

Abstract
A subset of superoxide dismutase 1 (Cu/Zn-SOD1) mutants that cause familial amyotrophic lateral sclerosis (FALS) have heightened reactivity with −ONOO and H2O2 in vitro. This reactivity requires a copper ion bound in the active site and is a suggested mechanism of motor neuron injury. However, we have found that transgenic mice that express SOD1-H46R/H48Q, which combines natural FALS mutations at ligands for copper and which is inactive, develop motor neuron disease. Using a direct radioactive copper incorporation assay in transfected cells and the established tools of single crystal x-ray diffraction, we now demonstrate that this variant does not stably bind copper. We find that single mutations at copper ligands, including H46R, H48Q, and a quadruple mutant H46R/H48Q/H63G/H120G, also diminish the binding of radioactive copper. Further, using native polyacrylamide gel electrophoresis and a yeast two-hybrid assay, the binding of copper was found to be related to the formation of the stable dimeric enzyme. Collectively, our data demonstrate a relationship between copper and assembly of SOD1 into stable dimers and also define disease-causing SOD1 mutants that are unlikely to robustly produce toxic radicals via copper-mediated chemistry.
Amyotrophic lateral sclerosis (ALS),3 which is characterized by progressive muscle weakness and motor neuron loss, presents as both sporadic and familial (FALS) illness. A subset of FALS cases is caused by missense mutations in the superoxide scavenging enzyme, Cu/Zn-superoxide dismutase 1 (SOD1) (1, 2). To date, over 100 different point mutations, and >5 early termination mutations have been linked to FALS (www.alsod.org) (for reviews see Refs. 2-4). Early studies of FALS-SOD1 enzymes demonstrated that some mutants retain high levels of activity and relatively long half lives (5). Moreover, mutant proteins that are inactive or short-lived do not exhibit evidence of dominant negative action with regard to the superoxide-scavenging activity of enzyme derived from the normal allele (6). In transgenic mice, the hyperexpression of the G93A and G37R variants of FALS-SOD1 increases superoxide scavenging activity, kills motor neurons, and causes paralysis (7, 8). SOD1 knock-out mice do not develop ALS-like phenotypes but do show sensory and motor neuropathy (9). Together, these studies establish that SOD1 mutations cause ALS through a gained toxic property. Although expressed ubiquitously (8, 10), mutant SOD1 selectively damages motor neurons, by mechanisms yet to be fully understood.
Each subunit of the mature homodimeric SOD1 enzyme binds one atom of copper and one atom of zinc and contains a single oxidized disulfide bond between Cys-57 and Cys-146 (11-13). Because copper can participate in many types of potentially deleterious reactions, the role of the copper cofactor of SOD1 in the toxicity associated with mutant protein has been intensely studied (for review see Refs. 4 and 14). To dissect the role of copper in the toxicity of mutant SOD1, we have examined the impact of mutations of the four histidine residues that are the primary copper ligands of the enzyme. Two of the four histidine residues that coordinate copper have been documented as targets of natural FALS mutations; His-46 to Arg and His-48 to Gln (www.alsod.org). Both of these mutants are almost completely devoid of superoxide-scavenging activity (15). Combining these two mutations into one protein produces a molecule that also lacks demonstrable activity but retains high toxicity to motor neurons (16). Additional substitutions at the two other copper ligands (His-63 to Gly and His-120 to Gly), eliminate the copper-binding ligands, generating a protein that remains capable of inducing motor neuron disease in transgenic mice (17). Studies of SOD1-H46R suggest that this single mutation interferes with copper binding (18, 19), whereas the H48Q mutant can be made to bind copper in the correct site, although with an altered coordination geometry (20). The copper-binding abilities of H46R/H48Q or SOD1-Quad are also predicted to be severely compromised, but this has not yet been demonstrated experimentally.
To fill this gap in knowledge, we here study copper binding of four SOD1 variants, H46R, H48Q, H46R/H48Q, and H46R/H48Q/H63G/H120G (Quad), using a direct radioactive copper incorporation assay. In transfected cell models, we show that none of these variants possess high affinity for copper. We also use single crystal x-ray diffraction to examine directly the copper binding site of SOD1-H46R/H48Q protein expressed in yeast and isolated by standard non-denaturing biochemical methods. This analysis reveals that the H46R/H48Q mutant protein does not bind copper in either of the metal-binding sites of the protein. We also noted that mutation of copper ligands correlated with reduced ability to form stable dimers, using native gel electrophoresis and a yeast two-hybrid assay. We interpret these findings as evidence that the loss of copper-binding His ligands in SOD1 reduces the stable binding of copper, and that the lack of such binding may underlie the inability of these mutants to mature into stable dimeric enzymes. We note that nearly all of SOD1-H46R/H48Q or SOD1-Quad proteins found in spinal cord tissues display an electrophoretic migration pattern of monomeric enzyme, suggesting that the poor incorporation of copper in these mutants also occurs in spinal cord tissues.
EXPERIMENTAL PROCEDURES
Expression Plasmids
cDNA of human SOD1 harboring the following mutations, A4V, G37R, G85R, G93A, I113T, H46R, H48Q, H46R/H48Q, and Quad were inserted into the pEF.Bos vector. Each of these plasmids has been described and used in prior studies (5, 6, 15-17).
Copper Incorporation Studies
The general methods used in metabolic radiolabeling with 64Cu have been described previously (21). Briefly, 64CuCl2 was obtained from Michael Welch at Washington University School of Medicine with a specific activity of 50–200 mCi/μg. CHO cells were transfected 48 h prior to copper metabolic labeling with 5 μg of DNA of the constructs noted using Lipofectamine 2000 (Invitrogen). Cells were labeled in Opti-Mem (Invitrogen) containing 50 μCi/ml 64Cu for 3 h at 37 °C. Cells were washed, harvested, and lysed in Nonidet P-40 lysis buffer (50 mM HEPES, 250 mM NaCl, 0.1% Nonidet P-40, 5 mM EDTA, pH 7.6) containing protease inhibitors. The lysate, 100 μg, was mixed with Laemmli sample buffer (final SDS concentration 1%) (22) and electrophoresed on a non-reducing 10% polyacrylamide gel containing 0.1% SDS. Samples were not heated prior to electrophoresis. The gel was exposed to PhosphorImager plates (Amersham Biosciences) overnight; plates were analyzed in the instrument as described by the manufacturer. For SOD1 immunoblot, the gel from the copper labeling study was transferred at 400 mA for 1 h then blocked in 5% nonfat milk in phosphate-buffered saline with Tween-20 (standard protocol, Pierce Biotechnology). Primary α-SOD1 antibody (SOD1 100, Stressgen Bioreagents Corp.-Nventa Biopharmaceuticals Corp., San Diego, CA) at 1:5,000 was incubated overnight at 4 °C. The blot was washed in phosphate-buffered saline with Tween-20 followed by secondary antibody (goat α-rabbit-horseradish peroxidase, Pierce) at 1:10,000 and washed prior to development with the Pico Kit (Pierce).
Yeast Two-hybrid Assessment of SOD1 Dimer Formation
To assess how FALS mutations effect homodimeric interactions, we utilized a yeast two-hybrid assay to measure interactions between mutant and wild-type subunits. The variants tested included the following: the G37R, G93A, and I113T mutations, which have been previously established to form stable dimers (5, 6, 23); the H46R, H48Q, H46R/H48Q double mutant, and the Quad variants, which affect residues critical for the coordinated binding of copper (13); and the A4V mutant, which has been suggested to be prone to monomerization (24). The A4V, G37R, G85R, G93A, H46R/H48Q, and Quad variants have all been expressed in transgenic mice to produce mouse models of FALS (7, 8, 16, 25-27).
The procedure for developing the assay involved the following, using the DupLEX-A system (OriGene Technologies, Rockville, MD). First, each of the 10 different mutants, and the wild-type cDNAs, were fused to the LexA DNA-binding domain of the bait-fusion protein (plasmid pEG202 between EcoR1 and BamH1 sites). Wild-type SOD1 cDNA was cloned into the target plasmid pJG4–5 between EcoR1 and XhoI sites located at 3′ of the activation domain. Bait plasmids were transformed into the EGY48 strain of yeasts, and the target plasmid was transformed into the mating strain RFY206. Before mating, yeasts harboring the bait constructs were screened for sufficient expression of the bait-fusion protein to repress transcription of a reporter gene in the plasmid pJK101 encoding β-galactosidase under the transcriptional control of a constitutive promoter interrupted by the LexA operator. Yeast cells were cultured on media containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, Sigma-Aldrich). For each bait construct, we identified three or four independent yeast clones that were white; indicating complete repression of β-galactosidase expression. Low expression of β-galactosidase in each colony was verified by liquid assay. This approach established that each clone expressed the bait-fusion protein at levels sufficient to saturate the DNA-binding site recognized by the LexA domain of the fusion protein.
Each of the three clones harboring the bait-fusion and reporter constructs were then made ready for mating to the RFY206 yeast strain (harboring the SOD1-wt target fusion proteins) by culturing the yeast on media containing 5-fluoroorotic acid (Sigma-Aldrich). The pJK101 repressor-reporter plasmids utilize URA-3 as the selectable marker gene, allowing the use of media containing 5-fluoroorotic acid to force the segregation of the bait-fusion constructs and the reporter construct (URA-3 is an enzyme in the uracil biosynthetic assay, which acts upon 5-fluoroorotic acid to produce a toxin). Only cells that have lost the repressor-reporter construct will grow. Again, three independent colonies were isolated, and these clones were screened by growth on media lacking uracil to ensure loss of pJK101 plasmid. These strains were then mated to SOD1-wt/RFY206 yeast that also harbored a plasmid (pSH18–34) encoding β-galactosidase behind a promoterless LexA DNA-binding domain. Initially, yeast cells harboring all three constructs were selected solely on the basis of selectable-marker genes within the plasmids. Again, three independent clones were isolated and cultures of yeast harboring both genes were then grown for lysis and liquid assay of β-galactosidase activity. Yeast cells were lysed mechanically, and the total amount of protein in each lysate was measured by a BCA kit (Pierce). The liquid assay contained a colorimetric substrate, 2-nitrophenyl-β-D-galactopyranoside, at a concentration of 0.67 mg/ml, and lysate from ~1 × 106 cells or 0.2 A600 units. Reactions were incubated at 28 °C for 15 min before absorbance was read by A420 on a standard spectrophotometer (Amersham Biosciences).
Structure Determination of SOD1-H46R/H48Q
The H46R/H48Q double mutant protein was expressed in yeast, purified, and characterized as previously described (28) with the addition of a DEAE-Sephadex chromatography step between the hydrophobic interaction chromatography and gel-filtration column steps. The metal content of the double mutant prior to crystallization screens was determined using inductively coupled plasma-mass spectrometry. Crystals suitable for x-ray diffraction work were grown by the hanging drop, vapor-diffusion method. H46R/H48Q SOD1 at 26 mg/ml in 2.25 mM potassium phosphate, pH 7.0, was mixed with a precipitating solution consisting of 20% polyethylene glycol 1000, 0.1 M phosphate, pH 6.2, 0.2 M NaCl at 25 °C. Hexagonal plates appeared within 2 months. A suitable specimen was flash-cooled in liquid nitrogen using mother liquor made 20% (w/v) in polyethylene glycol 400 as a cryoprotectant. Diffraction data were gathered at beam line 4.2.2 at the Advanced Light Source, Berkeley, CA. All diffraction data were processed using the program d*TREK (29).
The structure was determined by molecular replacement using the program MOLREP (30). A monomer of human G37R SOD1 (31) was used as the search model. Crystallo-graphic refinement was performed initially in CNS (32) and, in later stages, in SHELX-97 (33). The program COOT (34) was used for manual adjustment of the molecular models.
RESULTS
Previous studies had established that natural pathogenic mutations at histidines 46 (H46R) and 48 (H48Q) drastically reduce enzyme activity (15). Not surprisingly, combining the H46R/H48Q mutations into one variant or adding experimental mutations H63G/H120G did not restore activity (16, 17). However, whether any of these mutants stably bind copper has not been directly assayed.
In previous work, we demonstrated that SOD1-H46R/H48Q is relatively stable, inactive, and capable of inducing motor neuron disease in transgenic mice (16). To examine the copper-binding site of this mutant, purified protein was crystallized and analyzed as described under “Experimental Procedures.” Analysis of the copper- and zinc-binding sites of the double mutant superimposed on SIGMAA electron density contoured at 1.5 σ demonstrated that the mutations preclude copper ion binding at the copper site without spurious binding in the zinc site (Fig. 1). This observation agrees with data obtained from inductively coupled plasma-mass spectrometry on the protein sample prior to crystallization, which returned values of 0.05 equivalents of copper and 1.5 equivalents of zinc, per dimer.
FIGURE 1. The metal-binding sites of the human FALS SOD1 double mutant H46R/H48Q (left) and the wild-type enzyme (right).

The structure surrounding the copper-binding site of one subunit of SOD1-H46R/H48Q is compared with SOD1-wt (right). All subunits in the crystal of SOD1-H46R/H48Q showed perturbed copper-binding sites. The structure shown is that of the crystal subunits that most closely resemble SOD1-wt. Arg-143 and residues corresponding to metal ions ligands in the wild-type enzyme (metal ligand 46, 48, 63, 71, 80, 83, and 120) are labeled. The metal ions are represented by spheres. Metal ligand and hydrogen bonds are shown as dotted lines. In the left image, the side chain of the Arg residue substituted at position 46 donates a hydrogen bond to the carbonyl oxygen of Thr-137 on the opposite side of the active site channel, preventing the binding of copper ion (see text).
To assess copper binding by these variants, and others, in mammalian cells, we used a cell transfection model to express high levels of mutant SOD1 in CHO cells. 48 h after transfection, cells were incubated with 64Cu, then lysed in buffers with non-ionic detergent and chromatographed on non-reducing, non-denaturing 10% polyacrylamide gels, containing 0.1% SDS and standard Laemmli buffers (22), before exposure to Phosphor-Imager plates (21). Cells transfected with human SOD1-wt expression plasmids contained abundant levels of radiolabeled homodimer enzyme, whereas in cells transfected with the H46R, H48Q, H46R/H48Q, or Quad variants, the only labeled protein evident was the endogenous CHO protein (Fig. 2A). Because these gels allow for assay of crude cell lysates and because there are currently no radioactively labeled forms of zinc that are available to us, whether any of the proteins visualized on these gels contain zinc could not be determined. Thus apo/holo designation refers to the absence/presence of copper only, respectively, here and throughout this report.
FIGURE 2. FALS mutations at copper-binding histidine residues of SOD1 dramatically reduce affinity for copper.

A, CHO cells were transfected to express human SOD variants before being metabolically labeled with 50 μCi/ml of 64Cu for 3 h. 100 μg of each cell lysate was separated on a non-reducing 10% polyacrylamide gel containing 0.1% SDS. The 64Cu autoradiogram shows Cu-labeled endogenous hamster SOD1 dimer (solid arrow) in all samples, and Cu-labeled human SOD1 dimer (solid arrowhead) only in the WT sample. B, a duplicate of the gel used for 64Cu autoradiogram was analyzed by SOD1 immunoblot, which reveals endogenous hamster SOD1 monomer and human SOD1 monomers that are not labeled by 64Cu. Note: apo refers to the absence or presence of copper.
To confirm expression of the mutant proteins and to determine the relative position of migration in these gels, after exposure to the PhosphorImager plates, the gels were electrotransferred to nitrocellulose for immunoblotting as described under “Experimental Procedures.” The only SOD1 found in the lysates of cells transfected with the histidine mutants displayed an electrophoretic migration consistent with the previously reported electrophoretic migration of monomeric SOD1 (21, 35), which migrates much faster than the holo, dimeric enzyme (also see supplemental Fig. S2). Note the lack of 64Cu labeling (in Fig. 2A) at positions on the gel corresponding to the position of the four histidine variants (Fig. 2B).
To further address the effect of FALS mutations on dimer interactions, we developed a yeast two-hybrid assay to measure interactions between mutant and wild-type subunits. The variants tested included the four histidine variants described above as well as the A4V mutant, which has been reported to be prone to monomerization (24), and the G37R, G85R, G93A, and I113T mutants, which we have previously established to form stable active dimers (5, 6, 23, 36). The assay involved two steps described in detail under “Experimental Procedures.” In the first step, yeast cells were transfected with plasmids that harbor the bait fusion constructs (each mutant cDNA fused to a LexA DNA-binding domain) and a reporter plasmid to assess the stable expression of the bait-fusion protein. Three independent yeast clones showing complete repression of reporter transcription were isolated for each bait-fusion construct. This assay established that the expression of the various bait proteins was at a level sufficient to saturate binding of the promoter elements of reporter constructs. Each of these clones was then cultured in selection media to screen for segregation of the bait-fusion constructs and the repression-reporter construct. Three independent clones for each were isolated and cultured in media to confirm loss of the reporter construct. In the second step, these strains were mated to yeast harboring the target-fusion construct (SOD1-wt/transcriptional activator domain) and a promoterless β-galactosidase reporter plasmid before selection solely on the basis of selectable-marker genes within the plasmids. Assays for β-galactosidase activity in yeast lysates revealed significant variation in the strength of bait-target fusion interaction to promote β-galactosidase production (Fig. 3). The wt, G37R, G85R, G93A, and I113T bait-fusion constructs showed strong activation of β-galactosidase synthesis. Whereas, the A4V construct, and all variants harboring mutations at copper-binding histidines, showed very poor activation of β-galactosidase production (Fig. 3).
FIGURE 3. FALS mutations at copper-binding histidine residues of SOD1 dramatically reduce the strength of normal dimer interactions.
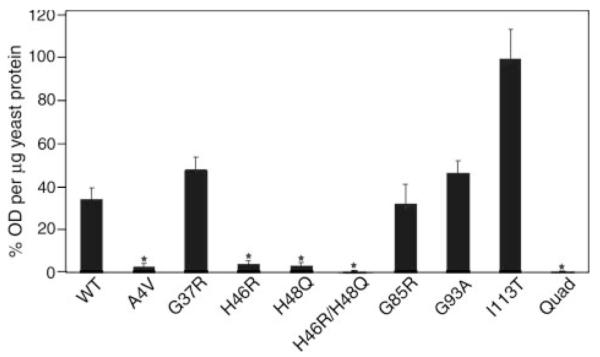
Interactions between the human WT SOD1 and mutant subunits were measured by a yeast two-hybrid assay (see “Experimental Procedures”). Yeasts expressing mutant-SOD1 bait-fusion proteins were mated with yeasts expressing wild-type SOD1 target-fusion proteins. The interactions between two subunits were recorded by induction of a β-galactosidase reporter gene. Enzyme activity was measured by optical density values per microgram of yeast protein extract and then normalized totheI113T,whichhadthehighestlevels ofβ-galactosidase production. The datarepresentmeans±S.E.Threeor fourindependentbaitfusion yeast colonies, all of which express sufficient proteins to pass a repression assay, were used for each SOD1 variant (total number of measurements for each variant, n = 9–12). Significance levels were determined by one-way analysis of variance with Bonferroni corrections; the asterisks mark interaction measurements that are significantly lower than that of the wild-type to wild-type interaction values and mutant to wild-type interaction values with p < 0.01.
Given the apparent correlation between mutation of copper ligand residues and loss of stable homodimer interactions, we prepared extracts of spinal cords from transgenic mice that express wt, G37R, G85R, G93A, H46R/H48Q, and Quad variants of human SOD1. These extracts were then electrophoresed on native gels for SOD1 activity assay (5, 37) or were transferred to nitrocellulose and immunoblotted with a human SOD1-specific antiserum (Fig. 4, A and C). To determine the levels of SOD1 in each sample, fractions were also mixed with 2× Laemmli buffer and analyzed by standard SDS-PAGE and immunoblot with an antiserum that detects both mouse and human SOD1 (Fig. 4B). In the extracts from wt, G37R, and G93A mice, the pattern of activity in the assay gel (Fig. 4A) matches well with the pattern of immunoreactivity on the immunoblot of the native gel (Fig. 4C). However, for mice expressing H46R/H48Q and SOD1-Quad, there is no evidence of transgene-derived superoxide-scavenging activity (Fig. 4A), and the migration pattern of these proteins (Fig. 4C) resembles that of wild-type enzyme reduced in the presence of chelating agents (see supplemental Fig. S2). A small amount of these mutants co-migrated with SOD1-wt and SOD1-G93A homodimers and may therefore be in a homodimeric state. Clearly, however, the majority migrates much more rapidly, similar to metal-deficient/reduced wild-type protein.
FIGURE 4. The electrophoretic migration of mutant SOD1 (H46R/H48Q and Quad-His) isolated from spinal cord resembles that of metal-deficient/reduced wild-type SOD1.

Spinal cords from pre-symptomatic transgenic animals (2–3 months of age) were removed and homogenized in PBS by probe sonication for 30 s at 50% output (70 watts, Tekmar, Cincinnati, OH) and centrifuged at 100,000 × g for 5 min in a Beckman Airfuge (Beckman Coulter, Inc., Fullerton, CA). A, 100 μg of supernatant protein was separated by native gel electrophoresis and assayed for superoxide dismutase activity by gel assay as described under “Experimental Procedures.” Amounts (0.125–2.0 μg) of purified human SOD1 proteins were used as standards. Only dimeric holoenzymes show activity. In contrast to the WT, G37R, and G93A human SOD1 that show abundant active human holoenzymes (hDimer, solid arrow), H46R/H48Q, Quad, and G85R proteins show neither detectable activity nor affect the migration of mouse SOD1 homodimer (mDimer, open arrow), confirming their inability to form heterodimers (m/hDimer, solid arrowhead). NTg = non-transgenic sample. B, 0.5 μg of supernatant protein from mouse spinal cords was assayed for SOD1 protein levels by the standard SDS-PAGE using an antiserum against a conserved region in human SOD1 (hSOD1, solid arrow) and mouse SOD1 protein (mSOD1, open arrow). Note: G85R human protein runs slightly above the mouse SOD1. C, supernatant protein (5 μg) from mouse spinal cords was separated by native gel electrophoresis and immunoblotted using an antiserum that recognizes the human SOD1 but not the mouse protein. The homodimers (solid arrow) formed by WT, G37R, or G93A are consistent with those in A. The majorities of H46R/H48Q and Quad mutants migrate at positions similar to reduced and de-metallated hSOD1-WT (open arrow). The less abundant mutant G85R appears to migrate at a position expected for dimeric enzyme.
Analysis of extracts from the mice expressing the G85R variant reveal data consistent with the yeast two-hybrid assay in that G85R SOD1 is capable of forming a dimeric protein (Fig. 4C). G85R is reported to retain partial activity in a solution assay, but its activity is undetectable in the gel assay (15). It is possible that copper was initially loaded in the dimeric enzyme and then lost during some part of the processing and gel electrophoresis. Alternatively, the G85R variant may have copper bound, stabilizing the homodimer, but for some other reason is less able to catalyze superoxide disproportionation in the gel assay.
DISCUSSION
In previous work, we established that transgenic mice expressing SOD1-H46R/H48Q develop motor neuron disease typical of FALS (16, 17). Although this mutant lacks detectable superoxide disproportionation activity (16), whether this mutant stably binds copper was not known. Here, we provide three lines of evidence that indicate that this protein does not stably bind copper. First, the crystal structure of SOD1-H46R/H48Q purified from yeast indicates an absence of copper ions in the copper or zinc sites and substantial rearrangement of the copper-binding pocket. Expression of SOD1-wt in the same system yields correctly metallated, active, homodimeric enzyme (28). Second, inductively coupled plasma-mass spectrometry analysis of the purified protein isolated from yeast indicates <0.05 equivalent of copper per unit of purified protein (prior to crystallization). Third, cells transfected with expression plasmids for SOD1-H46R/H48Q demonstrate little or no 64Cu associated with this protein as assayed by autoradiography of non-reducing, non-denaturing gels. Together, these data establish that SOD1-H46R/H48Q does not possess a high affinity for copper.
Moreover, we extend our analyses to other SOD1 variants harboring mutations at Cu-ligand histidine residues. We find that single mutations at H46R or H48Q, when expressed in CHO cells, also diminish the binding of radioactive copper. Not surprisingly, the experimental mutant SOD1-Quad also lacks evidence of copper binding. We also find an additional property shared by SOD1 variants with mutations at His Cu-ligands. In native gels, SOD1 variants expressed in CHO cells that encoded mutations at histidine ligands showed electrophoretic mobilities similar to that of the monomeric protein. In addition, the majority of mutant SOD1 in spinal cord extracts from mice expressing both the H46R/H48Q and Quad mutants migrates at a position resembling monomeric protein. We therefore conclude that SOD1-H46R/H48Q and SOD1-Quad have greatly diminished ability to stably bind copper, which appears to also affect the formation of the normal dimeric enzyme.
Metal Binding and Dimerization of Mutant SOD1
The absence of copper in the copper-binding site of the H46R/H48Q mutant is consistent with what is observed in two different crystal structures of singly substituted H46R SOD1 that reside in the protein data bank, which contains a combined total of twelve H46R SOD1 subunits (19, 20). In each case, the side chain of Arg-46 in these subunits donates a hydrogen bond to an acceptor across the active site channel. These hydrogen bond acceptors include the indole nitrogen of His-63, the carbonyl or side-chain oxygen of Thr-137 (as in Fig. 1), or a side-chain oxygen of Asp-124 (19, 20). Taken together, these data suggest that the H46R substitution alone markedly disrupts the binding of copper in the copper site.
In contrast, the singly substituted human H48Q SOD1 protein has been shown to bind copper ions at the copper site when expressed in the presence of high levels of copper (38) or when re-folded in vitro (20). Spectroscopic analysis of SOD1-H48Q, expressed in insect cells grown in media supplemented with copper and zinc sulfate (up to 300 μM), suggested that the copper is coordinated in a geometry that deviates from the distorted tetragonal arrangement found in wild-type toward one that is more regular (38). The coordination of copper ion bound to yeast H48Q SOD1 reconstituted with two equivalents of copper and zinc per dimer was observed directly in the x-ray crystal structure refined to high resolution (PDB code 1F1A). In this case, the copper coordination geometry was found to be square pyramidal, with a water molecule and the indole nitrogens of His-46, His-63, and His-120 in the square plane and an axial water molecule acting as a fifth ligand. Thus, all of the studies mentioned above provide evidence that H48Q SOD1 is capable of binding metal ions in the copper site in a non-native conformation.
Our cell culture labeling studies are generally in agreement with the structural data in that SOD1 proteins with the H46R mutation fail to show binding of radiolabeled copper. In contrast to the studies cited above that used purified protein, we find that the H48Q variant, when expressed in CHO cells, does not bind radiolabeled copper with significant affinity. The loading buffers and gels used in 64Cu labeling experiments contained SDS at concentrations that do not affect the binding of copper to wild-type SOD1 (21). Although we cannot rule out the possibility that the SDS removed loosely bound copper from the H48Q mutant, the electrophoretic migration of SOD1-H48Q in these gels was identical to that of the other histidine mutants (see Fig. 2), which together are similar to monomeric wild-type protein (see below and supplemental Fig. S2).
In yeast and human SOD1, the oxidation of the intrasubunit disulfide bond between Cys-57 and Cys-146 stabilizes the monomeric subunit structure, facilitating the dimerization of enzyme. This oxidation of the disulfide bond appears to be dependent upon the loading of copper (39, 40). When analyzing purified wild-type human enzyme that has an oxidized disulfide and zinc bound in the zinc site, the removal of copper alone is not sufficient to dissociate homodimeric enzyme (28). To produce monomers, removal of both the metal ions and the reduction of the normal intramolecular disulfide bond are required (28). Therefore, for wild-type SOD1, monomerization is associated with both loss of metal and reduction of the disulfide. However, for the mutants we study here, we do not know whether monomerization also requires loss of the disulfide bond, although it seems likely. One simple interpretation of the data would be that the His mutants monomerize, because they bind copper poorly. This interferes with the maturation of the protein to generate structures that allow for normal dimeric interactions (39).
Assays of SOD1 Dimerization
Native gel electrophoresis has routinely been used to distinguish dimeric and monomeric species of SOD1, with monomeric enzyme running significantly faster than dimeric enzyme (21, 35). A potential caveat in using native gels is that some FALS mutations can affect the electrophoretic migration of dimeric SOD1 (5). For example, SOD1-G37R dimers migrate more slowly than SOD1-wt (see Figs. 4 and 5), whereas SOD1-G41D migrates more rapidly (5). In the analysis of protein expressed in CHO cells (see Fig. 2), the sample and gel buffers contained SDS, which masks the small charge effects of amino acid substitutions on gel migration (21). In the study of proteins extracted from mouse tissues, native gels lacking SDS were used, where charge can have a greater effect on electrophoretic migration. However, the most robust effect would occur by the substitution of His-46 for Arg (pI of Arg is 11.15 versus 7.41 for His), which should slow, rather than speed migration as it occurs for the G37R substitution (5). Moreover, other studies of SOD1-H46R purified from Sf9 insect cells and analyzed by native gel (no SDS) also reported migration consistent with monomeric protein (38). Overall, we believe that the histidine mutants are less able to form stable dimers.
Whether the monomeric forms of these mutants bind zinc is unknown. The crystals of SOD1-H46R/H48Q contained forms of the protein that resembled normal dimers and contained zinc bound correctly in the zinc site (see Fig. 1 and supplemental Fig. S1). However, the role of zinc binding in the maturation of the protein in cell cytosol is uncertain and requires further study.
The yeast two-hybrid assay we developed to assess dimer interactions provides additional experimental validation of the role Cu-ligand residues have in dimer formation. As described in under both “Experimental Procedures” and “Results,” we demonstrated that the bait fusion proteins of each mutant were expressed to sufficient levels to saturate the DNA-binding domains of reporter constructs. Thus, the lack of production of β-galactosidase is indicative of poor interaction with the target SOD1-wt fusion protein. The bait fusions of SOD1-wt (positive control) and three of the FALS mutant enzymes behaved as predicted by other studies. Studies by us (5) and others (7, 23, 36) have collectively established that the G85R, G93A, and I113T mutants can form stable dimers. We also note that the electrophoretic migration pattern of G85R protein extracted from mouse tissues was consistent with dimeric enzyme (see Fig. 4C). The A4V variant did not interact with wild-type subunits efficiently in the two-hybrid assay; data consistent with previous reports that the A4V variant monomerizes at low concentrations (24). Moreover, other studies have demonstrated that if A4V fails to bind copper and oxidize the intramolecular disulfide, then the protein is essentially an unfolded monomer (41). Therefore, the data on SOD1-wt, SOD1-A4V, SOD1-G85R, SOD1-G93A, and SOD1-I113T in the yeast two-hybrid assay are consistent with other biochemical data on these mutants. The histidine mutants uniformly show very poor interactions in the two-hybrid assay, data that are corroborated by the electrophoretic migration of these mutants in native gels. Notably, in the high protein concentrations used in crystallization, some of the H46R/H48Q subunits formed dimers resembling the mature enzyme (PDB accession code 2NNX). However, at the concentrations present in cell cytosol or transgenic mouse tissues, our data indicate that the majority of the protein is monomeric. We interpret these data as an indication that the four variants we have analyzed here, harboring mutations at Cu-ligand His residues, fail to adopt a structure compatible with the formation of stable dimers.
In yeast, human SOD1 acquires copper via interactions with the yeast copper chaperone for SOD1 or from reduced glutathione (42), and we therefore expect that the fusion proteins used in the yeast two-hybrid experiments could acquire copper. Unfortunately, we found that the levels of SOD1-fusion protein expression in the yeast strains used in our assay were too low to determine whether any of the proteins bound copper (i.e. were active in assay gels, data now shown). We therefore cannot be certain that the binding of copper by the wt, G93A, G85R, or I113T bait-fusion proteins is responsible for the ability to interact with SOD1-wt target-fusion proteins or that the lack of copper binding by the His mutants is responsible for the inability to for these bait-fusion proteins to interact with SOD1-wt. Because the Cu-ligand residues are not known to be directly involved in bonding at the dimer interface (13, 43), we believe it likely that poor binding of copper by the fusion proteins is one factor in determining the quaternary structure and the ability to of subunits to dimerize.
CONCLUSIONS
In summary, we demonstrate that mutations at histidine residues critical for the coordinated binding of copper, particularly H46R, dramatically reduce the ability of these enzymes to stably bind copper, which affects subsequent post-translational folding to achieve dimeric structure. It is of interest that previous studies have associated the H46R mutation in humans with a very slowly progressing form of the disease (44, 45), which could be construed as consistent with the idea that Cu-mediated toxicity plays a role in disease, because this mutant would bind copper poorly and thus be less toxic. However, in transgenic mouse models, the dose of protein required for Cu-deficient variants to induce disease is similar to that of variants that stably bind copper (16, 17). To explain this observation, mutants that bind copper weakly would have to somehow possess toxicity equivalent to mutants that bind copper far better, and presumably are far more active in toxic Cu-mediated chemistry. The simplest interpretation in our view is that Cu-mediated chemistry, when possible to occur, plays a secondary role in disease pathogenesis with other mechanisms driving the basic disease process. Based on the observation that all mice expressing mutant forms of SOD1, including the mutants examined here and very unstable C-terminal truncation mutants, accumulate aggregated species of mutant protein as symptoms progress (17, 46-48), we believe that aggregation of the mutant protein is likely to be one of the central mechanisms of toxicity.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NIH44464 (to J. G.), by the ALS Association (to D. R. B.), by the Muscular Dystrophy Association (to D. R. B.), by Robert A. Welch Foundation Grant AQ-1399 (to P. J. H.), by the Packard Center for ALS Research at JHU (to D. R. B.), and by NINDS, NIH Grants P01 NS049134 (to D. R. B. and P. J. H.) and R01 NS39912 (to P. J. H.). The production of 64Cu at the Washington University School of Medicine is supported by NCI, NIH Grant R24 CA86307. The X-ray Crystallography Core Laboratory is supported by the VPR and the Executive Research Council at the University of Texas Health Science Center at San Antonio.
The on-line version of this article (available at http://www.jbc.org) contains supplemental text, references, and Figs. S1 and S2.
- ALS
- amyotrophic lateral sclerosis
- FALS
- familial ALS
- SOD1
- Cu/Zn-superoxide dismutase 1
- CHO
- Chinese hamster ovary
- Quad
- quadruple mutant H46R/H48Q/H63G/H120G.
The atomic coordinates and structure factors (code 2NNX) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
REFERENCES
- 1.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Andersen PM, Morita M, Brown RH., Jr. In: Amyotrophic Lateral Sclerosis. Brown RH Jr., Meininger V, Swash M, editors. Martin Dunitz Ltd.; London: 2000. pp. 223–250. [Google Scholar]
- 3.Cleveland DW, Rothstein JD. Nat. Rev. Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 4.Hart PJ. Curr. Opin. Chem. Biol. 2006;10:131–138. doi: 10.1016/j.cbpa.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 5.Borchelt DR, Lee MK, Slunt HH, Guarnieri M, Xu Z-S, Wong PC, Brown RH, Jr., Price DL, Sisodia SS, Cleveland DW. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borchelt DR, Guarnieri M, Wong PC, Lee MK, Slunt HS, Xu Z-S, Sisodia SS, Price DL, Cleveland DW. J. Biol. Chem. 1995;270:3234–3238. doi: 10.1074/jbc.270.7.3234. [DOI] [PubMed] [Google Scholar]
- 7.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 8.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 9.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr., Scott RW, Snider WD. Nat. Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 10.Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. Proc. Natl. Acad. Sci. U. S. A. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartz JW, Deutsch HF. J. Biol. Chem. 1972;247:7043–7050. [PubMed] [Google Scholar]
- 12.Fridovich I. Adv. Enzymol. Relat. Areas Mol. Biol. 1974;41:35–97. doi: 10.1002/9780470122860.ch2. [DOI] [PubMed] [Google Scholar]
- 13.Parge HE, Hallewell RA, Tainer JA. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valentine JS, Hart PJ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR. Hum. Mol. Genet. 1999;8:1451–1460. doi: 10.1093/hmg/8.8.1451. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Neurobiol. Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Hum. Mol. Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 18.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Nat. Struct. Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 19.Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, Rodriguez JA, Hayward LJ, Valentine JS, Hart PJ, Hasnain SS. Protein Sci. 2005;14:1201–1213. doi: 10.1110/ps.041256705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liochev SI, Chen LL, Hallewell RA, Fridovich I. Arch. Biochem. Biophys. 1997;346:263–268. doi: 10.1006/abbi.1997.0298. [DOI] [PubMed] [Google Scholar]
- 21.Bartnikas TB, Gitlin JD. J. Biol. Chem. 2003;278:33602–33608. doi: 10.1074/jbc.M305435200. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli UK. Nature (Lond.) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Rumfeldt JA, Stathopulos PB, Chakrabarrty A, Lepock JR, Meiering EM. J. Mol. Biol. 2006;355:106–123. doi: 10.1016/j.jmb.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 24.Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr., Walz T, Lansbury PT., Jr. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 25.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Proc. Natl. Acad. Sci. U. S. A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 27.Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV, Siddique T. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doucette PA, Whitson LJ, Cao X, Schirf V, Demeler B, Valentine JS, Hansen JC, Hart PJ. J. Biol. Chem. 2004;279:54558–54566. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 29.Pflugrath JW. Acta Crystallogr. D. Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 30.Vagin A, Teplyakov A. Acta Crystallogr. D. Biol. Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 31.Hart PJ, Liu H, Pellegrini M, Nersissian AM, Gralla EB, Valentine JS, Eisenberg D. Protein Sci. 1998;7:545–555. doi: 10.1002/pro.5560070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. D. Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Malinowski DP, Fridovich I. Biochemistry. 1979;18:237–244. doi: 10.1021/bi00568a037. [DOI] [PubMed] [Google Scholar]
- 36.Vassall KA, Stathopulos PB, Rumfeldt JA, Lepock JR, Meiering EM. Biochemistry. 2006;45:7366–7379. doi: 10.1021/bi0600953. [DOI] [PubMed] [Google Scholar]
- 37.Beauchamp C, Fridovich I. Anal. Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 38.Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH., Jr. J. Biol. Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 39.Furukawa Y, Torres AS, O’Halloran TV. EMBO J. 2004;23:2872–2881. doi: 10.1038/sj.emboj.7600276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown NM, Torres AS, Doan PE, O’Halloran TV. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5518–5523. doi: 10.1073/pnas.0401175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll MC, Girouard JB, Ulloa JL, Subramaniam JR, Wong PC, Valentine JS, Culotta VC. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5964–5969. doi: 10.1073/pnas.0308298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. J. Mol. Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 44.Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Nat. Genet. 1993;5:323–324. doi: 10.1038/ng1293-323. [DOI] [PubMed] [Google Scholar]
- 45.Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. J. Neurol. Sci. 1994;126:77–83. doi: 10.1016/0022-510x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 46.Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C, Copeland NG, Jenkins NA, Borchelt DR. Hum. Mol. Genet. 2005;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- 48.Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Rehnmark A, Marklund SL. J. Neurosci. 2006;26:4147–4154. doi: 10.1523/JNEUROSCI.5461-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.