

Abstract
The myriad functions of complex carbohydrates include modulating interactions between bacteria and their eukaryotic hosts. In humans and other vertebrates, variations in the activity of glycosyltransferases of CAZy family 6 generate antigenic variation between individuals and species that facilitates resistance to pathogens. The well characterized vertebrate glycosyltransferases of this family are multidomain membrane proteins with C-terminal catalytic domains. Genes for proteins homologous with their catalytic domains are found in at least nine species of anaerobic commensal bacteria and a cyanophage. Although the bacterial proteins are strikingly similar in sequence to the catalytic domains of their eukaryotic relatives, a metal-binding Asp-X-Asp sequence, present in a wide array of metal ion-dependent glycosyltransferases, is replaced by Asn-X-Asn. We have cloned and expressed one of these proteins from Bacteroides ovatus, a bacterium that is linked to inflammatory bowel disease. Functional characterization shows it to be a metal-independent glycosyltransferase with a 200-fold preference for UDP-GalNAc as substrate relative to UDP-Gal. It efficiently catalyzes the synthesis of oligosaccharides similar to human blood group A and may participate in the synthesis of the bacterial O-antigen. The kinetics for GalNAc transfer to 2′-fucosyl lactose are characteristic of a sequential mechanism, as observed previously for this family. Mutational studies indicate that despite the lack of a metal cofactor, there are pronounced similarities in structure-function relationships between the bacterial and vertebrate family 6 glycosyltransferases. These two groups appear to provide an example of horizontal gene transfer involving vertebrates and prokaryotes.
The structures of complex glycans are determined by the specificities of the glycosyltransferases (GTs)2 that catalyze their biosynthesis. GTs fall into two groups that differ in mechanism, based on whether the anomeric configuration of the donor substrate (α for most UDP-sugars) is retained or inverted in the product (1–3). They are classified into 90 different families in the CAZy data base based on sequence similarities (4, 5), but the majority of those that have been structurally characterized fall into one of two fold types, designated GT-A and GT-B (2). The retaining GTs of CAZy family 6 (GT6) have a GT-A fold and catalyze the transfer of either galactose or GalNAc into an α-linkage with the 3-OH group of β-linked galactose or GalNAc. GT6 includes the histo-blood group A and B GTs (GTA and GTB), the α-galactosyltransferase (α3GT) that catalyzes the synthesis of the xenoantigen or α-gal epitope, Forssman glycolipid synthase, isogloboside 3 synthase, and their homologues from other vertebrates (6). GT6 enzymes from vertebrates are type-2 membrane proteins with N-terminal cytosolic domains, a transmembrane helix, a spacer, and a C-terminal catalytic domain (6). Crystallographic studies of recombinant catalytic domains of GTA, GTB, and α3GT have provided detailed information about their interactions with substrates, metal cofactor, and inhibitors (7–9). Most GT-A fold GTs, including those in the GT6 family, require divalent metal ions, such as Mn2+, for catalytic activity; their metal dependence is linked to a shared DXD sequence motif. Residues of this motif interact with the metal ion and both the ribose and phosphates of the donor substrate to produce an appropriate substrate orientation and conformation for catalysis and to stabilize the UDP leaving group (3, 7–10).
Mammalian members of GT6 are responsible for variations in glycan structures between different species and individuals as the result of selective enzyme inactivation in certain species (α3GT, Forssman glycolipid synthase, and isogloboside 3 synthase) or the inheritance of multiple alleles at one locus that encode enzymes with different substrate specificity (GTA and GTB) or are inactive (11–14). The presence of circulating antibodies against glycan structures that are subject to interspecies and individual variability has been linked to resistance to pathogens that also carry the glycans; these antibodies are thought to arise from exposure to potential pathogens, including enveloped viruses and bacteria that carry structurally similar glycans (11).
In addition to the well characterized enzymes discussed previously, atypical members of the GT6 family have been identified in mammals that have sequence changes in highly conserved regions of the active site, including the DXD motif (6). However, no glycosyltransferase activity was detected in recombinant forms of two of these, and their functions are unclear (6). Although GT6 members are widely distributed among vertebrates, no homologues have been found in other eukaryotes (6). However, GT6 members have been identified in several bacterial species (15–17). GT6 enzymes from Escherichia coli O86, and Helicobacter mustelae that appear to function in the biosynthesis of the lipopolysaccharide O-antigen have been cloned and expressed by Wang and co-workers (16, 17) and found to have specificities similar to those of human GTB and GTA, respectively. These enzymes have been applied in the enzymatic synthesis of oligosaccharides. Other homologues are encoded by Hemophilus somnus, Psychroacter sp., PRwf-1 (15), Francisella philomiragia, and three Bacteroides species, Bacteroides ovatus, Bacteroides caccae, and Bacteroides stercoris, as well as a cyanophage, PSSM-2 (15). Genes for other homologues from unidentified species are present in the marine metagenome (18, 19) and human gut metagenome (20, 21). The phage and bacterial enzymes are substantially truncated at the N terminus relative to the catalytic domains of vertebrate GT6 representatives and are smaller than the reported minimal functional unit of a primate α3GT (22). When bacterial and vertebrate GT6 amino acid sequences are aligned (Fig. 1 and supplemental Figs. S1 and S2), it can be seen that the metal-binding DXD of the eukaryotic GTs is replaced by NXN (where X is Ala, Gly, or Ser) in the bacterial homologues. The cyanophage GT6 member and related proteins in the marine metagenome, however, retain the DXD motif. This conspicuous difference in the bacterial proteins is particularly interesting, since, in the mammalian enzymes, the aspartates of the DXD and adjacent residues are crucial for catalytic activity (10, 23).
FIGURE 1.

An alignment of selected bacterial, cyanophage and mammalian GT6 amino acid sequences. Abbreviations and Interpro sequence IDs (in parentheses) are as follows. HuA, human histo-blood group A synthase (A1EAJ6); Bova, bovine α1,3-galactosyltransferase (P14769); PSSM2, cyanophage PSSM-2 (Q58M87); Bs, B. stercoris (B0NSM3); Bo1, B. ovatus GT1 (A7LVT2); Bo2, B. ovatus GT2 (A7M0P3); Bc, B. caccae (A5ZC71). The boxed regions in the alignment identify regions that have been shown to be involved in interactions with substrates and cofactor and in catalysis in bovine α1,3-galactosyltransferase and histo-blood group A and B enzymes. These are labeled (below) as follows. A, interactions with uracil; B, interactions with the galactose moiety of UDP-Gal; C, interactions with Mn2+, phosphates, and galactose; D, interactions with acceptor substrate; E, interactions with Gal or GalNAc of donor substrate; F, interactions with monosaccharide of donor substrate and acceptor and catalysis; The arrow (above) denotes the intron/exon boundary in vertebrate GT6s, and the asterisks indicate the residues in BoGT6a that were subjected to mutagenesis.
B. ovatus is a Gram-negative commensal bacterium that inhabits the distal mammalian gut and has been implicated in the pathology of inflammatory bowel disease in humans (24). The B. ovatus genome contains two genes that encode GT6 representatives (Fig. 1). We selected one of these for initial investigation, and designate it BoGT6a (family 6 glycosyltransferase 1 of Bacteroides). The gene for this protein was amplified by PCR and cloned and expressed in His-tagged form in E. coli BL21(DE3). Assays with a variety of substrates show that its substrate specificity is similar to that of human GTA. Previous studies of the activities of bacterial enzymes were conducted in the presence of Mn2+ (16, 17), but we find that the B. ovatus enzyme does not require divalent metal ions for activity and is fully active in EDTA. Despite this striking difference, BoGT6a is similar to its metal-dependent relatives in catalytic properties; also, the effects of amino acid substitutions for residues corresponding to several that act in substrate binding and catalysis in vertebrate GT6 glycosyltransferases suggest that they have similar structure-function relationships. These results indicate that the metal cofactor is not a conserved feature in the GT6 family. They also raise questions about the catalytic mechanism of prokaryotic GT6 members and the evolutionary relationship between bacterial, phage, and vertebrate enzymes.
EXPERIMENTAL PROCEDURES
Amplification, Cloning, and Expression of BoGT6a
Freeze-dried B. ovatus cells were purchased from ATCC. Forward and reverse synthetic oligonucleotide primers (Invitrogen) that introduce NdeI and BamHI coding sites were designed to amplify full-length and C-terminally truncated forms of the gene: forward, 5′-AAA AAA CAT ATG (NdeI) AGA ATT GGT ATA TTA TAT ATC TGT ACT GGC-3′; reverse full-length, 5′-AAA AAA GGA TCC (BamHI) TCA ATC AGC CGA TTT AAA TTT TTG GCA GAT TAG-3′; reverse truncated, 5′-AAA AAA GGA TCC (BamHI) TCA GTT TTT TCT TCG CAA TAA TTC ATG CCC GCC-3′.
B. ovatus cells (1 mg) were suspended in water (100 μl). A PCR mixture (49 μl) containing 1 μl of the B. ovatus cell suspension, 1 nmol each of the forward and reverse primers, dNTPs, and 5 μl of Thermo Buffer for Polymerase (New England Biolabs) was heated at 100 °C for 10 min. The mixture was allowed to cool to room temperature, and 2 units (1 μl) of Vent polymerase (New England Biolabs) was added. The B. ovatus gene was amplified under the following conditions: 94 °C for 3 min, followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1.5 min with a final incubation at 72 °C for 10 min. The PCR product was gel-purified (Qiagen) and digested with 40 units (2 μl) each of NdeI and BamHI in 6 μl of BamHI buffer (New England Biolabs) and 1 μl of 100× bovine serum albumin at 37 °C for 3 h. The digestion product was gel-purified and mixed in a 1:1 ratio with pET42b vector that had been previously digested with NdeI and BamHI, together with 2.5 μl of 10× T4 ligase buffer and 800 units (2 μl) of T4 DNA ligase in a total volume of 25 μl. The mixture was incubated in a thermocycler under the following conditions: 30 cycles of 10 °C for 3 min, 12 °C for 3 min, 14 °C for 3 min, 16 °C for 3 min, and 18 °C for 1 min followed by a final incubation at 65° for 10 min. 10 μl of the ligation product was transformed into E. coli DH5α-competent cells using heat shock. Transformants were grown in 1 ml of SOC medium with rapid shaking (250 rpm) at 37 °C for 60 min before being plated on LB agar plates containing 50 μg/ml kanamycin. The plates were placed incubated at 37 °C for 18–20 h. A single colony was inoculated in 8 ml of LB (kan) medium and grown overnight, and the vector DNA (pET42b_BoGT6a) was extracted and sequenced (Davis Sequencing, LLC).
Bacterial Expression and Purification
Cultures of E. coli BL21(DE3) cells were transformed with pET42b_BoGT6a and grown in LB medium containing 50 μg/ml kanamycin with rapid shaking (250 rpm) at 37 °C. The temperature was reduced to 24 °C when the A600 nm of the culture reached 0.8–1.0, and incubation was continued overnight to allow leaky expression at the lower temperature. Bacterial cells were harvested by centrifugation at 4,000 rpm for 20 min, and the cell pellets were washed with 20% sucrose containing 20 mm Tris-HCl, pH 8.0, suspended in 30 ml of lysis buffer (50 mm Tris-HCl buffer, pH 8.0, containing 1 mm EDTA and 0.1 m NaCl), and disrupted using a French press. Insoluble material was removed by centrifugation at 30,000 rpm for 20 min, and the supernatant was applied to an Ni2+-nitrilotriacetic acid column (Qiagen), which had been equilibrated with 50 volumes of 20 mm Tris-HCl, pH 7.9, containing 0.5 m NaCl. The column was subsequently washed with 10 volumes of 20 mm Tris buffer containing 0.5 m NaCl and 5 mm imidazole, pH 7.9, followed by 20 volumes of 20 mm Tris buffer containing 0.5 m NaCl and 60 mm imidazole, pH 7.9, and finally eluted with 10 volumes of 20 mm Tris-HCl, pH 7.9, containing 0.5 m NaCl and 500 mm imidazole. Fractions containing the purified protein were dialyzed against two changes of 50 volumes of 20 mm Tris-HCl, pH 7.9, containing 0.1 m NaCl and 2 mm dithiothreitol; 10 mm EDTA was added to the buffer for storage. All steps in enzyme purification were conducted at 4 °C.
Mutagenesis
Seven mutants (D95N, D97N, A155M, A155Q, E192Q, R299A, and K231A) (see Fig. 2) were constructed using the PCR megaprimer method with previously described modifications (25) with pET42b_BoGT6a as a template. In the first amplification, a 50-μl mixture of (forward) mutagenic primers (Invitrogen) template, T7 terminator (Invitrogen), dNTPs (Eppendorf), Thermo Buffer, and Vent polymerase (New England Biolabs) was used under the following incubation conditions: 3 min at 94 °C followed by 30 cycles of 94 °C for 1 min, 60 °C for 1 min, 72 °C for 1 min, followed by 10 min at 72 °C. The product was gel-purified (Qiagen) and used as the reverse primer in a second reaction that also included T7 promoter, dNTPs, template, Thermo Buffer, and Vent polymerase under the following conditions: 94 °C for 3 min followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min and a final 10-min incubation at 72 °C.
FIGURE 2.

The amino acid sequence of BoGT6a showing regions corresponding to parts of the active sites of mammalian GT6 glycosyltransferases and sites of mutagenesis. Residues subjected to mutagenesis have shaded backgrounds, and the boxes correspond to active site regions A–F (see legend of Fig. 1 for details). The site of truncation in the recombinant protein is denoted by the arrow.
The double mutant, R244A/R245A, was constructed by PCR in one step. The amplification reaction contained the (reverse) mutagenic primer, template, T7 promoter, dNTPs, Thermo Buffer, and Vent polymerase and was incubated under the following conditions: 3 min at 94 °C followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min with a final 10-min incubation at 72 °C.
The following primers were used for mutagenesis: N95D (forward), 5′-CTATTTTTCTTCGATGCCAATCTCTTATTCACC-3′; N97D (forward), 5′-CTATTTTTCTTCAATGCCGATCTCTTATTCACC-3′; A155M (forward), 5′-CGATATTATTACATGGGAGGGCTTTCAGGTGGA-3′; A155Q (forward), 5′-CGATATTATTACCAGGGAGGGCTTTCAGGTGGA-3′; E192Q (forward), 5′-CCAATTTGGCACGACCAATCTCTAATCAATAAA-3′; R229A (forward), 5′-CCAATAATCCTCATTGCAGACAAAAATAAA-3′; K231A (forward), 5′-ATCCTCATTCGAGACGCAAATAAACCCCAATAT-3′; R244A/R245A (reverse), 5′-AAAAAAGGATCCTCAGTTTTTTGCTGCCAATAATTCATGCCCGCC-3′.
Molecular Size
The native molecular size of BoGT6a was investigated by medium pressure gel filtration with a column (10 × 300 mm) of Superdex 200 (Tricorn; GE Healthcare), equilibrated and eluted with 0.1 m sodium phosphate buffer, pH 6.8, containing 0.4 m NaCl and 10 mm sodium azide. Protein samples were applied in 100 μl of sample buffer. The column was calibrated with standard proteins of known molecular weight, thyroglobulin (670,000), catalase (250,000), immunoglobulin G (158,000), transferrin (79,500), ovalbumin (44,000), myoglobin (17,000), and α-lactalbumin (14,000), and the molecular weight was estimated from a regression analysis of a plot of elution volume versus log(molecular weight) of the standards.
Enzyme Assays
Glycosyltransferase activities were measured using a radiochemical assay (10) with the potential acceptor substrates 2′-fucosyllactose, fucosyl α1-2 galactose, blood group H type II trisaccharide (Fucα1-2Galβ1-4GlcNAc) (V-Labs), N-acetyl-lactosamine, and Forssman synthase acceptor (GalNAc β1-3Gal α1-4Gal), by following the transfer of radioactive sugars from UDP-[3H]N-acetylgalactosamine (UDP-GalNAc), UDP-[3H]Gal, or UDP-[3H]Glc (Sigma). Standard assays contained 0.021 μg of enzyme, 50 mm Tris-HCl buffer, pH 7.0, 10 mm EDTA, and 0.1% bovine serum albumin in a total volume of 50 μl and were incubated at 37 °C for 10–15 min. For more detailed characterization of enzyme kinetics, acceptor concentrations were 0.1–0.5 mm for 2′-fucosyllactose, 0.1–3.0 mm for Fucα1-2Gal, and 0.2–1.0 mm for Fucα1-2Galβ1-4GlcNAc. The concentration of UDP-[3H]GalNAc (specific activity, 666 cpm/nmol) was varied from 0.06 to 0.3 mm. Reactions from which the acceptor substrate is omitted served as controls. A higher concentration of enzyme (3 μg) and a longer incubation time (20 min) were used in assays of UDP-GalNAc hydrolase activity. Because the BoGT6a-catalyzed reactions could not be terminated by the addition of EDTA or by cooling on ice (since BoGT6a has a significant level of activity at 0 °C), the reaction mixture was immediately applied to a 1-ml Dowex (1 × 800) anion exchange column, followed by 0.5 ml of water to wash the tube. The product was subsequently eluted with 1 ml of water to elute the product; the eluate was collected in a plastic vial, mixed with 10 ml of Ecolume (ICN Biomedicals, Costa Mesa, CA); and radioactivity was measured with a liquid scintillation counter (LKB).
Kinetic data were analyzed by non-linear regression using Sigma PlotTM. Data were fitted to Michaelis-Menten equations for a single substrate reaction (Equation 1) and a general two substrate sequential reaction (Equation 2),
 |
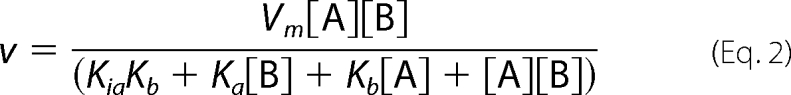 |
where, in Equation 1, [S] represents the concentration of the varied substrate, and Vm and Km are the apparent maximum velocity and Michaelis constants. In Equation 2, [A] is the concentration of UDP-GalNAc, and [B] is the concentration of 2′-fucosyllactose. Ka and Kb are the cognate Michaelis constants, and Kia is the dissociation constant substrate A. Data were also fitted to variants of Equation 2 lacking a Ka or Kia term, respectively. Mutant enzymes were characterized by varying each substrate individually at a fixed concentration of the second substrate, and the data were fitted to Equation 1 to give apparent Km and Vm values.
RESULTS
Cloning and Expression of BoGT6a
The sequences of 11 bacterial GT6 family members are in the following data bases: NR data base, H. somnus, E. coli O86, and Psychrobacter sp. PRf-1; Whole-Genome Shotgun Reads, B. ovatus (two sequences), B. stercoris, B. caccae, and Subdoligranulum variable (two sequences). The single putative viral protein sequence from Prochlorococcus cyanophage PSSM-2 is also in the NR data base. The bacterial sequences are also listed in Interpro (available on the World Wide Web), entry IPR005076. An alignment of the GT6 protein sequences from the three Bacteroides species with that from the cyanophage PSSM-2 and the catalytic domains of human GTA and bovine α3GT generated using MUSCLE (available on the World Wide Web) is shown in Fig. 1, and an alignment of all of the bacterial GTs is shown in supplemental Fig. S1. The Environmental Samples data base contains six additional sequences from the Human Gut Metagenome that encode proteins that are similar to the bacterial GT6s (including one that is identical with the B. stercoris sequence) and more than 90 sequences with high levels of sequence similarity to the putative cyanophage protein are found in the Marine Metagenome. There are high levels of sequence similarity between the bacterial, phage, and vertebrate GT6s, amounting to 35% amino acid sequence identity for some pairwise comparisons, and relatively few gaps need to be inserted to align their sequences. Sections of sequence identified as parts of the active site in α3GT, GTA, and GTB by structural and mutational studies (25–27) are enclosed in boxes in Fig. 1 (A–F). These are similar to ligand binding regions assigned by Heissigerova et al. (28) in vertebrate GT6s. The sequences within and adjacent to some of these regions are well conserved, but in regions D and E, the sequences are less conserved because these regions are responsible for specificity differences for acceptor and donor substrates (6, 9, 27). In the vertebrate GT6 glycosyltransferases, an additional region at the C terminus is also important for activity (region G; supplemental Fig. S2). However, because the C-terminal regions of the vertebrate and bacterial enzymes do not align well, we have not marked this region in Fig. 1. Despite their strong overall similarity in sequence, the bacterial and phage proteins are shorter than the catalytic domains of the vertebrate enzymes by 47 residues. The truncation is at the N terminus and includes, in the known structures of the vertebrate enzymes, GTA, GTB, and α3GT, an α-helix, and a β strand (8, 9). This region does not appear to be a separate domain or subdomain, since it has a large number of interactions with the rest of the catalytic domain. The C-terminal regions of the bacterial sequences, which align less well with the eukaryotic enzymes and with each other, contain a large proportion of basic amino acids.
Region C corresponds to the DXD motif that, in all of the bacterial proteins, including those listed above, is replaced by NXN (Fig. 1). It is interesting that in all homologues from the marine metagenome, the DXD motif is conserved; these sequences are probably from unidentified cyanophage species, whereas those from the human gut metagenome have an NXN sequence, and appear to originate from unidentified species of commensal bacteria (see supplemental Figs. S1 and S2).
We selected one of the two B. ovatus glycosyltransferases (designated BoGT6a) for characterization. The coding sequence was amplified by PCR of B. ovatus DNA and was cloned into the pET42b vector. Sequence analysis showed that it is identical with the sequence in the data base. Two forms of the enzyme were cloned into the pET42b vector with an N-terminal His tag, a full-length form and a truncated form that is missing the C-terminal 17 residues and terminates at Asn246 (see Fig. 2). Both were expressed in E. coli BL21(DE3) as soluble proteins and were purified by nickel-chelate chromatography in yields of 12 mg/liter of culture. Their levels of catalytic activity are closely similar, and the truncated enzyme was used for detailed characterization. SDS-PAGE of both preparations under reducing conditions showed a single band of the expected size (31 kDa) for the protein stored in the presence of dithiothreitol. During storage in the absence of reducing agent, the activity declined, and the protein aggregated; SDS gel electrophoresis showed a large proportion of what appears to be a disulfide-linked dimer (∼60 kDa) and a small fraction of higher oligomers, all of which were converted to the monomer on reduction (data not shown). The native molecular weight of the preparation (stored under standard conditions in the presence of dithiothreitol) that was used for characterizing the specificity and kinetic properties was investigated by medium pressure gel filtration as described. BoGT6a eluted from the column as a single peak with an elution time corresponding to an apparent Mr of about 25,000. This is somewhat lower than the molecular weight calculated from the amino acid sequence, including the His tag (31,050) but supports the view that the protein is monomeric.
Catalytic Properties
The catalytic activity of BoGT6a was tested with different UDP-sugars (UDP-Gal, UDP-GalNAc, UDP-Glc) as potential donor substrates and glycans that are acceptor substrates of vertebrate GT6 glycosyltransferases as potential acceptor substrates. Unlike its vertebrate counterparts whose activities are completely dependent on divalent metal ions, particularly Mn2+ (10), the activity of BoGT6a does not require exogenous metal ions. The addition of either EDTA or Mn2+ produces a small progressive decline in activity, whereas bovine α3GT is inactive in the absence of exogenous Mn2+ or in the presence of EDTA and is progressively activated by metal ion (Fig. 3). BoGT6a is fully active in the presence of 10 mm EDTA, which was included in the kinetic analyses reported here. Among the various substrates, high levels of activity were obtained only with UDP-GalNAc as donor and 2′-fucosyllactose, 2′-fucosyl-LacNAc, or 2′-fucosylgalactose as acceptors, indicating that the enzyme has a substrate specificity similar to that of human GTA. Very low levels of activity were obtained with UDP-Gal or UDP-Glc as donor substrates (<1% of that with UDP-GalNAc) in combination with 2′-fucosyllactose or 2′-fucosyl-LacNAc as acceptor substrates. Similarly, when UDP-GalNAc was used as the donor substrate donor together with lactose, LacNAc, or GalNAc β1-3Gal α1-4Gal (Forssman synthase substrate) as acceptor substrate, the activity level was also <1% of that obtained with 2-fucosyllactose. The three effective acceptor substrates were compared by varying their concentrations at a fixed concentration (0.3 mm) of UDP-GalNAc. Their respective apparent Km and kcat values (Table 1) show that 2′-fucosyllactose and 2′-fucosyl-LacNAc are the best substrates and have very similar apparent kinetic parameters. However, the saturation curve for 2′-fucosyl galactose is essentially linear up to a concentration of 3 mm. Because of the lack of curvature, Km and Vm could not be determined, but the value of kcat/Km, calculated from the slope of the line, indicates that 2′-fucosyl galactose is the weakest of the three substrates.
FIGURE 3.

Metal independence of catalytic activity in BoGT6a contrasted with metal dependence of bovine α3GT indicated by the effects of exogenous EDTA and Mn2+ on activity. The preparation of BoGT6a contained 10 mm EDTA, contributing 0.6 mm to the assay mixtures; it was assayed using 0. 3 mm UDP-GalNAc and 0.3 mm 2′-fucosyllactose, and the activity is expressed relative to the activity determined in the absence of exogenous Mn2+ or EDTA. α3GT was assayed using 0.3 mm UDP-Gal and 10 mm lactose, and the activity is expressed relative to the extrapolated activity at saturating [Mn2+]. Circles, activity of BoGT6a; squares, activity of α3GT. Filled symbols, effects of increasing Mn2+; open symbols, effects of increasing EDTA.
TABLE 1.
Kinetic parameters of BoGT6a for different acceptors with UDP-GalNAc as donor substrate
| Acceptor substrate | Parametera | Value |
|---|---|---|
| 2′-Fucosyllactose | Km | 0.64 mm |
| kcat | 14.1 s−1 | |
| kcat/Km | 22 s−1 mm−1 | |
| 2′-Fucosyl-LacNAc | Km | 0.65 mm |
| kcat | 14.3 s−1 | |
| kcat/Km | 22 s−1 mm−1 | |
| 2′-Fucosylgalactose | kcat/Km | 1.81 s−1 mm−1 |
a These are apparent values, determined by varying acceptor substrate concentration at a fixed concentration (0.3 mm) of UDP-GalNAc.
A steady state kinetic study was conducted in which the concentration of UDP-GalNAc was varied at a series of fixed concentrations of 2′-fucosyllactose. The data fit best to the rate equation for a sequential mechanism (Equation 2), displayed as patterns of intersecting lines in a double reciprocal plot of velocity versus [UDP-GalNAc] or [2′-fucosyllactose] (see supplemental Fig. S3. Fitting the data to this equation generates Km values for the two substrates (Ka and Kb), a Kia value, and the turnover number, kcat. UDP-GalNAc was assigned as substrate A and 2′-fucosyllactose as B, and their Km values as Ka and Kb, respectively, based on their designations in mammalian GT6 transferases, which have ordered mechanisms. However, although this rate equation implies a mechanism in which both substrates bind prior to catalysis, it does not indicate that substrates bind in a specific order. The kinetic parameters are comparable with those previously reported for GTA and bovine α3GT (10, 29) (Table 2), except that the kcat value is higher. BoGT6a also catalyzes GalNac transfer to water (UDP-GalNAc hydrolysis) at a low but significant rate, analogous to the hydrolysis of UDP-Gal catalyzed by bovine α3GT (10); this suggests that, for the transferase reaction, UDP-GalNAc binds prior to acceptor, or substrate binding is random but appears to be inconsistent with ordered binding of acceptor prior to donor substrate. UDP-GalNAc hydrolysis was characterized at a higher enzyme concentration by varying the concentration of donor substrate, and the data were fitted to Equation 1. The results show reasonable agreement between the Km for UDP-GalNAc hydrolysis and the Kia for GalNAc transfer; the kcat value for hydrolysis, which amounts to 0.06% of the transferase activity, is lower than the corresponding relative rate (0.25%) observed with α3GT (10).
TABLE 2.
Steady state kinetic parameters of BoGT6a in comparison with human GTA and bovine α3GT
| Parameter | GTAa | α3GTb | BoGT6ac |
|---|---|---|---|
| Donor substrate | UDP-GalNAc | UDP-Gal | UDP-GalNAc |
| Transferase activity | |||
| Ka (μm) | 13 | 430 | 335 ± 46 |
| Kb (μm) | 15 | 19 × 103 | 1170 ± 150 |
| Kia (μm) | 5 | 140 | 67 ± 5 |
| kcat (s−1) | 4.9 | 6.4 | 38 ± 3.8 |
| Hydrolase activity | |||
| Ka (μm) | NDd | 100 | 97 |
| kcat (s−1) | ND | 0.016 | 0.024 |
a Data reported by Seto et al. (29), measured in the presence of 20 mm Mn2+. The acceptor substrate was Fucα(1–2)Galβ-O-(CH2)7-CH3.
b Determined with lactose as substrate at 10 mm Mn2+ (10).
c Assays conducted using 2′-fucosyllactose as acceptor substrate in the presence of 10 mm EDTA.
d ND, not determined.
To determine whether the low catalytic activity with UDP-Gal arises from a deficiency in substrate binding or rate of catalysis, galactose transfer from UDP-Gal to 2′-fucosyllactose was characterized in more detail by varying the concentration of each substrate at a fixed concentration of the second to obtain apparent Vm and Km values. These results (see Table 3) suggest that the low activity with UDP-Gal as donor substrate mainly arises from the low kcat value (0.14% of that for UDP-GalNAc).
TABLE 3.
Effects of mutations on kinetic parameters of BoGT6a
| Parametera | Ka | Kb | kcat | Relative kcat |
|---|---|---|---|---|
| mm | mm | s−1 | ||
| UDP-GalNAc substrate | ||||
| Wild type | 0.11 ± 0.02 | 0.69 ± 0.04 | 34 ± 1.5 | 1 |
| N95D | 4.0 ± 1.6 | 3.3 ± 0.58 | 8 ± 0.8 × 10−3 | 2.3 × 10−4 |
| N97D | 0.08 ± 0.01 | 6.4 ± 0.9 | 4.9 ± 0.4 | 0.14 |
| A155M | 0.85 ± 0.06 | 0.27 ± 0.03 | 0.08 ± 0.003 | 2.3 × 10−3 |
| A155Q | 1.35 ± 0.37 | 1.3 ± 0.3 | 0.027 ± 0.003 | 8 × 10−4 |
| E192Q | 0.52 ± 0.21 | 1.7 ± 1.0 | 1.5 ± 0.5 × 10−3 | 4.4 × 10−5 |
| R229A | 0.61 ± 0.06 | 1.7 ± 0.2 | 11.9 ± 0.8 | 0.34 |
| K231A | 0.24 ± 0.04 | 3.4 ± 0.1 | 0.15 ± 0.003 | 3.8 × 10−2 |
| R243A/R244A | 0.23 ± 0.02 | 2.4 ± 0.25 | 3.4 ± 0.17 | 0.1 |
| UDP-Gal substrate | ||||
| Wild type | 0.45 ± 0.04 | 1.35 ± 0.20 | 0.023 ± 0.002 | 6.8 × 10−4 (1)b |
| A155M | 1.27 ± 0.15 | 1.43 ± 0.35 | 0.11 ± 0.007 | 3.2 × 10−3 (4.7) |
| A155Q | 0.98 ± 0.10 | 0.27 ± 0.2 | 0.090 ± 0.004 | 2.6 × 10−3 (3.9) |
a These are apparent values, determined by varying the concentration of one substrate at a fixed concentration (1 mm) of the second. Ka is the apparent Km for UDP-GalNAc or UDP-Gal, and Kb is the apparent Km for 2′-fucosyllactose; kcat (app) is calculated from the apparent Vm determined by varying 2′-fucosyllactose at 1 mm UDP-GalNAc or UDP-Gal.
b Activity relative to that of wild-type enzyme with UDP-Gal as substrate.
Investigation of Potential Active Site Residues in BoGT6a
To test the hypothesis that BoGT6a is similar to mammalian GT6 glycosyltransferases in structure-function relationships, we have investigated the functional effects of mutating residues in BoGT6a that correspond to those that have key roles in catalysis and substrate binding in its mammalian homologues. The residues selected for mutation are located in regions C, E, and F and in the C-terminal region (see Fig. 2). Specifically, asparagines 95 and 97 of the NAN sequence were replaced individually with aspartates. Alanine 155 corresponds to histidine 280 in α3GT and leucine or methionine 266 in human GTA and GTB, respectively, residues that are a key to donor substrate specificity; this residue was substituted by methionine and glutamine based on residues present at the corresponding site in vertebrate and prokaryotic GT6 family members. Glutamate 192, which corresponds to a key residue in the catalytic mechanism of α3GT, Glu317 (26), was substituted by glutamine. Lys359 and Arg365 in the C-terminal region of α3GT interact with the phosphates of the UDP moiety of the donor substrate (26); this region undergoes large structural changes on binding UDP or donor substrate (8, 26). Because the C-terminal sequences differ considerably between bacterial and vertebrate GT6 glycosyltransferases (Fig. 2), it is difficult to reliably align their sequences in this region so that the equivalence of residues between α3GT and BoGT6a is uncertain. In fact, as shown in supplemental Fig. S1, the bacterial enzymes as a group have a high level of sequence diversity in this region. Therefore, we used alanine scanning to investigate all basic amino acids of BoGT6a in the C-terminal region that show some conservation in the bacterial GT6 group specifically: Arg229, Lys243, Arg244, and Arg244 (Fig. 1). The first two were separately mutated, and the adjacent residues, 244 and 245, were probed by a double substitution. These eight variants were all produced in yields similar to those of the wild-type enzyme and, after nickel-chelate column chromatography, showed, in each case, a single band on SDS-PAGE with a mobility corresponding to that of wild-type BoGT6a. Steady state kinetic characterization of each mutated protein was conducted by varying each substrate at a fixed concentration of the second using UDP-GalNAc as acceptor substrate, but the Ala155 mutants were characterized using both UDP-GalNAc and UDP-Gal. The results, summarized in Table 3, show that conservative substitutions for either Asn95 or Glu192 have very unfavorable effects on enzyme activity, largely resulting from reductions in kcat of 4400-fold and 22,000-fold, respectively. The S.E. values for the kinetic parameters of these variants are higher than those for the other mutants because of their low activities. The Asn95 to Asp mutant was less stable than the other mutants, and assays of this protein were conducted at 30 °C instead of 37 °C. In this mutant, the low kcat was accompanied by an increase in the apparent Ka value that is not observed with the other mutants; however, the kinetic parameters are not strictly comparable with those of the other variants because of the different assay conditions. In contrast, the Asn97 to Asp mutation has less effect on activity, producing an ∼7-fold reduction in kcat and 5-fold increase in Kb. Substitution of Met or Gln for Ala155 produced variants with greatly reduced GalNAc transferase activity and a preference for UDP-Gal over UDP-GalNAc as donor substrate reflected by 5- and 4-fold increases in the kcat for galactosyltransferase activity relative to wild type. Alanine mutagenesis of the conserved basic amino acids in the C-terminal region indicates that Arg229 has little influence on activity, whereas mutation of Lys231 generates a >200-fold decrease in kcat; the effect of mutating both Arg244 and Arg245 produced a significant but relatively modest (10-fold) reduction in kcat.
DISCUSSION
Amino acid sequence alignments substantiate the common ancestry of the genes for GT6 family members from vertebrates, bacteria, and cyanophage (Fig. 1 and supplemental Figs. S1 and S2). Molecular phylogeny analyses of the translated amino acid sequences of the GT6s from bacteria, the cyanophage, PSSM-2, the Human Gut Metagenome, and representatives from the Marine Metagenome together with those of the catalytic domains of selected vertebrate enzymes using Phylogeny.fr (30) show that they form three main branches: (i) bacteria and gut metagenome sequences, (ii) cyanophage and marine metagenome, and (iii) vertebrate sequences. Searches have failed to identify additional homologues in the currently known genomes of invertebrates, fungi, protozoa, plants, or archaea, suggesting that GT6 enzymes have a notably discontinuous distribution. Among prokaryotes and viruses, they are confined to nine known bacterial strains and one known T4-type phage, but their presence in additional unidentified commensal bacteria and many (presumed) bacteriophage species is indicated by sequences from the human gut and marine metagenomes. The distribution of the GTs and phylogenetic trees generated using their sequences suggest that lateral gene transfer involving vertebrates and prokaryotes (31) has played a role in the evolution of this family. With respect to this, it should be noted that the bacterial enzymes correspond closely in length to an exon of the vertebrate genes (see the arrow denoting the location of the exon/intron boundary in Fig. 1).
BoGT6a differs from previously characterized GT6 glycosyltransferases in having metal-independent catalytic activity. This may be linked to the change of the DXD motif, found in the vertebrate GT6 branch and many other GTA fold GTs, to NXN. Coutinho et al. (5) have pointed out, appropriately, that a DXD motif is not a unique feature of metal-dependent glycosyltransferases, since it is present in 51% of sequences in the SwissProt data base, so it is of questionable diagnostic significance with respect to metal-dependent glycosyltransferase activity. Nevertheless, in the vertebrate GT6s, the role of region C (Fig. 1) in binding the metal cofactor and donor substrate and in catalysis has been definitively established by structural and mutational studies (2, 10, 23). The precise alignment of the bacterial NXN sequence with the conserved DXD sequence of the vertebrate catalytic domains and putative phage GTs is supported by identities at multiple sites in the surrounding sequence (Fig. 1). The bacterial type GT6 sequences from the human gut metagenome as well as those from known bacterial species share the NXN sequence, suggesting that they may be metal-independent, but this requires experimental investigation. The biological significance of this substantial difference in properties is unclear. Metal independence could reflect a feature of the ancestor of the GT6 family, metal dependence being a later development, or vice versa. BoGT6a and other bacterial GT6s are intracellular proteins, and cellular levels of Mn2+ and other transition metals in bacteria are closely regulated by metal uptake and efflux systems so that intracellular concentrations of free Mn2+ are low (32, 33). Most are commensal bacteria that inhabit the distal intestinal tracts of vertebrates, an environment that is likely to be deficient in metal ions as the result of intestinal absorption processes. Thus, metal independence in the bacterial GTs could result from adaptation to their intracellular environment in these species.
To compare structure-function relationships in BoGT6a with those of the well characterized vertebrate enzymes, we investigated the effects of mutations in BoGT6a on activity. The roles of asparagines 95 and 97 in the activity of BoGT6a were probed by structurally conservative substitutions with aspartate. The results indicate that mutation of Asn95 had a large effect on catalytic activity, much greater than the corresponding substitution for Asn97. The >4000-fold reduction in kcat and 30-fold increase in the Km for UDP-GalNAc generated by the Asn95 to Asp mutation indicate that this substitution perturbs the interaction of the enzyme with donor substrate. However, structural studies are needed to determine the role of this region in donor substrate binding.
Residues in regions B and F (Fig. 1) have key roles in the catalytic activity of vertebrate GT6s. Sterically conservative substitutions of Asn and Gln, respectively, for residues corresponding to Asp191 or Glu192 in BoGT6a region F (Fig. 1) in bovine α3GT produce a major loss of catalytic activity (25, 26, 34). Crystallographic structures of bovine α3GT show that the Asp in region F interacts with the Arg in region B (also conserved in all GT6 glycosyltransferases) to stabilize an active site structure that allows both side chains to interact with hydroxyl groups of the galactosyl moiety of the donor substrate so that UDP-Gal is bound in an appropriate manner for catalysis (34). The glutamate was initially proposed to function as a catalytic nucleophile in α3GT that forms a covalent bond with the transferred sugar in a double displacement mechanism (7). Later studies have not supported this mechanism but indicate that this residue functions in transition state stabilization and in interacting with the acceptor substrate (26). Mutagenesis of the corresponding residue of BoGT6a, Glu192, to Gln results in a 10-fold greater reduction of kcat (22,000-fold) than that produced by the same substitution for the corresponding residue, Glu317, of α3GT (26), indicating that Glu192 is a key residue in catalysis in BoGT6a. Substitutions of Met and Gln for Ala155 (region E) were found to greatly reduce GalNAc transferase activity and significantly increase the low level of galactosyltransferase activity in BoGT6a. In the structurally characterized vertebrate GT6 enzymes, α3GT, and the histo-blood group glycosyltransferases, GTA and GTB, the residue corresponding to Ala 155 and the next 2 residues in the sequence form part of the binding site for the monosaccharide moiety of the donor substrate (9, 28), and substitutions for them can modify donor substrate specificity (27, 29). The size of the side chain at the site corresponding to Ala155 appears to determine if the 2-acetamido group of the GalNAc moiety of UDP-GalNAc can be accommodated (27). Although the BoGT6a mutants with substitutions for Ala155 have relatively low activities, their properties are consistent with a general similarity in structure specificity relationships between the prokaryotic and vertebrate GT6 representatives. Other bacterial glycosyltransferases have Ala, Met, or Gln at this site (supplemental Fig. S1), and it seems likely that Ala denotes GalNAc transferase activity, whereas Met and Gln are markers of Gal transferase activity.
The conservation of active site residues in BoGT6a (and other bacterial GT6 enzymes), as well as our mutational studies, suggests that it is similar in structure-function relationships to other well characterized mammalian members of the GT6 family. This suggests that some part of the protein may perform the role of the metal cofactor of the vertebrate enzymes in catalysis (3). Leukocyte type core 2 β1,6-GalNAc transferase, a GT-A fold enzyme of CAZy family 14, is metal-independent and is not significantly similar in sequence with metal-dependent GT-A fold glycosyltransferases. However, based on a comparison of its three-dimensional structure with those of metal-dependent GT-A fold glycosyltransferases, Rini and co-workers (35) have proposed that basic amino acids close to the C terminus might stabilize the nucleotide diphosphate leaving group in a manner similar to that of the metal ion in GT-A fold metal-dependent GTs (3, 35). The C-terminal regions of BoGT6a and other bacterial GT6s, unlike the rest of the sequences, show limited similarity to those of their vertebrate homologues or to the cyanophage protein (Fig. 1) and contain a high proportion of basic amino acids. Alanine scanning of these residues indicates that mutation of Lys231, which is conserved in all of the bacterial enzymes, produces a >200-fold reduction in kcat, whereas substitutions for the other residues have much smaller effects. The effect of this mutation is comparable with the 400-fold reduction in kcat produced by an Ala substitution for Lys359 of bovine α3GT (36), which could correspond to Lys231 of BoGT6a (Fig. 1). Therefore Lys231 of BoGT6a may function similarly to Lys359 of α3GT; however, the magnitude of the activity loss produced by Ala substitution is not comparable with the complete loss of activity in α3GT produced by removal of the metal cofactor. Therefore, the function of the metal cofactor may be replaced by some other structural component of BoGT6a, such as a helix macrodipole (37).
Steady state kinetic studies indicate that BoGT6a resembles vertebrate GT6s in utilizing a sequential mechanism (10), although whether substrate binding is ordered, as in the vertebrate enzymes, or random is not known; BoGT6a and vertebrate GT6 members also catalyze monosaccharide transfer to water (Table 1). These results show that the catalytic properties of BoGT6a and metal-dependent GT6s are closely similar. Although this may be contrary to expectation, it is not unique; for example, bacterial 3-deoxy-d-manno-octulosonate 8-phosphate synthases can be interconverted between obligate metal dependence and metal independence by substitution of Asn for a metal-binding Cys (38, 39).
In summary, B. ovatus GT6a is smaller than its vertebrate homologues and is metal-independent. It catalyzes the formation of blood group A-like structures that are expected to be part of the O-antigen on the bacterial surface that modulates interactions with human and other vertebrate hosts (40). Despite its differences from the vertebrate GT6 enzymes, it mirrors them in catalyzing a sequential mechanism, having low but significant UDP-sugar hydrolase activity, and in utilizing some equivalent amino acid residues from conserved regions of sequence in substrate binding and catalysis. A homology-based model for BoGT6a, based on the three-dimensional structure of bovine α3GT, was generated through the Swiss-Model Web site (41). The locations of residues subjected to mutagenesis are shown in this model in supplemental Fig. S3. Not surprisingly, the active site, identified by the residues that exert a major influence on function, has a location similar to those in structurally characterized mammalian homologues, whereas residues that have minor functional effects, including the C-terminal region (subsequent to Arg243, the C-terminal residue in the model), are distant from this putative active site. We conclude that in the GT6 family, metal dependence and metal independence are not associated with major changes in either active site structure or mechanism.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant AR40994.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- GT
- glycosyltransferase
- GT6
- CAZy family 6
- GTA and GTB
- human histo-blood group A and B synthase, respectively
- α3GT
- UDP-galactose, N-acetyllactosamine, α1,3-galactosyltransferase
- LacNAc
- N-acetyllactosamine.
REFERENCES
- 1.Unligil U. M., Rini J. M. (2000) Curr. Opin. Struct. Biol. 10, 510–517 [DOI] [PubMed] [Google Scholar]
- 2.Breton C., Snajdrová L., Jeanneau C., Koca J., Imberty A. (2006) Glycobiology 16, 29R–37R [DOI] [PubMed] [Google Scholar]
- 3.Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 4.Campbell J. A., Davies G. J., Bulone V., Henrissat B. (1997) Biochem. J. 326, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003) J. Mol. Biol. 328, 307–317 [DOI] [PubMed] [Google Scholar]
- 6.Turcot-Dubois A. L., Le Moullac-Vaidye B., Despiau S., Roubinet F., Bovin N., Le Pendu J., Blancher A. (2007) Glycobiology 17, 516–528 [DOI] [PubMed] [Google Scholar]
- 7.Gastinel L. N., Bignon C., Misra A. K., Hindsgaul O., Shaper J. H., Joziasse D. H. (2001) EMBO J. 20, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boix E., Swaminathan G. J., Zhang Y., Natesh R., Brew K., Acharya K. R. (2001) J. Biol. Chem. 276, 48608–48614 [DOI] [PubMed] [Google Scholar]
- 9.Patenaude S. I., Seto N. O., Borisova S. N., Szpacenko A., Marcus S. L., Palcic M. M., Evans S. V. (2002) Nat. Struct. Biol. 9, 685–690 [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y., Wang P. G., Brew K. (2001) J. Biol. Chem. 276, 11567–11574 [DOI] [PubMed] [Google Scholar]
- 11.Galili U., Shohet S. B., Kobrin E., Stults C. L., Macher B. A. (1988) J. Biol. Chem. 263, 17755–17762 [PubMed] [Google Scholar]
- 12.Xu H., Storch T., Yu M., Elliott S. P., Haslam D. B. (1999) J. Biol. Chem. 274, 29390–29398 [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto F., Clausen H., White T., Marken J., Hakomori S. (1990) Nature 345, 229–233 [DOI] [PubMed] [Google Scholar]
- 14.Keusch J. J., Manzella S. M., Nyame K. A., Cummings R. D., Baenziger J. U. (2000) J. Biol. Chem. 275, 25308–25314 [DOI] [PubMed] [Google Scholar]
- 15.Sullivan M. B., Coleman M. L., Weigele P., Rohwer F., Chisholm S. W. (2005) PLoS Biol. 3, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi W., Shao J., Zhu L., Li M., Singh M., Lu Y., Lin S., Li H., Ryu K., Shen J., Guo H., Yao Q., Bush C. A., Wang P. G. (2005) J. Am. Chem. Soc. 127, 2040–2041 [DOI] [PubMed] [Google Scholar]
- 17.Yi W., Shen J., Zhou G., Li J., Wang P. G. (2008) J. Am. Chem. Soc. 130, 14420–14421 [DOI] [PubMed] [Google Scholar]
- 18.Venter J. C., Remington K., Heidelberg J. F., Halpern A. L., Rusch D., Eisen J. A., Wu D., Paulsen I., Nelson K. E., Nelson W., Fouts D. E., Levy S., Knap A. H., Lomas M. W., Nealson K., White O., Peterson J., Hoffman J., Parsons R., Baden-Tillson H., Pfannkoch C., Rogers Y. H., Smith H. O. (2004) Science 304, 66–74 [DOI] [PubMed] [Google Scholar]
- 19.Yooseph S., Sutton G., Rusch D. B., Halpern A. L., Williamson S. J., Remington K., Eisen J. A., Heidelberg K. B., Manning G., Li W., Jaroszewski L., Cieplak P., Miller C. S., Li H., Mashiyama S. T., Joachimiak M. P., van Belle C., Chandonia J. M., Soergel D. A., Zhai Y., Natarajan K., Lee S., Raphael B. J., Bafna V., Friedman R., Brenner S. E., Godzik A., Eisenberg D., Dixon J. E., Taylor S. S., Strausberg R. L., Frazier M., Venter J. C. (2007) PLoS Biol. 5, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill S. R., Pop M., Deboy R. T., Eckburg P. B., Turnbaugh P. J., Samuel B. S., Gordon J. I., Relman D. A., Fraser-Liggett C. M., Nelson K. E. (2006) Science 312, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurokawa K., Itoh T., Kuwahara T., Oshima K., Toh H., Toyoda A., Takami H., Morita H., Sharma V. K., Srivastava T. P., Taylor T. D., Noguchi H., Mori H., Ogura Y., Ehrlich D. S., Itoh K., Takagi T., Sakaki Y., Hayashi T., Hattori M. (2007) DNA Res. 14, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henion T. R., Macher B. A., Anaraki F., Galili U. (1994) Glycobiology 4, 193–201 [DOI] [PubMed] [Google Scholar]
- 23.Persson M., Letts J. A., Hosseini-Maaf B., Borisova S. N., Palcic M. M., Evans S. V., Olsson M. L. (2007) J. Biol. Chem. 282, 9564–9570 [DOI] [PubMed] [Google Scholar]
- 24.Saitoh S., Noda S., Aiba Y., Takagi A., Sakamoto M., Benno Y., Koga Y. (2002) Clin. Diagn. Lab. Immunol. 9, 54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Deshpande A., Xie Z., Natesh R., Acharya K. R., Brew K. (2004) Glycobiology 14, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Swaminathan G. J., Deshpande A., Boix E., Natesh R., Xie Z., Acharya K. R., Brew K. (2003) Biochemistry 42, 13512–13521 [DOI] [PubMed] [Google Scholar]
- 27.Tumbale P., Jamaluddin H., Thiyagarajan N., Acharya K. R., Brew K. (2008) Glycobiology 18, 1036–1043 [DOI] [PubMed] [Google Scholar]
- 28.Heissigerova H., Breton C., Moravcova J., Imberty A. (2003) Glycobiology 13, 377–386 [DOI] [PubMed] [Google Scholar]
- 29.Seto N. O., Compston C. A., Evans S. V., Bundle D. R., Narang S. A., Palcic M. M. (1999) Eur. J. Biochem. 259, 770–775 [DOI] [PubMed] [Google Scholar]
- 30.Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., Gascuel O. (2008) Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozupone C. A., Hamady M., Cantarel B. L., Coutinho P. M., Henrissat B., Gordon J. I., Knight R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 15076–15081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hantke K. (2001) Curr. Opin. Microbiol. 4, 172–177 [DOI] [PubMed] [Google Scholar]
- 33.Rosch J. W., Gao G., Ridout G., Wang Y. D., Tuomanen E. I. (2009) Mol. Microbiol. 72, 12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tumbale P., Jamaluddin H., Thiyagarajan N., Brew K., Acharya K. R. (2008) Biochemistry 47, 8711–8718 [DOI] [PubMed] [Google Scholar]
- 35.Pak J. E., Arnoux P., Zhou S., Sivarajah P., Satkunarajah M., Xing X., Rini J. M. (2006) J. Biol. Chem. 281, 26693–26701 [DOI] [PubMed] [Google Scholar]
- 36.Jamaluddin H., Tumbale P., Withers S. G., Acharya K. R., Brew K. (2007) J. Mol. Biol. 369, 1270–1281 [DOI] [PubMed] [Google Scholar]
- 37.Hol W. G. (1985) Adv. Biophys. 19, 133–165 [DOI] [PubMed] [Google Scholar]
- 38.Shulami S., Furdui C., Adir N., Shoham Y., Anderson K. S., Baasov T. (2004) J. Biol. Chem. 279, 45110–45120 [DOI] [PubMed] [Google Scholar]
- 39.Kona F., Xu X., Martin P., Kuzmic P., Gatti D. L. (2007) Biochemistry 46, 4532–4544 [DOI] [PubMed] [Google Scholar]
- 40.Comstock L. E., Kasper D. L. (2006) Cell 126, 847–850 [DOI] [PubMed] [Google Scholar]
- 41.Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.