

Abstract
Exocytosis of the acrosome (the acrosome reaction) relies on cAMP production, assembly of a proteinaceous fusion machinery, calcium influx from the extracellular medium, and mobilization from inositol 1,4,5-trisphosphate-sensitive intracellular stores. Addition of cAMP to human sperm suspensions bypasses some of these requirements and elicits exocytosis in a protein kinase A- and extracellular calcium-independent manner. The relevant cAMP target is Epac, a guanine nucleotide exchange factor for the small GTPase Rap. We show here that a soluble adenylyl cyclase synthesizes the cAMP required for the acrosome reaction. Epac stimulates the exchange of GDP for GTP on Rap1, upstream of a phospholipase C. The Epac-selective cAMP analogue 8-pCPT-2′-O-Me-cAMP induces a phospholipase C-dependent calcium mobilization in human sperm suspensions. In addition, our studies identify a novel connection between cAMP and Rab3A, a secretory granule-associated protein, revealing that the latter functions downstream of soluble adenylyl cyclase/cAMP/Epac but not of Rap1. Challenging sperm with calcium or 8-pCPT-2′-O-Me-cAMP boosts the exchange of GDP for GTP on Rab3A. Recombinant Epac does not release GDP from Rab3A in vitro, suggesting that the Rab3A-GEF activation by cAMP/Epac in vivo is indirect. We propose that Epac sits at a critical point during the exocytotic cascade after which the pathway splits into two limbs, one that assembles the fusion machinery into place and another that elicits intracellular calcium release.
During fertilization in eutherian mammals, the spermatozoon must penetrate the zona pellucida to reach the oolema. Only sperm that have completed the acrosome reaction (AR)4 can successfully accomplish this task (1). The AR is a regulated exocytosis where the membrane of the acrosome, the single dense core secretory granule in sperm, fuses to the plasma membrane surrounding the anterior portion of the head. This process releases hydrolytic enzymes stored in the granule. These enzymes, together with the physical thrust derived from strong flagellar beating, enable sperm to penetrate the zona pellucida (1, 2). Physiological agonists accomplish the AR by inducing an influx of calcium from the extracellular medium and the assembly of a conserved proteinaceous fusion machinery that includes Rab3A, α-SNAP/NSF, synaptotagmin, complexin, and neurotoxin-sensitive SNAREs; the AR also requires an efflux of calcium from inside the acrosome through IP3-sensitive channels (reviewed in Refs. 3, 4).
In certain neurons, neuroendocrine and exocrine acinar cells, cAMP potentiates calcium-dependent exocytosis. Either cAMP-dependent protein kinase (PKA) or the exchange protein directly activated by cAMP (Epac) can be the targets of cAMP in the cAMP-regulated exocytosis. On the other hand, cAMP is the principal trigger of regulated secretion in various non-neuronal cells (5–7). Likewise, an elevation of cAMP alone is sufficient to trigger exocytosis in human sperm. Moreover, calcium relies on endogenous cAMP to accomplish acrosomal release, and it does so through a PKA-insensitive pathway involving Epac. The stimulation of endogenous Epac by the selective cAMP analogue 8-(p-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pCPT-2′-O-Me-cAMP) is sufficient to trigger the AR even in the absence of extracellular calcium. Furthermore, when Epac is sequestered with specific antibodies, cAMP, calcium (8), and recombinant Rab3A (this study) are unable to elicit exocytosis.
Epac1 and Epac2 are multidomain proteins that consist of an N-terminal regulatory region and a C-terminal catalytic region (9–11). The regulatory domain harbors the cAMP-binding site, which auto-inhibits the catalytic activity in the absence of cAMP (12–15). The catalytic portion bears a guanine-nucleotide exchange factor (GEF) activity specific for Rap1 and Rap2 (16, 17). Like all small G proteins, Raps cycle between an inactive GDP-bound and an active GTP-bound conformation. The GDP-GTP cycle is regulated by GEFs that induce the release of the bound GDP to be replaced by the more abundant GTP and by GTPase-activating proteins that coax the intrinsic GTPase activity to rapidly hydrolyze bound GTP, returning the G proteins to the inactive GDP-bound state (18, 19). Most small G proteins are linked to biological membranes via lipid modifications at their C terminus; for instance, Rap2A is farnesylated, and Rap1A/B, Rap2B, and Rabs are geranylgeranylated (20, 21). Guanine nucleotide dissociation inhibitors (GDIs) remove Rabs from membranes by sequestration of their lipid tails (22).
Extracellular stimuli often result in the activation of cellular adenylate cyclases and an increase in cAMP levels. By serving as a cAMP-binding protein with intrinsic GEF activity, Epac couples cAMP production to a variety of Rap-mediated processes such as the control of cell adhesion and cell-cell junction formation, water resorption, cell differentiation, inflammatory processes, etc. (9–11). Many are the effectors of Epac and Epac-Rap signaling. Of particular interest to us is the observation that Epac stimulates phospholipase Cϵ (PLCϵ) through the activation of Rap1 and -2, resulting in IP3-mediated release of calcium from internal stores (23, 24). PLCϵ is an unusual enzyme with two catalytic activities as follows: the typical phosphatidylinositol 4,5-bisphosphate hydrolyzing PLC activity plus a Rap-GEF activity. Thus, PLCϵ acts both downstream and upstream of Ras-like GTPases, perhaps to guarantee sustained Rap signaling (25).
During membrane fusion, Rab proteins direct the recognition and physical attachments of the compartments that are going to fuse (26, 27). This association, or tethering, represents one of the earliest known events in membrane fusion and is accomplished through the recruitment of tethering factors. Rab3A localizes to vesicles and secretory granules and is one of the isoforms directly implicated in regulated exocytosis of neurotransmitters and hormones (28). Rab3A interacts in a GTP-dependent manner with at least two effector proteins, rabphilin and Rim (29–31). Rab3A is present in the acrosomal region of human (32), rat (33), and mouse sperm (34). Rab3A (full-length recombinant protein or a synthetic peptide corresponding to the effector domain) stimulates human (32, 35) and ram (36) and inhibits rat sperm AR (33). Rab3A is required for the AR triggered by calcium (37, 38) and cAMP (8).
Epac is a multifunctional protein in which cAMP exerts its effects not only by promoting the exchange of GDP for GTP on Rap but also by allosterically regulating other molecules (10). In exocytosis for instance, a number of Rap-independent, Epac-linked signaling pathways have been described. They include the interaction of Epac2 with Rim2 (39) and the Rim2-related protein Piccolo (40). Epac2 also stimulates exocytosis by interacting with SUR1 (41). Finally, Epac2 controls ryanodine-sensitive calcium channels that are involved in calcium-induced calcium release (CICR) from internal stores in insulin-secreting cells (42).
In this study, we piece together the analysis of two phenomena as follows: calcium mobilization and protein-protein interactions preceding exocytosis. To the best of our knowledge, this constitutes the first integrated molecular model that includes both the assembly of the fusion and intravesicular calcium release protein machineries during regulated exocytosis. By enquiring further into the signaling pathways operating during sperm exocytosis, we have found more players than previously suspected, and we discovered that the key components of these cascades are not arranged in a linear sequence. Epac sits at a central point of the signaling cascade after which the exocytotic pathway splits into two limbs as follows: one that assembles the fusion machinery into place, and another that elicits the release of calcium from the acrosome; both need to act in concert to achieve exocytosis. Our results identify Rab3A for the first time as a downstream target for Epac and place this small GTPase as an early component of the “fusion machinery” branch of the pathway. They also show that Epac stimulates the exchange of GDP for GTP on Rap1 and that this protein, as well as a PLC, drives intracellular calcium mobilization. Finally, our data reveal that a soluble adenylyl cyclase (sAC) (43, 44) synthesizes the cAMP that activates Epac. Again, we believe that this is the first report linking sAC to an exocytotic event.
EXPERIMENTAL PROCEDURES
Reagents
Recombinant streptolysin O (SLO) was obtained from Dr. Bhakdi (University of Mainz, Mainz, Germany). Spermatozoa were cultured in human tubal fluid media (as formulated by Irvine Scientific, Santa Ana, CA) supplemented with 0.5% bovine serum albumin (HTF media). The rabbit polyclonal antibodies to Rap1A/B and Rab3A (purified IgG) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The rabbit polyclonal anti-NSF (whole serum) was from Synaptic Systems (Göttingen, Germany), and rabbit polyclonal antibody to Rab11 was from Invitrogen. The rabbit polyclonal antibodies against Epac were generated by Genemed Synthesis, Inc. (San Francisco), using the synthetic peptide LREDNCHFLRVDK, and affinity-purified on immobilized Epac peptide (8). Anti-Rap 1A/B rabbit polyclonal antibodies were raised against the peptide EDERVVGKEQGQNLC and affinity-purified on immobilized peptide (GenScript Corp., Piscataway, NJ). Horseradish peroxidase-conjugated goat anti-rabbit IgG (anti-γ chain) was from Jackson ImmunoResearch (West Grove, PA). Glutathione S-transferase (GST)-tagged, human recombinant cAMP-specific phosphodiesterase 4D (PDE), and Rap1A were from Abnova Corp. (NeiHu, Taipei, Taiwan). KH7 was purchased from ChemDiv, Inc. (San Diego). 8-pCPT-2′-O- Me-cAMP, N6-benzoyladenosine-3′,5′-cyclic monophosphate (6-Bnz-cAMP), and 2′,3′-O-(N′-methylanthraniloyl)-GDP(mGDP) were from Biolog-Life Science Institute (Bremen, Germany). U73122 (1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione) and the inactive analogue U73343 (1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidine-dione) were from Biomol International L.P. (Plymouth Meeting, PA). Adenophostin A, hexasodium salt, and 2-aminoethoxydiphenyl borate (2-APB) from Calbiochem were purchased from Merck Química Argentina S.A.I.C. (Buenos Aires, Argentina). O-Nitrophenyl EGTA-acetoxymethyl ester (NP-EGTA-AM) and Fluo-3-AM were purchased from Molecular Probes (Eugene, OR). Prestained molecular weight markers were from Boston BioProducts, Inc. (Worcester, MA). Glutathione-Sepharose and nickel-nitrilotriacetic acid-agarose were from GE Healthcare. All other chemicals were purchased from Sigma, Genbiotech, or Tecnolab (all from Buenos Aires, Argentina).
Recombinant Proteins
Plasmid pGEX-2T containing the cDNA-encoding human Rab3A was provided by Dr. P. Stahl (Washington University, St. Louis, MO). The Rap1-GTP binding cassette Ral-GDS-RBD fused to GST (45) was a kind gift from Dr. O. Coso (Universidad de Buenos Aires, Buenos Aires, Argentina). The Rab3-GTP binding cassette RIM-RBD fused to GST (46) was generously provided by Dr. R. Regazzi (University of Lausanne, Lausanne, Switzerland). cDNA encoding GDI-α was a kind gift from Dr. Y. Takai (Osaka University, Osaka, Japan). The expression plasmid pGEX-3X (GE Healthcare) encoding GST fused to amino acids 1–321 of PTP1B bearing the substrate-trapping mutation D181A was kindly provided by Dr. N. Tonks (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Plasmid pAC21 encoding a His6-tagged phosphomimetic mutant of NSF (NSF Y83E) was a gift from Dr. N. Bottini (The Burnham Institute, La Jolla, CA). The Epac1 construct is derived from human cDNA and the Epac2 construct from murine cDNA. Epac1 (amino acids 149–881) and Epac2 (amino acids 280–993) were expressed as GST fusion proteins from the pGEX-4T2 vector in the Escherichia coli strain CK600K as described previously (47). GST-Rab3A was expressed in E. coli BL21 (Stratagene) as described (48). GST-Ral-GDS-RBD, GST-RIM, GST-PTP1B D181A, GST-Rap1A, and GST-GDI were transformed into E. coli BL21, and protein expression was induced with isopropyl β-d-thio-galactoside (0.2 mm for the first two proteins and 0.1 mm for the rest) for 16 h at 22 °C. All GST-fused recombinant proteins were purified on glutathione-Sepharose following standard procedures, except GST-Ral-GDS-RBD and GST-RIM, where bacterial lysates were frozen until use. His6-NSF Y83E was expressed and purified on nickel-nitrilotriacetic acid-agarose as described for wild type NSF (49). Purified, recombinant His6-Rab3A (35) was a kind gift from Dr. C. López (IHEM, Mendoza, Argentina). Rab3A proteins were prenylated for all but the in vitro activation experiments and loaded with the appropriate guanosine nucleotides. Recombinant protein concentrations were determined by the Bio-Rad protein assay in 96-well microplates. Bovine serum albumin was used as a standard, and the results were quantified on a 3550 Microplate Reader (Bio-Rad).
In Vitro Activation of Rab3A
Experiments were performed as described (50, 51), using Rab3A in addition to Rap1. Purified Rab3A or Rap1 (200 nm, GST removed with thrombin) loaded with the fluorescent GDP analogue mGDP was incubated in the presence of 20 μm unlabeled GDP plus 150 nm purified Epac1 or Epac2. Cyclic AMP (100–500 μm) was added as indicated. The nucleotide exchange was measured in real time as decay in fluorescence using a Cary Eclipse spectrofluorometer (Varian, Varian B.V., Middelburg, The Netherlands). The decay is caused by the release of protein-bound mGDP, which shows higher fluorescence intensity in the hydrophobic environment of the protein than in the buffer solution. The decay in the fluorescence signal is equal to the rate of nucleotide dissociation, and nucleotide exchange should be accelerated in the presence of an active GEF. All data analysis, fitting, and plotting were done with the Grafit 3.0 program (Erithacus software).
SLO Permeabilization and AR Assay
Human semen samples were obtained from normal healthy donors. Semen was allowed to liquify for 30–60 min at 37 °C. We used a swim-up protocol to isolate highly motile sperm. Sperm concentrations were adjusted to 5–10 × 106/ml before incubating for at least 2 h under capacitating conditions (HTF, 37 °C, 5% CO2, 95% air). For experiments involving intact sperm, we added increasing concentrations of KH7 and 10 μm A23187 or 5 μm progesterone sequentially and incubated for 10–15 min at 37 °C after each addition. For experiments involving permeabilized cells, washed spermatozoa were resuspended in cold PBS containing 2.1 units/ml SLO for 15 min at 4 °C. Cells were washed once with PBS and resuspended in ice-cold sucrose buffer (250 mm sucrose, 0.5 mm EGTA, 20 mm Hepes-K, pH 7) containing 2 mm dithiothreitol. We added inhibitors and stimulants sequentially as indicated in the figure legends and incubated for 10–15 min at 37 °C after each addition. Where indicated, we preloaded SLO-permeabilized sperm with NP-EGTA-AM before incubating with inhibitors and/or calcium, carrying out all procedures in the dark. Photolysis was induced after the last incubation by exposing twice (1 min each time) to an UV transilluminator and mixing after each exposure. Intact and permeabilized sperm were spotted on Teflon-printed slides, air-dried, and fixed/permeabilized in ice-cold methanol for 1 min. Acrosomal status was evaluated by staining with FITC-coupled Pisum sativum (FITC-PSA) (52). At least 200 cells were scored using an upright Nikon microscope equipped with epifluorescence optics. Basal (no stimulation) and positive (0.5 mm CaCl2 corresponding to 10 μm free calcium estimated by MAXCHELATOR, a series of program(s) for determining the free metal concentration in the presence of chelators) controls were included in all experiments. Acrosomal exocytosis indexes were calculated by subtracting the number of spontaneously reacted spermatozoa from all values and expressing the results as a percentage of the AR observed in the positive control. Data were evaluated using one way analysis of variance. The Tukey-Kramer post hoc test was used for pairwise comparisons. Differences were considered significant at the p < 0.05 level.
Rab3-GTP and Rap1-GTP Precipitation Assays
Capacitated sperm (10–50 × 106 cells) were washed twice and suspended in PBS. Sperm were treated with 100 μm 2-APB (an IP3-sensitive calcium channel blocker) to prevent cytosol/membrane loss due to exocytosis before adding the AR inducers. Alternatively, sperm were permeabilized with SLO and treated with 2-APB and 20 μg/ml anti-Rap1 antibodies (GenScript) or 1 μg of His6-Rab3A before challenging with 50 μm 8-pCPT-2′-O-Me-cAMP or 0.5 mm CaCl2. After 10 min of incubation at 37 °C, cells were lysed in GST pulldown buffer (200 mm NaCl, 2.5 mm MgCl2, 1% (v/v) Triton X-100, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, 1× protease inhibitor mixture (P2714, Sigma), and 50 mm Tris-HCl, pH 7.4) by sonication on ice (two times for 15 s). We let proteins diffuse into the lysis buffer for 15 min at 4 °C. These whole cell detergent extracts were clarified by centrifugation at 12,000 × g for 5 min and used immediately.
Glutathione-Sepharose beads were washed twice with GST pulldown buffer and incubated with bacterial lysates containing GST-Ral-GDS-RBD or GST-RIM for 1 h at 4 °C under constant rocking. Beads were washed twice with PBS and once with GST pulldown buffer and used immediately. Twenty μl of glutathione-Sepharose containing 10 μg of the appropriate fusion protein was added to sperm lysates in a total volume of 0.6 ml and incubated by rotation at 4 °C for 30 min. The resin was recovered by centrifugation at 4 °C (5 min at 10,000 rpm) and washed three times with ice-cold GST pulldown buffer. The resin-bound fractions were resolved by SDS-PAGE, and cellular GTP-Rap1 and GTP-Rab3A levels were analyzed by immunoblotting as described later.
Triton X-114 Partition
Partition experiments were conducted following standard procedures (53, 54). Briefly, capacitated sperm (100 × 106 cells) were washed twice with cold PBS, and proteins were extracted in 1 ml of lysis buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10% glycerol, 5 mm MgCl2, and 1% Triton X-114 by sonication on ice (three times for 15 s with 10-s intervals). The lysates were rocked for 45 min at 4 °C and spun at 14,000 × g during 20 min at 4 °C. These whole cell detergent extracts were incubated for 15 min at 30 °C and centrifuged at 3,000 × g for 2 min. Hydrophilic proteins partitioned into the upper (aqueous) phase, whereas hydrophobic proteins were recovered from the lower (detergent) phase. Protein precipitation and removal of detergent were achieved via extraction with CCl3H-CH3OH. Precipitated proteins were dissolved in sample buffer by heating once at 60 °C for 10 min and once at 95 °C for 3 min.
SDS-PAGE and Western Blots
Proteins were separated on SDS-gel electrophoresis (55) and transferred to 0.22-μm nitrocellulose membranes (Hybond, GE Healthcare). Nonspecific reactivity was blocked by incubation for 1 h at room temperature with 5% skim milk dissolved in washing buffer (PBS, pH 7.6, 0.1% Tween 20). Blots were incubated with the anti-Rab3A (0.2 μg/ml) or the custom-made anti-Rap1 (0.5 μg/ml in blocking solution) antibodies in blocking solution for 1 h at room temperature or overnight at 4 °C. Horseradish peroxidase-conjugated goat anti-rabbit IgG was used as secondary antibody (3 μg/ml) with 1-h incubations. Excess first and second antibodies were removed by washing five times for 7 min in washing buffer. Detection was accomplished with a chemiluminescence system (Western Lightning, PerkinElmer Life Sciences, Migliore Laclaustra, Buenos Aires, Argentina) and subsequent exposure to CL-XPosure film (Tecnolab, Pierce) for 1–10 min.
Calcium Measurements
Calcium levels were measured in sperm suspensions using the intracellular fluorescent probe Fluo-3. Motile sperm were adjusted to a concentration of 5–10 × 106 cells/ml and loaded with the permeable form of the dye (Fluo-3-AM, 2 μm) and 0.02% of pluronic acid for 30 min at 37 °C. Cells were washed once and resuspended in nominally calcium-free (∼2 μm) medium (10 mm Hepes-Na, 120 mm NaCl, 4 mm KCl, 15 mm NaHCO3, 1 mm MgCl2, 5 mm d-glucose, 1 mm sodium pyruvate, 10 mm lactic acid, pH 7.4). Sperm suspensions were transferred to thermostated (37 °C) cuvettes for fluorescence measurements and stirred constantly. At the indicated times, 50 μm 8-pCPT-2′-O-Me-cAMP was added to the samples with or without previous incubation with 1 μm U73122 or U73343. Fluo-3 fluorescence (λEx = 505 nm, λEm = 525 nm emission) was recorded in an Aminco 8000 spectrofluorometer. Data were collected during 600 s at a frequency of 0.5 Hz. To calibrate the maximal response, [Ca2+]i was determined using Triton X-100 (0.1%). Measurements were performed at least five times with different batches of sperm. The criterion we used for inclusion of the results obtained with a given sample into the statistical analysis was the normal response of the said sample to 4 μm progesterone. The Tukey-Kramer post hoc test was used for pairwise comparisons. Differences were considered significant at the p < 0.05 level.
RESULTS
sAC Synthesizes cAMP Required for Human Sperm Exocytosis
Previous findings from our laboratory (8, 49) had led us to formulate the AR as summarized in Pathway 1, where single arrows indicate there is one step between the terms connected, and double arrows indicate that the number of steps taking place between the connected terms is unknown.
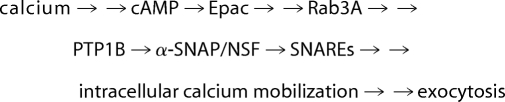 |
This pathway begins with the sequence “calcium →→ cAMP → Epac.” In nonpermeabilized sperm, calcium enters the cells from the extracellular milieu when channels open in the plasma membrane in response to physiological agonists or when a calcium ionophore transports it to the cytosol; in permeabilized sperm, calcium enters through the SLO-generated pores. Because depletion of endogenous cAMP by an active PDE prevents the AR induced by calcium (8), we know that this cyclic nucleotide is required to accomplish exocytosis. Where does this cAMP come from? We found that the pool of cAMP relevant for the AR is synthesized by sAC, because the ARs elicited by calcium in permeabilized human sperm, as well as that elicited by the calcium ionophore A23187 and progesterone in nonpermeabilized cells, were sensitive to the selective sAC blocker KH7 (56) (Fig. 1, A and B). In contrast, the exocytosis triggered by 8-pCPT-2′-O-Me-cAMP was insensitive to KH7 (Fig. 1A), pointing to the specificity of the effect of the inhibitor. These data also suggest that Epac is the ultimate target of the cAMP synthesized by sAC during the AR.
FIGURE 1.

AR relies on cAMP synthesized by sAC. A, intact (nonpermeabilized) sperm were exposed to the indicated concentrations of KH7 for 15 min at 37 °C. Acrosomal exocytosis was initiated by adding 10 μm A23187 (left), 15 μm progesterone (Pg, right), or 50 μm 8pCPT-2′-O-Me-cAMP (8pCPT, right) and incubating at 37 °C for a further 15 min. Cells were fixed; acrosomal exocytosis was evaluated by FITC-PSA binding, and data were plotted without normalization (mean ± S.E. of at least three independent experiments); **, p < 0.01 compared with A23187 (left) or progesterone (right). B, SLO-permeabilized human sperm were treated for 15 min at 37 °C with increasing concentrations of KH7. AR was subsequently initiated by adding 0.5 mm CaCl2 and incubating for 15 min at 37 °C. Exocytosis was evaluated by FITC-PSA binding, and data were normalized as described under “Experimental Procedures.” Plotted results represent the mean ± S.E. of at least three independent experiments.
Activation of Rap1 by cAMP through Epac Mediates Acrosomal Exocytosis
Epac is present and functionally important for human sperm exocytosis (8). The primary function of Epac is to act as a GEF for Rap proteins; therefore, we asked if Rap is present in human sperm and if its activation is required for exocytosis. We used a specific, custom-made polyclonal antibody to detect the presence of Rap1 in sperm extracts by Western blot. As shown in Fig. 2A, the anti-Rap1A/B antibodies recognized a single protein band; we inferred that Rap1 was geranylgeranylated in sperm because it partitioned into the detergent phase following Triton X-114 phase separation.
FIGURE 2.

Calcium and 8-pCPT-2′-O-Me-cAMP activate Rap1 during sperm exocytosis. A, proteins from whole sperm homogenates (100 × 106 cells) were partitioned in Triton X-114 and precipitated with organic solvents as described under “Experimental Procedures.” Proteins from the aqueous and detergent phases (normalized by initial cell counts) were resolved in 10% Tricine/SDS-polyacrylamide gels and immunoblotted with the custom-made anti-Rap1 antibody. Molecular mass markers are indicated on the left. B, top, SLO-permeabilized human sperm were treated with 20 μg/ml of an anti-Rap1 polyclonal antibody (Santa Cruz Biotechnology) for 15 min at 37 °C. Acrosomal exocytosis was subsequently initiated by adding 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT, black bar) and incubating for a further 15 min at 37 °C. Bottom, SLO-permeabilized human sperm were incubated for 15 min at 37 °C with 20 μg/ml of the commercial or the custom-made (CM) anti-Rap antibodies (black bars). Asterisks indicate that the antibodies had been pretreated with 205 μg/ml recombinant Rap (anti-Rap* → calcium) or 453 μg/ml Rap peptide (CM-anti-Rap* → calcium). Sperm were challenged with 0.5 mm CaCl2 and incubated for an additional 15 min at 37 °C. Control condition (gray bars) include the following: background AR in the absence of any stimulation (control), AR induced with 0.5 mm CaCl2 (calcium), or 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT); lack of effect on the AR of recombinant Rap1 and 200 μm 6-Bnz-cAMP alone; and unaffected exocytosis by 20 μg/ml anti-Rab11 and nonimmune antibodies (anti-Rab11/IgG → calcium). Cells were fixed; acrosomal exocytosis was evaluated by FITC-PSA binding, and data were normalized as described under “Experimental Procedures.” C, sperm suspensions (20 × 106 cells) were treated (+) or not (−) with 50 μm 8-pCPT-2′-O-Me-cAMP for 10 min at 37 °C in the presence of 100 μm 2-APB to prevent protein loss due to exocytosis. Cells were disrupted; whole cell lysates were subjected to pulldown assays using GST-Ral-GDS-RBD-Sepharose, and the levels of GTP-bound Rap1 were determined as described under “Experimental Procedures.” Shown is a blot representative of three repetitions. D, permeabilized spermatozoa were incubated with 1 μg/ml Ral-GDS-RBD or 1 μg/ml GST for 15 min at 37 °C. Acrosomal exocytosis was then initiated by adding 0.5 mm CaCl2 (calcium) or 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT) and a further 15-min incubation at 37 °C. Sperm were stained and the AR scored as in B. The data represent the mean ± S.E. of at least three independent experiments.
Incorporation of a 2′-O-methyl substitution on the ribose ring of cAMP impairs its ability to activate PKA but not Epac. Furthermore, the introduction of a pCPT substitution at position 8 on the adenine moiety of 2′-O-alkylated cAMP increases the affinity for Epac. On the other hand, cAMP analogues modified at position 6 are full PKA but poor Epac agonists (57). The 8-substituted analogue 8-pCPT-2′-O-Me-cAMP constitutes a unique, widespread tool to assess Epac-dependent, PKA-independent actions. One of the criteria to validate that 8-pCPT-2′-O-Me-cAMP is acting via Epac requires that N6-Bnz-cAMP does not mimic its actions (58). In human sperm, the activation of Epac by exogenously added 8-pCPT-2′-O-Me-cAMP suffices to elicit the AR (8). In contrast, treatment with the PKA-selective analogue N6-Bnz-cAMP had no effect (Fig. 2B).
We introduced a commercial anti-Rap1A/B antibody into SLO-permeabilized sperm to sequester the endogenous protein and investigate if Rap is implicated in the cAMP/Epac-initiated AR pathway. Pretreatment with the antibody inhibited 8-pCPT-2′-O-Me-cAMP-triggered AR by ≥90% (Fig. 2B, top). Next, we asked whether calcium also relies on Rap1 to fulfill its role as an AR inducer. When added to SLO-permeabilized sperm, two different anti-Rap1 antibodies effectively abrogated calcium-triggered AR (Fig. 2B, bottom). The effect of the custom-made anti-Rap1 polyclonal was diminished by preincubation with the synthetic peptide against which the antibody was raised. Likewise, preincubation of the commercial anti-Rap1 antibody with recombinant GST-Rap1 decreased the ability of the antibody to prevent the AR. Neither a nonimmune rabbit IgG nor an antibody to an unrelated small G protein (anti-Rab11) had any effect (Fig. 2B). These data suggest that the inhibitory effect of the anti-Rap1 antibodies was specific and due to binding to endogenous Rap1. Taken together, these results reveal that Rap1 is necessary for human sperm acrosomal exocytosis.
Many hormones and agonists that elevate intracellular cAMP levels increase the amount of active, GTP-bound Rap1 in responsive cells. We therefore asked whether the onset of the AR induces Rap1 activation in human sperm. This was indeed the case because when we pulled down GTP-bound Rap1 with immobilized Ral-GDS-RBD we found that the amount of active Rap1 increased in response to 8-pCPT-2′-O-Me-cAMP (Fig. 2C). Next, we introduced Ral-GDS-RBD into SLO-permeabilized sperm before challenging with AR inducers, hypothesizing that this cassette would sequester Rap1-GTP if formed in response to a stimulus. 8-pCPT-2′-O-Me-cAMP was our positive control because we knew that activating Epac exchanged GDP for GTP on Rap1 (Fig. 2C). Ral-GDS-RBD prevented the AR elicited by 8-pCPT-2′-O-Me-cAMP (Fig. 2D), indicating that Rap1-GTP was necessary for the AR and validating the use of the cassette to deplete the pool of active Rap1. Calcium was unable to achieve exocytosis in the presence of Ral-GDS-RBD (Fig. 2D), suggesting that this inducer also promoted the activation of Rap1 and required Rap1-GTP to elicit the AR. GST alone, used as negative control, had no effect on calcium-triggered exocytosis (Fig. 2D).
Rap1 Is Located Upstream of Calcium Mobilization from an Intracellular IP3-sensitive Store, Likely the Acrosome
The acrosome behaves as an internal store of releasable calcium (59, 60); efflux from this reservoir through IP3-sensitive channels is required for the AR initiated by calcium itself, Rab3A-GTP-γ-S, and cAMP (8, 35, 38, 59). To assess whether the requirement for Rap1 takes place prior to or following intra-acrosomal calcium efflux, we resorted to the photolabile calcium chelator NP-EGTA-AM. In our SLO-permeabilized human sperm model NP-EGTA-AM crosses the plasma and outer acrosomal membranes, accumulates inside the acrosome, and prevents the AR by sequestering intra-acrosomal calcium (59). UV photolysis of NP-EGTA-AM rapidly replenishes the acrosomal calcium pool, resuming exocytosis (Fig. 3A, gray bars). In combination with specific blockers, NP-EGTA-AM allowed us to place the requirement for the target of the blockers before or after the intra-acrosomal calcium-sensitive step. Briefly, we loaded SLO-permeabilized sperm with NP-EGTA-AM and challenged with an AR inducer (extracellular calcium in this case), allowing the fusion-related signaling pathways to advance up to the point where intra-acrosomal calcium release is required. Subsequently, we added AR blockers and illuminated the tubes. In this scenario, resistance to a blocker (revealed by unaffected exocytosis) implies that its target is required upstream of intra-acrosomal calcium efflux, whereas sensitivity to a blocker (revealed by inhibited exocytosis) suggests that its target is necessary after the intra-acrosomal calcium-sensitive step. The AR was unaffected by both anti-Rap1 antibodies and Ral-GDS-RBD (Fig. 3A, black bars), indicating that Rap1-GTP is necessary early in the fusion cascade, before calcium is released from the acrosome.
FIGURE 3.

Rap1 participates in a pathway that mobilizes calcium from an IP3-sensitive store. A, permeabilized spermatozoa were loaded with 10 μm NP-EGTA-AM (NP) for 10 min at 37 °C to chelate intra-acrosomal calcium. The AR was subsequently initiated by adding 0.5 mm CaCl2. After a further 15-min incubation at 37 °C to allow exocytosis to proceed up to the intra-acrosomal calcium-sensitive step, sperm were treated for 15 min at 37 °C with 20 μg/ml anti-Rap antibody (Santa Cruz Biotechnology) or 1 μg/ml Ral-GDS-RBD. All these procedures were carried out in the dark. UV flash photolysis of the chelator was induced at the end of the incubation period (hv), and the samples were incubated for 5 min (NP → calcium → inhibitor → hv; black bars). Several controls were run (gray bars) as follows: background AR in the absence of any stimulation (control); AR stimulated by 0.5 mm CaCl2 (calcium), inhibitory effect of NP-EGTA-AM in the dark (NP → calcium → dark), and the recovery upon illumination (NP → calcium → hv); and the effect of the inhibitors when present throughout the experiment (NP → inhibitor → calcium → hv). Sperm were fixed; the AR was measured by FITC-PSA binding, and the data were normalized as described under “Experimental Procedures.” B, permeabilized spermatozoa were incubated with 15 μm U73343 or U73122 for 15 min at 37 °C. Acrosomal exocytosis was then initiated by adding 0.5 mm CaCl2 (calcium) or 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT) and a further 15-min incubation at 37 °C (black bars). Sperm were stained, and the AR scored as in A. C, permeabilized spermatozoa were incubated with 20 μg/ml anti-Rap antibodies (Santa Cruz Biotechnology), 1 μg/ml Ral-GDS-RBD, or 15 μm U73122 for 10 min at 37 °C. Exocytosis was initiated by adding 0.5 mm CaCl2. To demonstrate that Rap1-GTP and an active PLC were located upstream of the intracellular calcium efflux, 5 μm adenophostin was added to mobilize calcium from IP3-sensitive stores, and incubations proceeded for an additional 10 min at 37 °C (black bars). Controls (gray bars) included the following: background AR in the absence of any stimulation (control), AR stimulated by 0.5 mm CaCl2 (calcium), AR inhibition by 20 μg/ml anti-Rap antibodies, 1 μg/ml Ral-GDS-RBD, or 15 μm U73122, and AR unaffected by 5 μm adenophostin. Sperm were stained, and the AR scored as in A. Plotted results represent the mean ± S.E. of at least three independent experiments.
In HEK-293 cells, cAMP increases intracellular calcium concentrations and IP3 synthesis by stimulating a PLCϵ activity in an Epac1/Rap2B-dependent manner (23). Likewise, the pathway Epac-Rap1-PLCϵ enhances calcium mobilization from the sarcoplasmic reticulum in ventricular cardiac myocytes (24). The AR triggered by 8-pCPT-2′-O-Me-cAMP relies on Epac, an active Rap1 (Fig. 2D), and on the mobilization of intravesicular calcium through IP3-sensitive receptors (8). To ask whether sperm exocytosis requires the activity of a PLC, we introduced U73122, an inhibitor of multiple phosphatidylinositol-specific phospholipase C (PI-PLC) isoforms, into permeabilized sperm before challenging with calcium or 8-pCPT-2′-O-Me-cAMP. Treatment with U73122 (but not the inactive control U73343) decreased the exocytotic response by >90% (Fig. 3B), suggesting that the AR requires an active PLC downstream of Epac activation.
We hypothesized that signaling through Rap1 and PLC, with the ensuing synthesis of IP3, drives the intracellular calcium mobilization required for the AR. If this was the case, then Rap (Fig. 2, B and D) and PLC (Fig. 3B) inhibitors blocked exocytosis because they prevented calcium mobilization. We tested this possibility by promoting intravesicular calcium release with the IP3 receptor agonist adenophostin. The sole addition of adenophostin rescued exocytosis impaired by anti-Rap1 antibodies, Ral-GDS-RBD, and U73122 (Fig. 3C), supporting the notion that the end point of the Rap-PLC pathway is the mobilization of intracellular calcium.
8-pCPT-2′O-Me-cAMP Augments the Concentration of Cytosolic Calcium in Human Sperm Suspensions
Human sperm bathed in a nominally calcium-free medium and loaded with the fluorescent calcium sensor Fluo-3-AM responded to 8-pCPT-2′-O-Me-cAMP with a transient increase in calcium concentration whose profile was similar to that elicited by progesterone under identical conditions (Fig. 4A). Preincubation with U73122, and to a much lesser degree with U73343, diminished the increase in calcium caused by the Epac activator (Fig. 4B). These data indicate that, in conditions identical to those in which it elicits exocytosis in sperm, 8-pCPT-2′-O-Me-cAMP increases the concentration of calcium in the cytosol through a mechanism involving an active PLC.
FIGURE 4.

Epac signaling mobilizes calcium in a PLC-dependent manner. Human sperm (nonpermeabilized) were loaded with 2 μm Fluo-3-AM and 0.02% pluronic acid and incubated for 30 min at 37 °C. At the indicated times (arrows) 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT) or 4 μm progesterone (Pg) were added with (B) or without (A) previous incubation with 1 μm U73122 or U73343. Maximal [Ca2+]i response was calibrated with 0.1% Triton X-100 (TX-100) at the end of the incubation period. Shown are traces representative of five experiments. The increase in fluorescence is expressed as (F/F0) − 1 ((maximum fluorescence intensity/initial fluorescence) − 1) versus time in seconds. Bars represent mean ± S.E. of five experiments; data were normalized against the calcium response to 4 μm progesterone (A) or 50 μm 8 pCPT-2′-O-Me-cAMP (B). ***, p < 0.001; *, p < 0.05 compared with 8 pCPT-2′-O-Me-cAMP.
Based on the data collected here we reasoned that, in addition to the sequence depicted in pathway 1, the AR can also be modeled to fit in a signaling pathway operating through the sequence shown in pathway 2,
|
In the following sections we will attempt to address the question of how do sperm signaling pathways summarized in pathways 1 and 2 integrate to accomplish exocytosis.
AR Triggered by Recombinant Rab3A-GTP-γ-S Requires cAMP, Epac, Rap1-GTP, and a PLC Activity
In addition to the early steps “calcium →→ cAMP → Epac,” pathways 1 and 2 share the late-acting steps “intracellular calcium mobilization →→ exocytosis.” We examined these pathways further by investigating whether the AR triggered by recombinant Rab3A (Rab3A is a term in pathway 1) requires cAMP, Rap-GTP, and PLC. In human sperm, prenylated, GTP-γ-S-loaded, recombinant Rab3A induces a strong exocytotic response that relies on the same fusion machinery characterized for calcium-triggered exocytosis (32, 35). Here we show that the Rab3A-GTP-γ-S-elicited AR also requires cAMP because pretreatment of permeabilized human sperm with KH7 or recombinant PDE impaired exocytosis (Fig. 5 A). The PKA blocker H89 did not abolish the AR, in agreement with previous observations that this kinase is not the target of cAMP during sperm exocytosis (8). To investigate if Rab3A signals through cAMP/Epac, as is the case when calcium serves as the AR inducer, we introduced anti-Epac antibodies into permeabilized cells. The antibodies (6.7 nm) efficiently blocked the AR induced by calcium (Fig. 5B) (8). The AR triggered by recombinant Rab3A-GTP-γ-S was also abolished by anti-Epac antibodies, albeit at a higher concentration (134 nm, Fig. 5B). Next, we analyzed the contribution of the other players depicted in pathway 2, and we found that sequestering endogenous Rap1 with specific antibodies or Rap1-GTP with Ral-GDS-RBD prevented the RA elicited by Rab3A-GTP-γ-S. The same was true when we inhibited phosphoinositide-specific PLC activity with U73122 (Fig. 5B). Taken together, these data suggest that Rab3A-GTP-γ-S requires cAMP synthesized by sAC to achieve exocytosis and that Epac is the relevant cAMP target. Active Rap1 and PLC are also part of the pathway initiated by Rab3A-GTP-γ-S. In other words, recombinant Rab3A-GTP-γ-S relies on all the players summarized in pathway 2 to achieve exocytosis.
FIGURE 5.

Rab3A-GTP-γ-S requires cAMP, Epac, Rap1, and an active PLC to induce exocytosis. A, SLO-permeabilized human sperm were treated for 15 min at 37 °C with 10 μm KH7, 2 μg/ml PDE, or 10 μm H89 before initiating acrosomal exocytosis with 300 nm GTP-γ-S-bound Rab3A (Rab3A) and incubating at 37 °C for 15 min. Sperm were fixed; the AR was measured by FITC-PSA binding, and the data were normalized as described under “Experimental Procedures.” B, SLO-permeabilized spermatozoa were incubated with 6.7 or 134 nm anti-Epac antibodies, 20 μg/ml anti-Rap antibodies (Santa Cruz Biotechnology), 1 μg/ml Ral-GDS-RBD, or 15 μm U73122 for 15 min at 37 °C. Acrosomal exocytosis was evaluated by FITC-PSA binding after an additional 15 min of incubation at 37 °C in the absence (control) or presence of 0.5 mm CaCl2 (calcium) or 300 nm GTP-γ-S-bound Rab3A (Rab3A). Plotted results represent the mean ± S.E. of at least three independent experiments.
Calcium Drives the AR through a Bifurcated Signaling Pathway
The two simplest models we can think of to represent the AR are as follows: (i) a linear cascade consisting of an arrangement of all players depicted in pathways 1 and 2; (ii) a pathway that bifurcates after some common early steps into parallel cascades that converge later to achieve fusion. We favor the second alternative and hypothesize that following Epac activation, the pathway splits into a branch that begins with Rap1 and ends in intracellular calcium mobilization and another that starts with Rab3A and finishes with the assembly of SNARE proteins in loose trans complexes (38), both being necessary for exocytosis (see model in Fig. 9). We tested the validity of this proposal with adenophostin, predicting that it should not be able to rescue the effect of inhibitors whose targets are early common steps of the pathway (i.e. cAMP and Epac) or those required to assemble SNAREs (i.e. Rab3A, PTP1B, α-SNAP, and NSF). Data depicted in Fig. 6 show that this was indeed the case. Adenophostin failed to reverse the AR block caused by recombinant PDE (2 μg/ml), KH7 (10 μm), and anti-Epac antibodies (134 nm) (Fig. 6A). The activity of NSF is inhibited by tyrosine phosphorylation and de-repressed by tyrosine phosphatases (PTPs) in macrophages (61) and sperm (49). In the latter, PTP1B disassembles SNARE complexes through tyrosine dephosphorylation of NSF. As expected, adenophostin failed to rescue the AR block imposed by the substrate trapping mutant PTP1B D181A (300 nm) and the phosphomimetic NSF mutant NSF Y83E (186 nm). Similarly, adenophostin did not rescue the anti-Rab3A (20 μg/ml) antibody block (Fig. 6B). Interestingly, adenophostin did rescue exocytosis when the anti-Epac antibodies were added at 6.7 nm (Fig. 6A). We interpret the concentration-dependent behavior as indicative that at 6.7 nm anti-Epac antibodies prevent calcium-triggered exocytosis because they block only the “Rap1” branch (the “Rab3A” branch proceeds normally), whereas at 134 nm they block both (Fig. 9).
FIGURE 9.

Working model for the biochemical cascade leading to the AR. Calcium enters the cell from the extracellular milieu when channels open in the plasma membrane (PM) in response to progesterone or other physiological inducers, when a calcium ionophore transports it to the cytosol, or through the SLO-generated pores. Cyclic AMP synthesized by sAC (or exogenously added 8-pCPT-2′-O-Me-cAMP) activates Epac, which directly activates Rap1. In turn, active Rap1 stimulates the synthesis of IP3 through the stimulation of a PLC activity. This IP3 (or exogenously added adenophostin) elicits the efflux of calcium from an IP3-sensitive store (likely the acrosome). 2-APB blocks calcium efflux through IP3-sensitive channels (data not shown). Epac-cAMP also indirectly activates Rab3A, triggering the tethering of the acrosome to the plasma membrane through the assembly of large macromolecular complexes. A reaction taking place during or as a consequence of tethering initiates the activation and/or recruitment of PTP1B, which in turns activates NSF. Next, NSF/α-SNAP render SNARE proteins fusion-competent. Both a local increase in calcium coming from the acrosome through IP3-sensitive channels and SNAREs converge to accomplish the final steps of membrane fusion. Recombinant Rab3A (rRab3A-GTP) initiates the AR through an alternative pathway (gray) that presumably begins with the recruitment of RIM to achieve tethering. The large quantities of RIM recruited by recombinant Rab3A-GTP-γ-S bind Epac and the intracellular calcium pathway proceeds as described. OAM, outer acrosomal membrane. Solid arrows mean there is one step between the terms connected, and dashed arrows mean that the number of steps is either unknown or not depicted for simplicity.
FIGURE 6.

Pivotal role of cAMP-Epac in governing two parallel but converging signaling pathways during the late steps of the AR. SLO-permeabilized human sperm were treated for 10 min at 37 °C with 2 μg/ml PDE, 10 μm KH7, or anti-Epac (6.7 or 134 nm) antibodies (A) or anti-Rab3A (20 μg/ml), PTP1B D181A (300 nm), or NSF Y83E (186 nm) (B) followed by 0.5 mm CaCl2 and an additional 15 min of incubation. To rescue the effect of the blockers, we added adenophostin (5 μm) and incubated for a further 10 min at 37 °C (black bars). Controls (gray bars) included the following: background AR in the absence of any stimulation (control), AR stimulated by 0.5 mm CaCl2 (calcium), and AR inhibition by 2 μg/ml PDE, 10 μm KH7, anti-Epac (6.7 or 134 nm), and anti-Rab3A (20 μg/ml) antibodies, 300 nm PTP1B D181A, and 186 nm NSF Y83E. Sperm were fixed; acrosomal exocytosis was evaluated by FITC-PSA binding, and data were normalized as described under “Experimental Procedures.” C, SLO-permeabilized spermatozoa were loaded with 6.7 nm anti-Epac antibodies for 10 min at 37 °C to selectively block the signaling pathway leading to IP3-sensitive calcium mobilization. AR was subsequently initiated by adding 0.5 mm CaCl2. After 10 min of incubation at 37 °C to allow exocytosis to proceed to the Epac-sensitive step, sperm were treated with antibodies that recognize Rab3A (20 μg/ml) or NSF (1:300) and incubated for an additional 10 min at 37 °C. Finally, we added 5 μm adenophostin to rescue the anti-Epac antibody block and incubated as before (black bars). We ran several controls in parallel (gray bars) as follows: background AR in the absence of any stimulation (control), AR stimulated by 0.5 mm CaCl2 (calcium), and the inhibitory effect of the antibodies when present throughout the experiment (anti-Epac → anti-Rab3A/NSF → calcium → adenophostin). Sperm were fixed, and AR was measured by FITC-PSA binding as described under “Experimental Procedures.” The data represent the mean ± S.E. of at least three independent experiments.
The reversibility of the inhibition of the anti-Epac antibodies (6.7 nm) by adenophostin supplied us with an alternative strategy to test the bifurcation hypothesis. When added after the AR inducer (extracellular calcium in this case) in the presence of anti-Epac antibodies (rescued at the end of the incubation by adenophostin), neither anti-Rab3A nor anti-NSF antibodies blocked the AR (Fig. 6C, black bars). These results suggest that the AR inducer had driven the Rab3A branch of the pathway to completion even though the Rap1 arm had been halted by the anti-Epac antibodies. In other words, the steps catalyzed by Rab3A/NSF had already taken place when we added the antibodies, and therefore the system was refractory to their inhibitory effect. In the control condition, anti-Rab3A and anti-NSF antibodies were added prior to extracellular calcium in the presence of anti-Epac antibodies; both blocked the AR despite the fact that the end point of the Rap1 branch was achieved by adenophostin (Fig. 6C, gray bars).
8-pCPT-2′-O-Me-cAMP Activates a GEF for Rab3A in Human Sperm
A central aspect of our model is based on the observation that the AR triggered by cAMP (8) and calcium (38) requires Rab3A. We reasoned that if Rab3 serves as a key element in cAMP/Epac-mediated signaling, it must be activated in response to 8-pCPT-2′-O-Me-cAMP. To determine whether Epac exhibits GEF activity toward Rab3A, we incubated 150 nm recombinant Epac1 and Epac2 with fluorescent mGDP-loaded Rab3A (200 nm) in the presence of excess unlabeled GDP. We followed the exchange of guanine nucleotides in real time as a decrease in fluorescence. As shown in Fig. 7, there was no difference between the GDP release rates in the presence of Epacs (squares) and the intrinsic exchange rate for mGDP-Rab3A (black circles) measured in the same experiment. In other words, neither Epac1 (Fig. 7, left) nor Epac2 (Fig. 7, center) served as a GEF for Rab3A.
FIGURE 7.

Epac is not a GEF for Rab3A. The dissociation of the Rab3A-mGDP complex was monitored by a time-dependent decrease in fluorescence intensity. Rab3A (200 nm) was loaded with the fluorescent nucleotide analogue mGDP and incubated in the presence (squares) or absence (circles) of inactive (□) or activated (500 μm cAMP treated; ■) 150 nm Epac1 (left) or Epac2 (center) after the addition (●) or not (○) of a 100-fold excess (20 μm) of unlabeled GDP, as indicated under “Experimental Procedures.” For clarity of presentation and to avoid the overlay of symbols, in the left panel the data points of □ and ■ were shifted by 0.05 and 0.1 relative units, respectively. Likewise, in the center panel data points of ●, □, and ■ were shifted by 0.05, 0.10, and 0.15 units, respectively. Please note that mGDP can be displaced very slowly from Rab3A (note the difference in time scales between the right panel and the rest) by an excess of GDP; thus the Rab protein is properly folded in a native, nucleotide-interacting state. The right panel demonstrates that the Epac1 (■) and Epac2 (▴) preparations were active and exhibited GEF activity toward Rap. Briefly, Rap1 (200 nm) was loaded with the fluorescent nucleotide analogue mGDP and incubated in the absence (○) or presence of activated (100 μm cAMP treated) 150 nm Epac1 (■) or Epac2 (▴) after the addition of a 100-fold excess (20 μm) of unlabeled GDP, as indicated under “Experimental Procedures.” The data represent the mean ± S.E. of at least two independent experiments.
Below we will attempt to show that the stimulation of Epac by 8-pCPT-2′-O-Me-cAMP leads, indirectly, to the activation of a Rab3A-GEF in a Rap-independent manner. The first approach we undertook consisted of sequestering endogenous Rab3A, presumably bound to GDP in resting sperm, with GDI before treating with recombinant Rab3A-GDP and 8-pCPT-2′-O-Me-cAMP. GDI inhibited the AR elicited by the cAMP analogue (Fig. 8A), likely because it maintained sperm Rab3A in its GDP-bound form, which is exocytosis-incompetent. Although recombinant Rab3A-GDP did not induce the AR by itself, it did in combination with 8-pCPT-2′-O-Me-cAMP (Fig. 8A), indicating that activation of Epac by cAMP stimulated a Rab3 exchange activity in sperm. In the second approach, we introduced a cassette consisting of the Rab3-GTP binding domain (amino acids 11–398) of RIM into SLO-permeabilized sperm before challenging with AR inducers. Calcium served as a positive control because it exchanges GDP for GTP on Rab3A (62). Calcium was unable to achieve exocytosis in the presence of RIM-(11–398) (Fig. 8B), indicating that Rab3A-GTP generated in response to calcium was necessary for the AR and validating the use of the cassette to sequester Rab3A-GTP. RIM-(11–398) also prevented the AR initiated by 8-pCPT-2′-O-Me-cAMP, suggesting that Rab3A-GTP was formed in response to this inducer and was required for Epac-dependent exocytosis. Recombinant Rab3A-GDP did not induce the AR by itself nor did 8-pCPT-2′-O-Me-cAMP accomplish exocytosis when endogenous Rab3A-GTP was sequestered by RIM-(11–398). However, normal levels of AR were achieved when combining all three (Fig. 8B), indicating once again that activation of Epac by cAMP stimulated a Rab3 exchange activity in sperm. Taken together, these results suggest that cAMP activates a GEF activity in spermatozoa that exchanges GDP for GTP in recombinant Rab3A, turning it into an AR inducer.
FIGURE 8.

Activation of Epac by 8-pCPT-2′-O-Me-cAMP stimulates a GEF for Rab3A during sperm exocytosis. A, permeabilized spermatozoa were incubated for 10 min at 37 °C with 4 μm GDI (in all the experiments using GDI, the buffer was supplemented with 0.7 mm MgCl2 and 0.2 mm GTP) followed or not by 300 nm Rab3A-GDP and further incubation for 10 min at 37 °C. Acrosomal exocytosis was initiated by adding 50 μm 8-pCPT-2′-O-Me-cAMP and incubating for an additional 10 min at 37 °C (black bars). Controls (gray bars) included the following: background AR in the absence of any stimulation (control), AR stimulated by 0.5 mm CaCl2 (calcium) and 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT), and lack of effect on the AR of 300 nm Rab3A-GDP and 4 μm GDI plus 300 nm Rab3A-GDP. Sperm were fixed; acrosomal exocytosis was evaluated by FITC-PSA binding, and data were normalized (mean ± S.E.) as described under “Experimental Procedures.” B, SLO-permeabilized spermatozoa were incubated for 10 min at 37 °C with 5 μg/ml RIM-(11–398) followed or not by 300 nm Rab3A-GDP and further incubation for 10 min at 37 °C. Acrosomal exocytosis was initiated by adding 0.5 mm CaCl2 or 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT) and incubating for an additional 10 min at 37 °C (black bars). Controls (gray bars) included the following background AR in the absence of any stimulation (control), AR stimulated by 0.5 mm CaCl2 (calcium) and 50 μm 8-pCPT-2′-O-Me-cAMP (8pCPT), and lack of effect on the AR of 300 nm Rab3A-GDP. Samples were processed, and the AR was scored as in A. C, capacitated human sperm were permeabilized with SLO, and the onset of the AR was prevented by 100 μm 2-APB. Sperm suspensions were subsequently incubated with (right) or without 20 μg/ml anti-Rap1 antibodies or with 1 μg of His6-Rab3A (top left). AR inducers 50 μm 8-pCPT-2′-O-Me-cAMP (top left and right) and 0.5 mm CaCl2 (bottom left) were added and incubations proceeded as described under “Experimental Procedures.” Whole sperm lysates were subjected to pulldown assay using GST-RIM-Sepharose, and the levels of GTP-bound Rab3A were determined by Western blot with an anti-Rab3A antibody. Shown are experiments representative of three repetitions; right, quantification (carried out with Image J, freeware from National Institutes of Health) is depicted above the immunoblot as mean ± S.E. from all replicates; different letters indicate statistical significance (p < 0.05).
We verified this capacity by conducting pulldown assays using GST-RIM-(11–398) and sperm extracts supplemented with geranylgeranylated His6-Rab3A. The Rab3A-GTP level increased 3–5-fold upon treatment with 8-pCPT-2′-O-Me-cAMP (Fig. 8C, top left), confirming that human sperm contain an activity that exchanges GDP for GTP in recombinant Rab3A in response to 8-pCPT-2′-O-Me-cAMP. Next, we applied the same strategy to examine the effects of AR inducers on endogenous Rab3A in sperm. As illustrated in Fig. 8C, the amount of GTP-bound Rab3A measured in lysates of cells treated with calcium (bottom left) was 1.4-fold higher than that in unstimulated controls. Likewise, addition of 8-pCPT-2′-O-Me-cAMP to sperm augmented the fraction of active Rab3A 1.6-fold with respect to that in untreated cells (Fig. 8C, right). Note that the threshold level of endogenous Rab3A-GTP in unstimulated sperm was high; therefore, the increase in active endogenous Rab3A (Fig. 8C, bottom left and right) was appreciably lower than the increase in recombinant Rab3A, whose basal level of GTP-bound form was negligible (Fig. 8C, top left). The increase in GTP-bound endogenous Rab3A we detected in human sperm in response to calcium and cAMP was similar to that reported for overexpressed Rab3D in amylase-secreting pancreatic acini in response to cholecystokinin (63). Because Epac proteins act as GEFs for Rap GTPases but not for Rab3, we next asked whether Epac-mediated Rab3 activation involved a signaling pathway downstream of Rap activation. To address this issue, we introduced anti-Rap antibodies into SLO-permeabilized human sperm before challenging with 8-pCPT-2′-O-Me-cAMP. As shown in Fig. 8C (right), the antibodies had no effect on 8-pCPT-2′-O-Me-cAMP -induced Rab3 activation, although they potently inhibited Rap signaling during the AR (Fig. 2B). Taken together, these data suggest that Epac plays a central role in cAMP-dependent Rab3 activation but that Rap activity is not essential for this signaling cascade. In other words, upon the onset of exocytosis, Epac activates a GEF for Rab3A through a still undefined mechanism.
DISCUSSION
Epac proteins exert diverse effects on a variety of cellular functions and are thought to mediate many of the PKA-independent effects of cAMP-regulated signaling (5, 9–11). These functions include the modulation of secretory processes in pancreatic β-cells (39, 42, 64–66), kidney collecting duct (67), melanotrophs (68), neurons (see Ref. 69 and references therein), and granulosa cells (70). We owe most of our understanding of the molecular mechanisms that couple cAMP/Epac to exocytosis to recent work conducted in insulin-secreting cells (reviewed in Refs. 5, 10). In these cells, the activation of Epac by cAMP sensitizes intracellular (ryanodine and IP3 receptors) calcium release channels, increasing cytosolic calcium concentrations and, subsequently, insulin secretion (42, 71). Direct sensitization of IP3 receptors to IP3 by cAMP has very recently been described in HEK-PR1 cells (72).
Epac is a GEF for Rap proteins. Rap1 belongs to the Ras subfamily of small GTP-binding proteins, which controls cell growth, differentiation, and survival. Although the best known Ras-independent Rap1 function is the control of adhesion-related events (73–75), there is evidence suggesting its involvement in insulin (76, 77) and amylase release (78). Until not too long ago, however, the reported protein-protein interactions linking Epac to insulin secretion did not include Rap (40, 66). We now know that Rap1 is required for cAMP/Epac-dependent potentiation of insulin secretion (79), pancreatic amylase release (80), and a nonamyloidogenic soluble form of the amyloid precursor protein release (81).
Epac proteins have been detected in mouse (82), hamster (83), and human sperm (8), as well as in mouse spermatids (84). In human sperm, calcium triggers exocytosis in a PKA-independent manner through a pathway involving cAMP and Epac. By activating Epac, cAMP drives the whole cascade of events necessary to bring exocytosis to completion. These events include sequential protein-protein interactions as well as the mobilization of calcium from an intracellular store (4). In insulin-secreting cells, cAMP potentiates CICR during the exocytotic cascade (42). Interplay between calcium and cAMP in human sperm appears to be different because cAMP achieves exocytosis in the presence of high concentrations of EGTA, which chelates extracellular calcium, preventing an influx from the outside, but not of low concentrations of 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester, which chelates intracellular calcium, preventing mobilization from stores (8). Interestingly, cAMP-elevating agents such as GLP-1 stimulate the release of calcium stored in insulin-containing secretory granules (85). In this study we show that cAMP/Epac achieve intracellular calcium mobilization in human sperm via a pathway involving Rap1 and a PLC. Briefly, Rap1 was present and required for regulated exocytosis in human sperm (Fig. 2, A and B). Three AR inducers, an Epac-selective cAMP analogue, recombinant Rab3A-GTP-γ-S, and calcium, promoted the activation of Rap1 in human sperm (Fig. 2, C and D, and Fig. 5B). Likewise, 8-pCPT-2′-O-Me-cAMP activates Rap1 in mouse germ cells (82, 84). The importance of Rap1 in male fertility has been highlighted by the discovery that transgenic mice expressing an inactive mutant in differentiating spermatids are severely subfertile (86). We and others have shown that the acrosome behaves as a calcium-storing organelle (59, 60). Release of intra-vesicular calcium takes place in response to AR inducers and is necessary for human sperm exocytosis (59). By using a photosensitive calcium chelator in combination with inhibitory antibodies and a Rap1-GTP sequestering cassette, we have determined that Rap1 is required before the release of calcium from the acrosome (Fig. 3A). Furthermore, this release is the end point of the pathway driven by Rap1, because adenophostin, by eliciting an efflux of intracellular calcium through IP3-sensitive channels, can bypass Rap1 blockers and rescue the AR (Fig. 3C).
A link between cAMP/Epac and calcium signaling implicating Rap2B and a PLCϵ activity in HEK-293 and neuroblastoma cells was reported a few years ago (23). More recently we learned that the pathway Epac-Rap1-PLCϵ enhances CICR from the sarcoplasmic reticulum in ventricular cardiac myocytes (24). PLCβ (87), PLCγ (88), PLCδ (89), and PLCζ (90) have been described in mammalian sperm (but not PLCϵ so far). An active PI-PLC operates downstream of Epac during the human sperm AR, because 8-pCPT-2′-O-Me-cAMP could not elicit exocytosis (Fig. 3B) or mobilize calcium (Fig. 4B) in the presence of U73122. A PI-PLC activity is also required by calcium (Fig. 3B) and Rab3A-GTP-γ-S (Fig. 5B) to achieve the AR. We believe this PI-PLC, whatever its molecular identity might be, is responsible for the synthesis of the IP3 that promotes efflux of calcium from an intracellular store in sperm, because adenophostin can rescue the block imposed by U73122 on the AR (Fig. 3C).
On reviewing the literature, we became aware of the existence of systems where sAC synthesizes the cAMP that culminates in a Rap1-activating cascade (91, 92). This calcium- and bicarbonate-stimulated cyclase is the predominant form in mammalian sperm (44, 56, 93, 94) and is critical for fertility (95, 96). It was therefore not surprising to discover that sAC was required for human sperm exocytosis. At low concentrations (10 μm), KH7 prevented calcium-elicited AR in permeabilized (Fig. 1B) as well as A23187-triggered (Fig. 1A)5 and progesterone-triggered (Fig. 1A) AR in nonpermeabilized human sperm. Likewise, KH7 (50–100 μm) inhibited the AR induced by egg jelly in sea urchin sperm (97). In contrast, KH7 did not alter the AR in response to solubilized zona pellucida in mouse sperm containing green fluorescent protein in the acrosome (56). Whether these discrepancies are because of an actual divergence of mechanisms in cAMP signaling among different species or because of different capacitation conditions (2-hydroxypropyl-β-cyclodextrin, no CaCl2 for mouse (56); bovine serum albumin, 1.7 mm CaCl2 for human, this study), AR measurement assays, or both, remain to be seen. Sperm from sAC null mice undergo normal exocytosis when challenged with solubilized zona pellucida or 20 μm A23187 for 1 h (56). However, when capacitated in the presence of bovine serum albumin and CaCl2 and challenged with 1–10 μm A23187 for 30 min, sperm from sAC−/− males shows decreased sensitivity to the ionophore (96). In brief, our current knowledge indicates that whereas sAC exhibits an important role in the AR in human and sea urchin sperm, its cyclase activity might be dispensable for the AR in the mouse. We do not yet know if sAC is being stimulated under our assay conditions or if its level of activity reached in response to the capacitation medium is sufficient to aid calcium to trigger the AR. In this regard, it is important to bear in mind that a signature of sAC is its ability to respond to cues more slowly and in a more sustained fashion than transmembrane ACs (98).
We have previously demonstrated by means of an AR assay in the presence of GDI that Rab3A is activated in sperm following calcium stimulation (62). Here we show, both with GDI and a protein cassette that binds Rab3A-GTP (46, 99, 100), that Rab3A activation relies on a novel signaling cascade involving cAMP/Epac (Fig. 8). Neither Epac1 nor Epac2 activated Rab3A directly (Fig. 7) (39), suggesting that an additional regulatory protein(s) is necessary to couple Epac activation to Rab3A signaling. The identity of this protein(s) is presently unknown.
Similarly to cAMP, recombinant Rab3A-GTP-γ-S-triggered AR was independent of PKA activity (Fig. 5A) (8, 35), relied on an efflux of calcium from an IP3-sensitive intracellular store, and did not require an influx of calcium from the extracellular milieu (8, 35, 59). We show here that calcium, cAMP, and Rab3A share a pathway that requires Epac, Rap1, and an active PLC to mobilize calcium and accomplish the AR (Fig. 2, B and D, Fig. 3C, and Fig. 5B). Rab3A-GTP-γ-S depended upon cAMP synthesized by sAC to accomplish the AR (Fig. 5A). At the moment, we lack evidence pointing to sAC stimulation by recombinant Rab3A-GTP-γ-S; we rather think that cAMP accumulated in sperm before the addition of this small G protein was sufficient to allow Rab3A-GTP-γ-S to elicit exocytosis, although higher concentrations were necessary to trigger the AR by themselves. In addition to intracellular calcium mobilization, the AR inducers calcium, cAMP, and recombinant Rab3A-GTP-γ-S utilize a proteinaceous fusion machinery that includes α-SNAP, NSF, and SNAREs (8, 38, 101, 102). Calcium and cAMP also required and activated Rab3A during the exocytotic cascade (Fig. 8).
The amount of anti-Epac antibodies required to block exocytosis was much smaller when the AR was induced by calcium or cAMP than with recombinant Rab3A-GTP-γ-S (8) (Fig. 5B). Because of the adenophostin rescue (Fig. 6A), we interpret that 6.7 nm anti-Epac antibody blocked only the “Rap-PLC-calcium” branch, likely by preventing the activation of Rap1 by the GEF activity of Epac. In contrast, when the AR was induced by recombinant Rab3A-GTP-γ-S, in much higher concentrations than the endogenous protein, gray pathway on Fig. 9, one can imagine that the amount of Epac recruited via RIM increased significantly. Because these interactions are stoichiometric rather than enzymatic, much higher concentrations of anti-Epac antibodies (134 nm) were required to block exocytosis (Fig. 5B). Adenophostin was not able to relieve the block imposed by 134 nm anti-Epac (Fig. 6A), which we interpret as indicative that at this concentration the antibodies were affecting an Epac function that went beyond calcium mobilization. The same was true for PDE, KH7, anti-Rab3A antibodies, PTP1B D181A, and NSF Y83E (Fig. 6, A and B). We could conceivably fit some of these data into a linear model whereby cAMP/Epac promoted a release of calcium from an intracellular store prior to (and not in parallel with) the tethering, priming, and docking steps. The results depicted in Fig. 6C go against this model because they suggest that, even while maintaining the intracellular calcium mobilization pathway artificially off with anti-Epac antibodies, calcium assembles the fusion machinery into place before anti-Rab3A and anti-NSF antibodies have the opportunity to prevent it.
Therefore, we suggest that the AR is organized as a bifurcated pathway, with two separate limbs that diverge downstream of cAMP/Epac (Fig. 9). The end point of one limb (Rap-PLC) is the mobilization of intracellular calcium, whereas the other (Rab3A-α-SNAP/NSF-SNAREs) assembles the fusion protein machinery so that the outer acrosomal and plasma membranes become physically connected. Both pathways operate in a concerted fashion and converge at/or downstream of intracellular calcium mobilization to accomplish exocytosis; once calcium is released into the cytosol, the protein machinery, likely through synaptotagmin (48), senses the rise in concentration, and the whole system evolves to open the fusion pores.
Acknowledgments
We thank M. Furlán and A. Medero for excellent technical assistance; F Rodriguez for assistance with the expression and purification of GST-Rap1; Dr. López for His6-Rab3A; Drs. Coso, Stahl, Regazzi, Bottini, Tonks, and Takai for plasmids; and Drs. Teves and Giojalas (Universidad Nacional de Córdoba, Córdoba, Argentina) for sharing unpublished results. We also thank Dr. R Branham for assistance with statistical analysis.
This work was supported in part by Consejo Nacional de Investigaciones Científicas y Técnicas Grant PIP 5951, Agencia Nacional de Promoción Científica y Tecnológica Grant PICT2006-1036, Argentina (to C. N. T.), Consejo Nacional de Ciencia y Tecnología (CONACyT, Mexico) Grants 49113 (to A. D.) and 42605 (to C. L. T.), and PAPIIT Grants IN204109 (to C. L. T.) and IN211809 (to A. D.).
E. Teves and L. Giojalas, personal communication.
- AR
- acrosome reaction
- 2-APB
- 2-aminoethoxydiphenyl borate
- 6-Bnz-cAMP
- N6-benzoyladenosine-3′,5′-cyclic monophosphate
- CICR
- calcium-induced calcium release
- Epac
- exchange protein directly activated by cAMP
- FITC-PSA
- fluorescein isothiocyanate-coupled Pisum sativum agglutinin
- GEF
- guanine-nucleotide exchange factor
- GDI
- guanine nucleotide-dissociation inhibitor
- mGDP
- 2′,3′-O- (N′-methylanthraniloyl)-GDP
- NSF
- N-ethylmaleimide-sensitive factor
- NP-EGTA-AM
- O-nitrophenyl EGTA acetoxymethyl ester
- 8-pCPT-2′-O-Me-cAMP
- 8-(p-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate
- PKA
- cAMP-dependent protein kinase
- PDE
- phosphodiesterase
- PLC
- phospholipase C
- PMSF
- phenylmethylsulfonyl fluoride
- PTP1B
- protein-tyrosine phosphatase 1B
- sAC
- soluble adenylyl cyclase
- SLO
- streptolysin O
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- IP3
- inositol 1,4,5-trisphosphate
- PBS
- phosphate-buffered saline
- GTP-γ-S
- guanosine 5′-3-O-(thio)triphosphate
- GST
- glutathione S-transferase
- SNARE
- soluble NSF attachment protein receptors
- PI-PLC
- phosphatidylinositol-specific phospholipase C.
REFERENCES
- 1.Florman H. M., Ducibella Elsevier-T. (2005) in Knobil and Neill's Physiology of Reproduction ( Neill J. D. ed) pp. 55–112, Elsevier Academic Press, San Diego, CA [Google Scholar]
- 2.Bedford J. M. (1998) Biol. Reprod. 59, 1275–1287 [DOI] [PubMed] [Google Scholar]
- 3.Mayorga L., Tomes C. N., Belmonte S. A. (2007) IUBMB Life 59, 286–292 [DOI] [PubMed] [Google Scholar]
- 4.Tomes C. N. (2007) Soc. Reprod. Fertil. Suppl. 65, 275–291 [PubMed] [Google Scholar]
- 5.Seino S., Shibasaki T. (2005) Physiol. Rev. 85, 1303–1342 [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne R. D., Morgan A. (2003) Physiol. Rev. 83, 581–632 [DOI] [PubMed] [Google Scholar]
- 7.Szaszák M., Christian F., Rosenthal W., Klussmann E. (2008) Cell. Signal. 20, 590–601 [DOI] [PubMed] [Google Scholar]
- 8.Branham M. T., Mayorga L. S., Tomes C. N. (2006) J. Biol. Chem. 281, 8656–8666 [DOI] [PubMed] [Google Scholar]
- 9.Bos J. L. (2006) Trends Biochem. Sci. 31, 680–686 [DOI] [PubMed] [Google Scholar]
- 10.Holz G. G., Kang G., Harbeck M., Roe M. W., Chepurny O. G. (2006) J. Physiol. 577, 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roscioni S. S., Elzinga C. R., Schmidt M. (2008) Naunyn Schmiedebergs Arch. Pharmacol. 377, 345–357 [DOI] [PubMed] [Google Scholar]
- 12.de Rooij J., Rehmann H., van Triest M., Cool R. H., Wittinghofer A., Bos J. L. (2000) J. Biol. Chem. 275, 20829–20836 [DOI] [PubMed] [Google Scholar]
- 13.Rehmann H., Rueppel A., Bos J. L., Wittinghofer A. (2003) J. Biol. Chem. 278, 23508–23514 [DOI] [PubMed] [Google Scholar]
- 14.Harper S. M., Wienk H., Wechselberger R. W., Bos J. L., Boelens R., Rehmann H. (2008) J. Biol. Chem. 283, 6501–6508 [DOI] [PubMed] [Google Scholar]
- 15.Rehmann H., Arias-Palomo E., Hadders M. A., Schwede F., Llorca O., Bos J. L. (2008) Nature 455, 124–127 [DOI] [PubMed] [Google Scholar]
- 16.de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998) Nature 396, 474–477 [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki H., Springett G. M., Mochizuki N., Toki S., Nakaya M., Matsuda M., Housman D. E., Graybiel A. M. (1998) Science 282, 2275–2279 [DOI] [PubMed] [Google Scholar]
- 18.Quilliam L. A., Rebhun J. F., Castro A. F. (2002) Prog. Nucleic Acid Res. Mol. Biol. 71, 391–444 [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y., Quilliam L. A. (2003) EMBO Rep. 4, 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leung K. F., Baron R., Seabra M. C. (2006) J. Lipid Res. 47, 467–475 [DOI] [PubMed] [Google Scholar]
- 21.Stork P. J. (2003) Trends Biochem. Sci. 28, 267–275 [DOI] [PubMed] [Google Scholar]
- 22.Bos J. L., Rehmann H., Wittinghofer A. (2007) Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 23.Schmidt M., Evellin S., Weernink P. A., von Dorp F., Rehmann H., Lomasney J. W., Jakobs K. H. (2001) Nat. Cell Biol. 3, 1020–1024 [DOI] [PubMed] [Google Scholar]
- 24.Oestreich E. A., Wang H., Malik S., Kaproth-Joslin K. A., Blaxall B. C., Kelley G. G., Dirksen R. T., Smrcka A. V. (2007) J. Biol. Chem. 282, 5488–5495 [DOI] [PubMed] [Google Scholar]
- 25.Bunney T. D., Katan M. (2006) Trends Cell Biol. 16, 640–648 [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez L., Jr., Scheller R. H. (1999) Cell 96, 755–758 [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer S. R. (1999) Nat. Cell Biol. 1, E17–E22 [DOI] [PubMed] [Google Scholar]
- 28.Lang T., Jahn R. (2008) Handb. Exp. Pharmacol. 184, 107–127 [DOI] [PubMed] [Google Scholar]
- 29.Regazzi R. (2007) in Molecular Mechanisms of Exocytosis ( Regazzi R. ed) pp. 28–41, Landes Bioscience, Austin, TX [Google Scholar]
- 30.Handley M. T., Haynes L. P., Burgoyne R. D. (2007) J. Cell Sci. 120, 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zerial M., McBride H. (2001) Nat. Rev. Mol. Cell Biol. 2, 107–117 [DOI] [PubMed] [Google Scholar]
- 32.Yunes R., Michaut M., Tomes C., Mayorga L. S. (2000) Biol. Reprod. 62, 1084–1089 [DOI] [PubMed] [Google Scholar]
- 33.Iida H., Yoshinaga Y., Tanaka S., Toshimori K., Mori T. (1999) Dev. Biol. 211, 144–155 [DOI] [PubMed] [Google Scholar]
- 34.Ward C. R., Faundes D., Foster J. A. (1999) Mol. Reprod. Dev. 53, 413–421 [DOI] [PubMed] [Google Scholar]
- 35.Lopez C. I., Belmonte S. A., De Blas G. A., Mayorga L. S. (2007) FASEB J. 21, 4121–4130 [DOI] [PubMed] [Google Scholar]
- 36.Garde J., Roldan E. R. (1996) FEBS Lett. 391, 263–268 [DOI] [PubMed] [Google Scholar]
- 37.Belmonte S. A., López C. I., Roggero C. M., De Blas G. A., Tomes C. N., Mayorga L. S. (2005) Dev. Biol. 285, 393–408 [DOI] [PubMed] [Google Scholar]
- 38.De Blas G. A., Roggero C. M., Tomes C. N., Mayorga L. S. (2005) PLoS. Biol. 3, e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozaki N., Shibasaki T., Kashima Y., Miki T., Takahashi K., Ueno H., Sunaga Y., Yano H., Matsuura Y., Iwanaga T., Takai Y., Seino S. (2000) Nat. Cell Biol. 2, 805–811 [DOI] [PubMed] [Google Scholar]
- 40.Fujimoto K., Shibasaki T., Yokoi N., Kashima Y., Matsumoto M., Sasaki T., Tajima N., Iwanaga T., Seino S. (2002) J. Biol. Chem. 277, 50497–50502 [DOI] [PubMed] [Google Scholar]
- 41.Eliasson L., Ma X., Renström E., Barg S., Berggren P. O., Galvanovskis J., Gromada J., Jing X., Lundquist I., Salehi A., Sewing S., Rorsman P. (2003) J. Gen. Physiol. 121, 181–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang G., Joseph J. W., Chepurny O. G., Monaco M., Wheeler M. B., Bos J. L., Schwede F., Genieser H. G., Holz G. G. (2003) J. Biol. Chem. 278, 8279–8285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steegborn C., Litvin T. N., Levin L. R., Buck J., Wu H. (2005) Nat. Struct. Mol. Biol. 12, 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamenetsky M., Middelhaufe S., Bank E. M., Levin L. R., Buck J., Steegborn C. (2006) J. Mol. Biol. 362, 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Triest M., de Rooij J., Bos J. L. (2001) Methods Enzymol. 333, 343–348 [DOI] [PubMed] [Google Scholar]
- 46.Coppola T., Perret-Menoud V., Gattesco S., Magnin S., Pombo I., Blank U., Regazzi R. (2002) Biochem. J. 362, 273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kraemer A., Rehmann H. R., Cool R. H., Theiss C., de Rooij J., Bos J. L., Wittinghofer A. (2001) J. Mol. Biol. 306, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 48.Roggero C. M., De Blas G. A., Dai H., Tomes C. N., Rizo J., Mayorga L. S. (2007) J. Biol. Chem. 282, 26335–26343 [DOI] [PubMed] [Google Scholar]
- 49.Zarelli V. E., Ruete M. C., Roggero C. M., Mayorga L. S., Tomes C. N. (2009) J. Biol. Chem. 284, 10491–10503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Berghe N., Cool R. H., Horn G., Wittinghofer A. (1997) Oncogene 15, 845–850 [DOI] [PubMed] [Google Scholar]
- 51.Rehmann H. (2006) Methods Enzymol. 407, 159–173 [DOI] [PubMed] [Google Scholar]
- 52.Mendoza C., Carreras A., Moos J., Tesarik J. (1992) J. Reprod. Fertil. 95, 755–763 [DOI] [PubMed] [Google Scholar]
- 53.Bordier C. (1981) J. Biol. Chem. 256, 1604–1607 [PubMed] [Google Scholar]
- 54.Pryde J. G., Phillips J. H. (1986) Biochem. J. 233, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 56.Hess K. C., Jones B. H., Marquez B., Chen Y., Ord T. S., Kamenetsky M., Miyamoto C., Zippin J. H., Kopf G. S., Suarez S. S., Levin L. R., Williams C. J., Buck J., Moss S. B. (2005) Dev. Cell. 9, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., Døskeland S. O. (2003) J. Biol. Chem. 278, 35394–35402 [DOI] [PubMed] [Google Scholar]
- 58.Holz G. G., Chepurny O. G., Schwede F. (2008) Cell. Signal. 20, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Blas G., Michaut M., Treviño C. L., Tomes C. N., Yunes R., Darszon A., Mayorga L. S. (2002) J. Biol. Chem. 277, 49326–49331 [DOI] [PubMed] [Google Scholar]
- 60.Herrick S. B., Schweissinger D. L., Kim S. W., Bayan K. R., Mann S., Cardullo R. A. (2005) J. Cell. Physiol. 202, 663–671 [DOI] [PubMed] [Google Scholar]
- 61.Huynh H., Bottini N., Williams S., Cherepanov V., Musumeci L., Saito K., Bruckner S., Vachon E., Wang X., Kruger J., Chow C. W., Pellecchia M., Monosov E., Greer P. A., Trimble W., Downey G. P., Mustelin T. (2004) Nat. Cell Biol. 6, 831–839 [DOI] [PubMed] [Google Scholar]
- 62.Michaut M., Tomes C. N., De Blas G., Yunes R., Mayorga L. S. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 9996–10001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohnishi H., Samuelson L. C., Yule D. I., Ernst S. A., Williams J. A. (1997) J. Clin. Invest. 100, 3044–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwan E. P., Xie L., Sheu L., Ohtsuka T., Gaisano H. Y. (2007) Diabetes 56, 2579–2588 [DOI] [PubMed] [Google Scholar]
- 65.Hatakeyama H., Takahashi N., Kishimoto T., Nemoto T., Kasai H. (2007) J. Physiol. 582, 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kashima Y., Miki T., Shibasaki T., Ozaki N., Miyazaki M., Yano H., Seino S. (2001) J. Biol. Chem. 276, 46046–46053 [DOI] [PubMed] [Google Scholar]
- 67.Balasubramanian L., Sham J. S., Yip K. P. (2008) Pflugers Arch. 456, 747–754 [DOI] [PubMed] [Google Scholar]
- 68.Sedej S., Rose T., Rupnik M. (2005) J. Physiol. 567, 799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gekel I., Neher E. (2008) J. Neurosci. 28, 7991–8002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chin E. C., Abayasekara D. R. (2004) J. Endocrinol. 183, 51–60 [DOI] [PubMed] [Google Scholar]
- 71.Kang G., Chepurny O. G., Rindler M. J., Collis L., Chepurny Z., Li W. H., Harbeck M., Roe M. W., Holz G. G. (2005) J. Physiol. 566, 173–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tovey S. C., Dedos S. G., Taylor E. J., Church J. E., Taylor C. W. (2008) J. Cell Biol. 183, 297–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Caron E. (2003) J. Cell Sci. 116, 435–440 [DOI] [PubMed] [Google Scholar]
- 74.Bos J. L. (2005) Curr. Opin. Cell Biol. 17, 123–128 [DOI] [PubMed] [Google Scholar]
- 75.Kooistra M. R., Dubé N., Bos J. L. (2007) J. Cell Sci. 120, 17–22 [DOI] [PubMed] [Google Scholar]
- 76.Leiser M., Efrat S., Fleischer N. (1995) Endocrinology 136, 2521–2530 [DOI] [PubMed] [Google Scholar]
- 77.Kowluru A., Li G., Rabaglia M. E., Segu V. B., Hofmann F., Aktories K., Metz S. A. (1997) Biochem. Pharmacol. 54, 1097–1108 [DOI] [PubMed] [Google Scholar]
- 78.D'Silva N. J., Jacobson K. L., Ott S. M., Watson E. L. (1998) Am. J. Physiol. 274, C1667–C1673 [DOI] [PubMed] [Google Scholar]
- 79.Shibasaki T., Takahashi H., Miki T., Sunaga Y., Matsumura K., Yamanaka M., Zhang C., Tamamoto A., Satoh T., Miyazaki J., Seino S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19333–19338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sabbatini M. E., Chen X., Ernst S. A., Williams J. A. (2008) J. Biol. Chem. 283, 23884–23894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maillet M., Robert S. J., Cacquevel M., Gastineau M., Vivien D., Bertoglio J., Zugaza J. L., Fischmeister R., Lezoualc'h F. (2003) Nat. Cell Biol. 5, 633–639 [DOI] [PubMed] [Google Scholar]
- 82.Amano R., Lee J., Goto N., Harayama H. (2007) J. Reprod. Dev. 53, 127–133 [DOI] [PubMed] [Google Scholar]
- 83.Kinukawa M., Oda S., Shirakura Y., Okabe M., Ohmuro J., Baba S. A., Nagata M., Aoki F. (2006) FEBS Lett. 580, 1515–1520 [DOI] [PubMed] [Google Scholar]
- 84.Berruti G. (2003) Cell. Mol. Biol. 49, 381–388 [PubMed] [Google Scholar]
- 85.Nakagaki I., Sasaki S., Hori S., Kondo H. (2000) Pflugers Arch. 440, 828–834 [DOI] [PubMed] [Google Scholar]
- 86.Aivatiadou E., Mattei E., Ceriani M., Tilia L., Berruti G. (2007) Mol. Biol. Cell 18, 1530–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walensky L. D., Snyder S. H. (1995) J. Cell Biol. 130, 857–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tomes C. N., McMaster C. R., Saling P. M. (1996) Mol. Reprod. Dev. 43, 196–204 [DOI] [PubMed] [Google Scholar]
- 89.Fukami K., Nakao K., Inoue T., Kataoka Y., Kurokawa M., Fissore R. A., Nakamura K., Katsuki M., Mikoshiba K., Yoshida N., Takenawa T. (2001) Science 292, 920–923 [DOI] [PubMed] [Google Scholar]
- 90.Nomikos M., Blayney L. M., Larman M. G., Campbell K., Rossbach A., Saunders C. M., Swann K., Lai F. A. (2005) J. Biol. Chem. 280, 31011–31018 [DOI] [PubMed] [Google Scholar]
- 91.Han H., Stessin A., Roberts J., Hess K., Gautam N., Kamenetsky M., Lou O., Hyde E., Nathan N., Muller W. A., Buck J., Levin L. R., Nathan C. (2005) J. Exp. Med. 202, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stessin A. M., Zippin J. H., Kamenetsky M., Hess K. C., Buck J., Levin L. R. (2006) J. Biol. Chem. 281, 17253–17258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wuttke M. S., Buck J., Levin L. R. (2001) JOP. 2, 154–158 [PubMed] [Google Scholar]
- 94.Litvin T. N., Kamenetsky M., Zarifyan A., Buck J., Levin L. R. (2003) J. Biol. Chem. 278, 15922–15926 [DOI] [PubMed] [Google Scholar]
- 95.Esposito G., Jaiswal B. S., Xie F., Krajnc-Franken M. A., Robben T. J., Strik A. M., Kuil C., Philipsen R. L., van Duin M., Conti M., Gossen J. A., Jaiswal B. S. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2993–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie F., Garcia M. A., Carlson A. E., Schuh S. M., Babcock D. F., Jaiswal B. S., Gossen J. A., Esposito G., van Duin M., Conti M. (2006) Dev. Biol. 296, 353–362 [DOI] [PubMed] [Google Scholar]
- 97.Beltrán C., Vacquier V. D., Moy G., Chen Y., Buck J., Levin L. R., Darszon A. (2007) Biochem. Biophys. Res. Commun. 358, 1128–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramos L. S., Zippin J. H., Kamenetsky M., Buck J., Levin L. R. (2008) J. Gen. Physiol. 132, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sakane A., Manabe S., Ishizaki H., Tanaka-Okamoto M., Kiyokage E., Toida K., Yoshida T., Miyoshi J., Kamiya H., Takai Y., Sasaki T. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10029–10034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y., Okamoto M., Schmitz F., Hofmann K., Südhof T. C. (1997) Nature 388, 593–598 [DOI] [PubMed] [Google Scholar]
- 101.Tomes C. N., De Blas G. A., Michaut M. A., Farré E. V., Cherhitin O., Visconti P. E., Mayorga L. S. (2005) Mol. Hum. Reprod. 11, 43–51 [DOI] [PubMed] [Google Scholar]
- 102.Tomes C. N., Michaut M., De Blas G., Visconti P., Matti U., Mayorga L. S. (2002) Dev. Biol. 243, 326–338 [DOI] [PubMed] [Google Scholar]