliferation of HSCs in a dose dependent manner. It has been previously reported that leptin facilitates HSC proliferation via activation of the MAPK/ERK1/2 and PI-3K/AKT pathways and by increased PDGF receptor expression.26,34 Activation of either the functional OB-Rb or OB-Ra receptors is able to trigger MAPK and PI-3K signaling25 and it therefore appears that the pro-proliferative effect of leptin on HSCs is more prominent than its profibrogenic effects. In our studies of the indirect profibrogenic effects of leptin that are mediated by KCs, we were unable to demonstrate any increase in PDGF protein in KC-conditioned medium, nor any increase in PDGF receptor expression following the addition of the KC medium to HSCs. These data suggest that HSC proliferation by leptin treated KC medium may be a complex phenomenon and must involve other pro-proliferative factors, the identity of which is unclear. In addition, it should be noted that activated HSCs elaborate leptin35, and this could in part, mediate HSC proliferation.
In contrast to the minimal direct profibrotic effects of leptin on HSCs, conditioned medium from leptin-treated KCs markedly induced gene expression of collagen I, TIMP1, TGFβ1 and CTGF/CCN2 compared to untreated KC-conditioned medium. Likewise, leptin-treated KC-conditioned medium augmented collagen I and αSMA protein. Conversely, leptin-treated SEC-conditioned medium did not enhance the expression of any of the profibrogenic genes tested. These results suggest that KCs play a major role in mediating the profibrogenic effects of leptin on HSCs by directly signaling to KCs.
OB-Rb expression in HSCs is still controversial.7,12,33 Ikejima et al. 12 noted that KCs harbored functional OB-Rb and that exposure to leptin activated the JAK-STAT signaling pathway of this receptor.12 Consistent with this result, we confirmed that OB-Rb is highly expressed in quiescent and activated KCs, and thus could mediate leptin signal transduction. In contrast, OB-Rb expression in activated, fibrogenic HSCs was dramatically decreased compared to that in quiescent HSCs, explaining why leptin treatment was unable to activate these cells. Having established that KCs express Ob-Rb receptors, we determined the downstream signaling pathways activated by leptin treatment. Our studies indicated that leptin treatment of KCs activates JAK/STAT3, MAPK/ERK1/2, PI-3K/Akt, and the transcription factors NF-κB and AP-1 in WT KCs.
Our results imply that upon ligand-binding to the leptin receptor, KCs release profibrogenic factor(s) that are able to activate HSCs. KCs produce a large array of cytokines and other mediators including TNFα, TGFβ1, CTGF/CCN2, IL-6 and ROS when activated, that may contribute to liver injury and fibrosis.24,36–40 We have shown that leptin-treated KCs increase production of TGFβ1 and CTGF/CCN2, both potent profibrogenic proteins.20–22, 41–43 The increased expression of TGFβ1 is consistent with data from Ikejima et al.12 and Leung et al.44 who demonstrated that leptin enhances TGFβ1 expression in KCs and mesothelial cells respectively. This finding is further supported by Sakaida et al. who noted decreased TGFβ1 expression in KCs with leptin receptor deficiency.13 As expected, we further demonstrated that CTGF/CCN2 production is TGFβ1 dependent as the sTGFβR fusion protein41 attenuated CTGF/CCN2 protein expression. Finally, by neutralizing TGFβ, we were able to confirm that this protein is indeed, a principal mediator of the profibrogenic effects of leptin on KCs. CTGF/CCN2 could also in part mediate the fibrogenic effects of leptin-treated KCs. Unfortunately, we were unable to unequivocally confirm this as potent and reliable CTGF/CCN2 blocking antibodies are not currently available. It should be noted that in our study, TGFβ1 treatment of HSCs did not increase αSMA protein expression, while leptin-treated KC conditioned media did. This suggests that collagen I, TIMP1 and CTGF/CCN2 are at least, in part, regulated by TGFβ1. In contrast, αSMA may be regulated by another soluble factor, possibly including CTGF/CCN2.
Finally, we sought to determine which of the signaling pathways activated by leptin is responsible for augmenting TGFβ1 expression in leptin-treated KCs and were able to demonstrate the STAT3 plays a critical role. Previous studies suggest that STAT3 enhances TGFβ1 production by binding to elements in the TGFβ1 promoter.45,46 Since we also observed increased AP-1 and NF-κB DNA binding activities, it is likely that AP-1 and NF-κB, together with STAT3 contribute to the observed increase in TGFβ1 production.
In conclusion, our results suggest that leptin mediates hepatic fibrosis mainly through actions on KCs. Increased TGFβ1 by KCs exposed to leptin is a major mediator of these effects. These mechanisms are likely to be important in mediating liver fibrosis associated with obesity and elevated leptin levels, such as that in persons with non-alcoholic fatty liver disease.
Acknowledgments
National Health & Medical Research Council of Australia (No: 358398) and the Robert W. Storr Bequest to the University of Sydney
We thank N. Subramaniam, J. Sesha, M. Kacevska, H. Yu and X. Wang for technical assistance. This work was supported by a grant from the National Health & Medical Research Council of Australia (No: 358398) and the Robert W. Storr Bequest to the University of Sydney.
Abbreviations
- αSMA
alpha-smooth muscle actin
- collagen I
collagen 1α1
- TGFβ1
transforming growth factor beta-1
- CTGF
connective tissue growth factor
- HSCs
hepatic stellate cells
- KC
Kupffer cells
- SECs
sinusoidal endothelial cells
- STAT3
signal transducer and activator of transcription-3
- ERK1/2
extracellular signal-related kinase 1 and 2
Footnotes
Financial disclosure: Authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 2.Chitturi S, Farrell G, Frost L, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: A manifestation of lipotoxicity? Hepatology. 2002;36:403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 3.Naveau S, Perlemuter G, Chaillet M, et al. Serum leptin in patients with alcoholic liver disease. Alcohol Clin Exp Res. 2006;30:1422–1428. doi: 10.1111/j.1530-0277.2006.00170.x. [DOI] [PubMed] [Google Scholar]
- 4.Testa R, Franceschini R, Giannini E, et al. Serum leptin levels in patients with viral chronic hepatitis or liver cirrhosis. J Hepatol. 2000;33:33–37. doi: 10.1016/s0168-8278(00)80156-7. [DOI] [PubMed] [Google Scholar]
- 5.Romero-Gómez M, Castellano-Megias VM, Grande L, et al. Serum leptin levels correlate with hepatic steatosis in chronic hepatitis C. Am J Gastroenterol. 2003;98:1135–1141. doi: 10.1111/j.1572-0241.2003.07450.x. [DOI] [PubMed] [Google Scholar]
- 6.Leclercq IA, Farrell GC, Schriemer R, et al. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 7.Saxena NK, Ikeda K, Rockey DC, et al. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikejima K, Honda H, Yoshikawa M, et al. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 2001;34:288–297. doi: 10.1053/jhep.2001.26518. [DOI] [PubMed] [Google Scholar]
- 9.Ding X, Saxena NK, Lin S, et al. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166:1655–1669. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda H, Ikejima K, Hirose M, et al. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology. 2002;36:12–21. doi: 10.1053/jhep.2002.33684. [DOI] [PubMed] [Google Scholar]
- 11.Cao Q, Mak KM, Ren C, et al. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292–4304. doi: 10.1074/jbc.M308351200. [DOI] [PubMed] [Google Scholar]
- 12.Ikejima K, Takei Y, Honda H, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 13.Sakaida I, Jinhua S, Uchida K, et al. Leptin receptor-deficient Zucker (fa/fa) rat retards the development of pig serum-induced liver fibrosis with Kupffer cell dysfunction. Life Sci. 2003;73:2491–2501. doi: 10.1016/s0024-3205(03)00653-2. [DOI] [PubMed] [Google Scholar]
- 14.Beljaars L, Molema G, Schuppan D, et al. Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J Biol Chem. 2000;275:12743–12751. doi: 10.1074/jbc.275.17.12743. [DOI] [PubMed] [Google Scholar]
- 15.Samarasinghe DA, Farrell GC. The central role of sinusoidal endothelial cells in hepatic hypoxia-reoxygenation injury in the rat. Hepatology. 1996;24:1230–1237. doi: 10.1002/hep.510240541. [DOI] [PubMed] [Google Scholar]
- 16.Teoh N, Dela Pena A, Farrell GC. Hepatic ischemic preconditioning in mice is associated with activation of NF-kappaB, p38 kinase, and cell cycle entry. Hepatology. 2002;36:94–102. doi: 10.1053/jhep.2002.33134. [DOI] [PubMed] [Google Scholar]
- 17.Steffen CL, Ball-Mirth DK, Harding PA, et al. Characterization of cell-associated and soluble forms of connective tissue growth factor (CTGF) produced by fibroblast cells in vitro. Growth Factors. 1998;15:199–213. doi: 10.3109/08977199809002117. [DOI] [PubMed] [Google Scholar]
- 18.Subramaniam N, Leong GM, Cock TA, et al. Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-beta signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. J Biol Chem. 2001;276:15741–15746. doi: 10.1074/jbc.M011033200. [DOI] [PubMed] [Google Scholar]
- 19.Disthabanchong S, Radinahamed P, Stitchantrakul W, et al. Chronic metabolic acidosis alters osteoblast differentiation from human mesenchymal stem cells. Kidney Int. 2007;71:201–209. doi: 10.1038/sj.ki.5002035. [DOI] [PubMed] [Google Scholar]
- 20.Ryu JH, Lee Y, Han SK, et al. The role of hydrogen peroxide produced by polychlorinated biphenyls in PMR1-deficient yeast cells. J Biochem. 2003;134:137–142. doi: 10.1093/jb/mvg121. [DOI] [PubMed] [Google Scholar]
- 21.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 22.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paradis V, Dargere D, Vidaud M, et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology. 1999;30:968–976. doi: 10.1002/hep.510300425. [DOI] [PubMed] [Google Scholar]
- 24.Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of Kupffer cells on stellate cells. Hepatology. 2006;44:1487–1501. doi: 10.1002/hep.21427. [DOI] [PubMed] [Google Scholar]
- 25.Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saxena NK, Titus MA, Ding X, et al. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aleffi S, Petrai I, Bertolani C, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 28.Eder AM, Dominguez L, Franke TF, et al. Phosphoinositide 3-kinase regulation of T cell receptor-mediated interleukin-2 gene expression in normal T cells. J Biol Chem. 1998;273:28025–28031. doi: 10.1074/jbc.273.43.28025. [DOI] [PubMed] [Google Scholar]
- 29.Cortez DM, Feldman MD, Mummidi S, et al. Interleukin-17 Stimulates MMP1 Expression in Primary Human Cardiac Fibroblasts Via p38 MAPK and ERK1/2-Dependent C/EBP{beta}, NF-{kappa}B, and AP-1 Activation. Am J Physiol Heart Circ Physiol. 2007;293:H3356–3365. doi: 10.1152/ajpheart.00928.2007. [DOI] [PubMed] [Google Scholar]
- 30.Lee KY, Ito K, Hayashi R, et al. NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J Immunol. 2006;176:603–615. doi: 10.4049/jimmunol.176.1.603. [DOI] [PubMed] [Google Scholar]
- 31.Fu M, Zhang J, Zhu X, et al. Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells byinterfering with Smad3. J Biol Chem. 2001;276:45888–45894. doi: 10.1074/jbc.M105490200. [DOI] [PubMed] [Google Scholar]
- 32.Bourgier C, Haydont V, Milliat F, et al. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smooth muscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut. 2005;54:336–343. doi: 10.1136/gut.2004.051169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang M, Potter JJ, Mezey E. Leptin enhances the effect of transforming growth factor beta in increasing type I collagen formation. Biochem Biophys Res Commun. 2002;297:906–911. doi: 10.1016/s0006-291x(02)02300-8. [DOI] [PubMed] [Google Scholar]
- 34.Lang T, Ikejima K, Yoshikawa M, et al. Leptin facilitates proliferation of hepatic stellate cells through up-regulation of platelet-derived growth factor receptor. Biochem Biophys Res Commun. 2004;323:1091–1095. doi: 10.1016/j.bbrc.2004.08.192. [DOI] [PubMed] [Google Scholar]
- 35.Potter JJ, Womack L, Mezey E, Anania FA. Transdifferentiation of rat hepatic stellate cells results in leptin expression. Biochem Biophys Res Commun. 1998 Mar 6;244(1):178–82. doi: 10.1006/bbrc.1997.8193. [DOI] [PubMed] [Google Scholar]
- 36.Matsuoka M, Tsukamoto H. Stimulation of hepatic lipocyte collagen production by Kupffer cell-derived transforming growth factor beta: implication for a pathogenetic role in alcoholic liver fibrogenesis. Hepatology. 1990;11:599–605. doi: 10.1002/hep.1840110412. [DOI] [PubMed] [Google Scholar]
- 37.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84:1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuoka M, Zhang MY, Tsukamoto H. Sensitization of hepatic lipocytes by high-fat diet to stimulatory effects of Kupffer cell-derived factors: implication in alcoholic liver fibrogenesis. Hepatology. 1990;11:173–182. doi: 10.1002/hep.1840110204. [DOI] [PubMed] [Google Scholar]
- 39.Han YP, Zhou L, Wang J, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279:4820–4828. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12:7413–7420. doi: 10.3748/wjg.v12.i46.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.George J, Roulot D, Koteliansky VE, et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA. 1999;96:12719–12724. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J Biol Chem. 2004;279:8848–8855. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- 43.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 44.Leung JC, Chan LY, Tang SC, et al. Leptin induces TGF β synthesis through functional leptin receptor expressed by human peritoneal mesothelial cell. Kidney Int. 2006;69:2078–2086. doi: 10.1038/sj.ki.5000409. [DOI] [PubMed] [Google Scholar]
- 45.Ogata H, Chinen T, Yoshida T, et al. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF β1 production. Oncogene. 2006;25:2520–2530. doi: 10.1038/sj.onc.1209281. [DOI] [PubMed] [Google Scholar]
- 46.Kinjyo I, Inoue H, Hamano S, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203:1021–1031. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]