

Abstract
In the molecular oscillatory mechanism governing circadian rhythms, positive regulators, including CLOCK and BMAL1, transactivate Per and Cry genes through E-box elements, and translated PER and CRY proteins negatively regulate their own transactivation. Like BMAL1, its paralog BMAL2 dimerizes with CLOCK to activate the E-box-dependent transcription, but the role of BMAL2 in the circadian clockwork is still elusive. Here we characterized BMAL2 function in NIH3T3 cells and found that the cellular rhythms monitored by Bmal1 promoter-driven bioluminescence signals were blunted by RNA interference-mediated suppression of Bmal2 as well as that of Bmal1. Transcription assays with a 2.1-kb mPer1 promoter revealed that CRY2 inhibited the transactivation mediated by BMAL1-CLOCK more strongly than that by BMAL2-CLOCK. In contrast, PER2 showed a stronger inhibitory effect on BMAL2-CLOCK than on BMAL1-CLOCK. The molecular link between BMAL2 and PER2 was further strengthened by the fact that PER2 exhibited a greater affinity for BMAL2 than for BMAL1 in co-immunoprecipitation experiments. These results indicate a functional partnership between BMAL2 and PER2 and reemphasize the negative role of PER2 in the circadian transcription. As a broad spectrum function, BMAL2-CLOCK activated transcription from a variety of SV40-driven reporters harboring various E/E′-box-containing sequences present in the upstream regions of clock and clock-controlled genes. Importantly, the efficiencies of BMAL2-CLOCK-mediated transactivation relative to that achieved by BMAL1-CLOCK were dependent heavily on the E-box-containing sequences, supporting distinguishable roles of the two BMALs. Collectively, it is strongly suggested that BMAL2 plays an active role in the circadian transcription.
A variety of organisms from bacteria to humans show circadian rhythms in physiology and behavior under the regulation of endogenous circadian clocks oscillating with an ∼24-h periodicity (1, 2). In mammals, the central clock is located in the hypothalamic suprachiasmatic nucleus, whereas peripheral clocks with self-sustainable oscillation machinery are located in many peripheral tissues (3). Even cultured fibroblasts were shown to retain the cellular clocks (4), and hence they have been used for studies on the oscillatory mechanism of peripheral clocks. The molecular clockwork in mammals centers on transcription/translation-based autoregulatory feedback loops of clock genes, to which bHLH3 -PAS proteins, BMAL1 and CLOCK, contribute as positive regulators of the transcription (2, 5). BMAL1-CLOCK complex activates transcription through CACGTG-type E-box and its related sequences found in promoter regions of clock genes and clock-controlled genes such as Per1 (6), Per2 (CACGTT E-box-like or E′-box sequence; see Ref. 7), plasminogen activator inhibitor-1 (PAI-1) (8), and Rev-Erbα/β (9, 10). The E-box-dependent transactivation mediated by the BMAL1-CLOCK complex is suppressed by an expanding number of negative regulators, including PER1 (11, 12), PER2 (13–15), PER3 (16, 17), CRY1, and CRY2 (18, 19). Translated PER and CRY proteins interact with each other to enter into the nucleus (11, 20–23), where these negative regulators form a multimeric complex, interact with the BMAL1-CLOCK complex, and suppress their own transactivation through the E-boxes (24, 25). In this way, transcription levels of these negative regulators exhibit robust circadian rhythms, closing the core circadian molecular loop (reviewed in Refs. 2, 5, 26).
Mice deficient in both Per1 and Per2 exhibited impaired circadian rhythmicity in locomotor activity, indicating important roles of these Per genes in the circadian molecular loop (27, 28). Similarly, mice deficient in either Cry1 or Cry2 exhibit abnormal rhythms, and their double knock-out mice show complete loss of circadian rhythmicity (19). In terms of protein function as the negative regulator, PER proteins inhibit the E-box-mediated transactivation to a degree much weaker than CRY proteins do when assessed by transcription assays (18), and PER proteins are suggested to play a more important role(s) as translocators and/or regulators of CRY proteins (2, 30).
BMAL2 (also termed CLIF, see Ref. 8, or MOP9, see Ref. 31), a member of bHLH-PAS superfamily, was identified in several vertebrates such as zebrafish (32), human (8, 31, 33, 34), chicken (34), rat, and mouse (35). Like BMAL1, BMAL2 interacts with CLOCK (8, 32), binds to E-box (8, 34), and induces E-box-dependent transactivation (8, 31, 32, 34, 36, 37). This functional parallelism is supported by the phylogenetic relationship between the two Bmal genes that are both orthologous to the Drosophila cyc (Bmal) gene. It is predicted that Bmal1 and Bmal2 were generated by gene duplication probably at a stage of a common ancestor of the vertebrates (35). The circadian rhythm of the locomotor activity is completely abolished in Bmal1-deficient mice (38), although the two Bmal genes are both expressed in the mouse suprachiasmatic nucleus (31, 39). These observations indicate that Bmal2 does not compensate for Bmal1, but it has been left undetermined whether Bmal2 plays an active role in the mechanism of rhythm generation.
In this study, physiological importance of BMAL2 was investigated in the cellular clock system by monitoring the circadian rhythms of bioluminescence signals driven by 2.8-kb Bmal1 promoter. The results illustrate nonredundant essential roles of the two BMALs in the cellular clock system. Molecular characterization by transcription assays together with physical interaction profiles among the relevant proteins revealed an intimate functional linkage between BMAL2 and PER2 and reemphasizes their roles as a positive and a negative regulator, respectively, in the E-box-dependent feedback loop of the molecular clock.
EXPERIMENTAL PROCEDURES
Plasmids
The coding region of mBmal2a (GenBankTM accession number AY005163) or mBmal2b (GenBankTM accession number AY014836) was subcloned into pcDNA 3.1/V5-His expression vector (Invitrogen) so as to produce the protein without tags. For producing an N-terminally FLAG-tagged protein, the mBmal2a or mBmal2b cDNA was subcloned into a pcDNA 3.1/FLAG expression vector that was modified from pcDNA 3.1/V5-His (Invitrogen). The nucleotide sequences were verified by sequencing. The mBmal1b cDNA (mJAP3/pBlueScript SK+) was kindly provided by Dr. Y. Fujii-Kuriyama and subcloned into the pcDNA3.1/V5-His or the pcDNA3.1/FLAG expression vector to produce BMAL1b protein without tags or with an N-terminal FLAG tag, respectively. The mCLOCK/pcDNA3.1, mCRY1-HA/pcDNA3.1, and mCRY2-HA/pcDNA3.1 were kindly provided by Dr. S. M. Reppert and Dr. K. Kume. The mPER2/pcDNA3 was kindly provided by Dr. H. Okamura and Dr. K. Yagita, and an HA tag-encoding sequence was subcloned into mPER2/pcDNA3 for expression of C-terminally HA-tagged PER2 protein in mammalian cells. The mPer2us1.6kb/pGL3 basic construct was kindly provided by Dr. P. Sassone-Corsi (40). To generate the mPer1us2.1kb/pGL3 basic construct, a 2.1-kb promoter region of mPer1 gene was isolated as described in Ref. 6 and subcloned into the pGL3 basic plasmid. The mBmal1us2.8kb/pGL3 basic construct was kindly provided by Dr. S. L. McKnight (41). The mCry1 E′-box SV40-luc reporter was constructed as follows. Three copies of mCry1 E′-box (E-box like) element, AACGTG with its flanking sequences within the promoter/enhancer region of mouse Cry1 gene (5′-TTCAGAAACGTGAGGTGC-3′), were linked in tandem and inserted into an SV40-driven luciferase reporter (pGL3-Promoter vector, Promega) that had been digested by NheI and BglII. Similarly, the other reporters harboring three tandem-linked copies of various E/E′-box elements were also constructed. The inserted sequences were as follows: for mDbp E-box, 5′-CCTCGCCACGTGAGTCCG-3′; for mDec1 E-box, 5′-CCTCGCCACGTGAGTCCG-3′; for mDec2 E′-box, 5′-TCGCATCACGTTGCCGGC-3′; for M34 E-box, 5′-GGACACGTGACC-3′ (reported in Ref. 31); for hPAI-1 E-box (“proximal E-box” in Ref. 37): 5′-GACAATCACGTGGCTGGC-3′; for mRev-erbα E-box1: 5′-CGGGCCCACGTGCTGCAT-3′; for mRev-erbα E-box2: 5′-GGGTGCCACGTGCGAGGG-3′; for mRev-erbβ E-box1: 5′-ACTGGCCACGTGCACGGT-3′; and for mRev-erbβ E-box2: 5′-CGGAGACACGTGAGGCCG-3′.
Cell Culture and Luciferase Reporter Assays
Luciferase-reporter assays were performed in HEK293EBNA (HEK293) cells that were maintained in Dulbecco's modified Eagle's medium (Nissui) supplemented with 10% fetal bovine serum (Biowest), 100 units/ml penicillin, and 100 μg/ml streptomycin. Briefly, HEK293 cells plated in 12-well (Fig. 7) or 6-well plates (the other figures) were transfected with the indicated reporter constructs (20 ng/well in Fig. 7; 50 ng/well in the other figures) and a combination of various expression plasmids using Lipofectamine Plus (Invitrogen). Renilla luciferase reporter plasmid pRL-SV40 (5 ng/well in Fig. 7, Promega) or pRL-CMV (0.5 ng/well in the other figures, Promega) was used as an internal control for normalization of the transfection efficiency. The total amount of the transfected plasmids was adjusted at a constant level (described in each experiment) by the addition of empty vector. The transfected cells were harvested 36 h (in Fig. 7) or 24 h (in the other figures) after the transfection, and the cell extracts were subjected to dual-luciferase assays according to the manufacturer's protocol (Promega).
FIGURE 7.

E/E′-box sequence-dependent transcriptional regulation by BMAL1 and BMAL2. A–J, transactivation ability of BMAL1-CLOCK (indicated by open squares) or BMAL2-CLOCK (solid circles) was examined with a reporter driven either by mCry1 E′-box (A), mDec2 E′-box (B), hPAI-1 E-box (C), mRev-erbα E-box1 (D), mRev-erbα E-box2 (E), mRev-erbβ E-box1 (F), mRev-erbβ E-box2 (G), mDbp E-box (H), mDec1 E-box (I), or M34 E-box (J). HEK293 cells seeded in 12-well plates were co-transfected with 100 ng of CLOCK expression plasmid and various amounts (0, 5, 25, 50 ng) of BMAL1 or FLAG-BMAL2 expression plasmid. The total amount of the transfected plasmids was adjusted to 525 ng by the addition of empty vector. The luciferase activity of 50 ng of BMAL1 expression plasmid normalized to that of control sample that was transfected without BMAL1 expression plasmid was set to 1.0. The luciferase activities (before setting to 1.0) were 30.12, 40.42, 23.33, 57.44, 30.71, 56.79, 16.62, 25.82, 32.77, and 10.73 for mCry1 E′-box, mDec2 E′-box, hPAI-1 E-box, mRev-erbα E-box1, mRev-erbα E-box2, mRev-erbβ E-box1, mRev-erbβ E-box2, mDbp E-box, mDec1 E-box, and M34 E-box, respectively. Data are means ± S.E. from three independent experiments (Student's t test, #, p < 0.05; ##, p < 0.01; ###, p < 0.001). The asterisk in B indicates the data point from two independent experiments (mean ± variation). K, ratio of BMAL2-CLOCK-dependent transactivation to BMAL1-CLOCK-dependent one when 50 ng of BMAL1 or FLAG-BMAL2 expression plasmid was transfected. L, a model for molecular loops of negative feedback regulation with PER2 and CRY. Both BMAL1-CLOCK and BMAL2-CLOCK regulate transcription of clock and clock-controlled genes including Per and Cry genes through the E/E′-box sequences. In this model, CRYs and PER2 are postulated to inhibit predominantly BMAL1-CLOCK and BMAL2-CLOCK, respectively.
Bioluminescence Monitoring of Cellular Circadian Rhythms
NIH3T3 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. For bioluminescence monitoring experiments, 7.5 × 105 cells were plated in 35-mm dishes, cultured for 24 h, and then transfected with 0.5 μg of mBmal1us2.8kb/pGL3 basic plasmid using Lipofectamine Plus (Invitrogen) according to the manufacturer's protocol. For overexpression-perturbation experiments, the cells were co-transfected with the indicated amounts of various expression plasmids, keeping the total amount of transfected DNA at 1.0 μg with pcDNA3.1/V5-His vector. For RNAi experiments, the cells were co-transfected with 50 pmol of Stealth RNAi (Invitrogen) against mBmal1 or mBmal2, which was designed by using BLOCK-iT RNAi designer. Stealth RNAi against firefly luciferase (as a positive control) and Stealth RNAi negative control medium GC duplex (Invitrogen; as a negative control) were also used. Their sequences were as follows: for mBmal1 sense, GGAAA UCAUG GAAAU CCACA GGAUA, and antisense, UAUCC UGUGG AUUUC CAUGA UUUCC; for mBmal2 sense, GUCCU GCUCA AAGAA GAAAG ACCAU, and antisense, AUGGU CUUUC UUCUU UGAGC AGGAC; and for luciferase sense, GCACU CUGAU UGACA AAUAC GAUUU, and antisense, AAAUC GUAUU UGUCA AUCAG AGUGC. Twenty four hours after the transfection, the cells in every experiment were treated with 0.1 μm dexamethasone (Sigma) for 2 h, and then the medium was replaced by 2 ml of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 10 mm HEPES buffer, pH 7.2, 0.1 mm luciferin (Promega), 100 units/ml penicillin, and 100 μg/ml streptomycin. The cellular bioluminescence signals driven by the mBmal1 promoter activity were continuously recorded with dish-type luminometer Kronos AB-2500 (ATTO) for a week. The amounts of proteins expressed in the cells were determined by immunoblot analysis.
Immunoprecipitation
HEK293 cells plated in 6-well plates were transfected with a combination of constant amounts of FLAG-BMAL1, FLAG-BMAL2a, FLAG-BMAL2b, and PER2-HA expression plasmids (75, 750, 750, and 250 ng, respectively) and lysed with 100 μl of IPB2-H buffer (20 mm Tris-HCl, pH 8.0, 4 °C, 2 mm EDTA, 400 mm NaCl, 0.3% Triton X-100, 5% glycerol, 1 mm dithiothreitol, 4 μg/ml aprotinin, 4 μg/ml leupeptin, 5 mm NaF, and 1 mm Na3VO4). The cellular lysate was centrifuged at 12,000 × g for 10 min, and the supernatant was diluted with 2 volumes of IPB2-L buffer (20 mm Tris-HCl, pH 8.0, 4 °C, 2 mm EDTA, 5.5 mm NaCl, 1.35% Triton X-100, 12.5% glycerol, 1 mm dithiothreitol, 4 μg/ml aprotinin, 4 μg/ml leupeptin, 72.5 mm NaF, and 1 mm Na3VO4). The diluted lysates containing constant amounts of proteins (120 μg) were incubated with 1 μg of anti-HA antibody overnight at 4 °C and then incubated additionally for 1 h after mixing with 10 μl of 50% slurry of protein G-Sepharose beads (Amersham Biosciences). The beads were washed three times with IPB2 buffer (20 mm Tris-HCl, pH 8.0, 4 °C, 2 mm EDTA, 137 mm NaCl, 1% Triton X-100, 10% glycerol, 1 mm dithiothreitol, 4 μg/ml aprotinin, 4 μg/ml leupeptin, 50 mm NaF, and 1 mm Na3VO4), and the bound proteins were immunoblotted with anti-FLAG and anti-HA antibodies.
Immunoblot Analysis
Relative levels of exogenously expressed FLAG-BMAL1 and FLAG-BMAL2 proteins were estimated by immunoblot analysis with anti-FLAG antibody (1:200, M2 monoclonal antibody, Sigma). HA-tagged CRY1, CRY2, and PER2 were detected by using anti-HA antibody (1:2,000, 12CA5 monoclonal antibody, Roche Applied Science). Horseradish peroxidase-conjugated anti-mouse IgG (0.2 μg/ml; Kirkegaard & Perry Laboratories) antiserum was used as the secondary antibody.
RESULTS
Transactivation of Period Promoters by BMAL1-CLOCK and BMAL2-CLOCK
We first asked whether BMAL2 contributes to transcriptional activation of mPer1 and mPer2 genes by examining regulation of luciferase reporters driven by 2.1-kb mPer1 promoter (Fig. 1A) and 1.6-kb mPer2 promoter (Fig. 1B) in HEK293 cells. When BMAL or CLOCK was expressed individually in HEK293 cells, no significant increase of the transcriptional activity was observed (Fig. 1, A and B, 1st to 4th bars), indicating that endogenous levels of BMALs and CLOCK proteins do not contribute to the transactivation. The reporter activities of both mPer1 and mPer2 promoters were stimulated with increasing amounts of not only mouse BMAL1 but also mouse BMAL2 expression plasmid in combination with a fixed amount of CLOCK expression plasmid, indicating that BMAL2 can substitute for BMAL1 as a positive regulator for E/E′-box-mediated transcriptional activation. With increasing BMALs, the maximal level of the transcriptional activation of the mPer1 promoter by BMAL1-CLOCK was 1.6-fold higher than that caused by BMAL2-CLOCK (Fig. 1A). Similarly, BMAL1-CLOCK-dependent transactivation of the mPer2 promoter was maximally 1.6-fold higher than BMAL2-CLOCK-dependent one (Fig. 1B), suggesting a difference in molecular properties between the two BMALs. Higher doses of either BMAL1 or BMAL2 expression plasmid caused slight reduction of transactivation level of the mPer1 promoter as observed for chicken BMAL2 (34).
FIGURE 1.

BMAL2 transactivates mPer1 and mPer2 promoters with CLOCK. Transactivation ability of BMAL1-CLOCK or BMAL2-CLOCK was examined with a reporter driven by 2.1-kb mPer1 promoter (A) or 1.6-kb mPer2 promoter (B). HEK293 cells were co-transfected with 250 ng of CLOCK expression plasmid and various amounts (0, 10, 20, 50, 100, and 250 ng) of BMAL1 or BMAL2 expression plasmid. The total amount of the transfected plasmids was adjusted to 1.0 μg by the addition of empty vector. The value of the control experiment in which pcDNA3.1/V5-His empty vector and the reporter vector were transfected was set to 1.0. Data are means ± S.E. from three independent experiments.
We previously found that a short variant of BMAL2 is expressed in the mouse hypothalamus, including the suprachiasmatic nucleus (35). The short isoform named BMAL2b (35) has an N-terminal third part of full-length BMAL2 (BMAL2a) and retains bHLH and PAS-A domains but lacks PAS-B domain (supplemental Fig. 1A). BMAL2b did not transactivate the Per1 promoter in combination with CLOCK, although it weakly inhibited the transactivation mediated by BMAL1-CLOCK (supplemental Fig. 1B). In this study, BMAL2 denotes the long isoform, BMAL2a.
Cellular Bioluminescence Rhythms Perturbed by RNAi-mediated Suppression of Bmal2
We then investigated the vulnerability of the cellular clock mechanism, especially toward the perturbation in BMAL2 level in NIH3T3 cells. To this end, we employed real time monitoring of the cellular bioluminescence rhythms generated by 2.8-kb Bmal1 promoter-driven luciferase activity. Basic properties of the bioluminescence rhythm were almost unaffected by overexpression of BMAL1 (Fig. 2, B and C) or BMAL2 (Fig. 2, E and F) under the condition where exogenously expressed BMAL protein levels overwhelmed endogenous BMALs (supplemental Fig. 2). On the other hand, overexpression of the negative regulator, PER2 (Fig. 2, H and I), CRY1 (J and K), or CRY2 (L and M), impaired the cellular rhythms very severely.
FIGURE 2.

Overexpression of PER2, CRY1, or CRY2 blunted cellular rhythms monitored by Bmal1 promoter-driven luciferase activity. The effects of overexpression of clock genes on the cellular clock were examined in NIH3T3 cells by real time monitoring of the bioluminescence signals derived from transiently transfected Bmal1us2.8kb/pGL3 basic plasmid. The reporter plasmid was co-transfected with 0, 0.25, or 1.0 μg of BMAL1 (A–C, respectively), BMAL2 (D–F, respectively), PER2-HA (G–I, respectively), CRY1-HA (G, J, and K, respectively), or CRY2-HA (G, L, and M, respectively) expression plasmid. The exogenous expression of BMAL1 and BMAL2 was confirmed by immunoblot analysis in parallel (see supplemental Fig. 2).
In contrast to the overexpression experiments, RNA interference-mediated knockdown of BMAL1 or BMAL2 significantly affected the Bmal1 promoter-driven bioluminescence rhythm. First, the knockdown efficiency was evaluated in HEK293 cells, in which transfection of siRNA against Bmal1 reduced the protein level of the exogenously expressed BMAL1 down to ∼38% of the control, leaving the BMAL2 level unaffected (Fig. 3A, left). On the other hand, transfection of siRNA against Bmal2 reduced the exogenously expressed BMAL2 protein level down to ∼31% of the control, while having no measurable effect on BMAL1 protein level (Fig. 3A, right). Then we examined the effect of the siRNA on the bioluminescent rhythms recorded from NIH3T3 cells, in which Bmal1 and Bmal2 are intrinsically transcribed (supplemental Fig. 3). The siRNA against Bmal1 blunted the circadian rhythmicity of NIH3T3 cells in all the six experiments (Fig. 3, B–G). These results demonstrate an essential role of BMAL1 in the circadian oscillation of the cellular clock and come into line with the arrhythmic behavior of Bmal1-deficient mice observed in constant darkness (38). Importantly, the siRNA against Bmal2 blunted the bioluminescence rhythms in four of six independent sets of experiments (Fig. 3, B–E), although diminished rhythmicities remained in the other two cases (Fig. 3, F and G). Thus, the siRNA against Bmal1 and Bmal2 had discernible effects on the cellular circadian rhythms of NIH3T3 cells, possibly because of their distinct action points on the clockwork (see “Discussion”). These results indicate that the two BMALs do not compensate for each other in the cellular clockwork and suggest that the point(s) affected by Bmal1 suppression is more central to the core oscillatory machinery than that by Bmal2 suppression.
FIGURE 3.

RNAi-mediated BMAL1 or BMAL2 deficiency blunted cellular rhythms monitored by Bmal1 promoter-driven luciferase activity. A, HEK293 cells seeded in 24-well plates were transfected with FLAG-tagged BMAL1 (10 ng) or BMAL2 (100 ng) expression plasmids in combination with 100 ng of CLOCK expression plasmid and 20 pmol of siRNA against Bmal1 or Bmal2. The cells were harvested 24 h after the transfection, and the cell extracts were subjected to immunoblotting with anti-FLAG antibody. The mean of the band intensities of samples from nontransfected cells is set to zero to show the protein levels of exogenously expressed FLAG-BMAL1 or FLAG-BMAL2. The mean of the normalized band intensities of samples from cells transfected with nonspecific oligonucleotide sequence (control siRNA) is set to 1. Data are means ± S.E. from three independent experiments (Student's t test, #, p < 0.05; ###, p < 0.001). WB, Western blot. B–G, effect of the RNAi-mediated suppression of BMAL1 or BMAL2 on the cellular bioluminescence rhythm was examined in NIH3T3 cells, which were transiently transfected with Bmal1us2.8kb/pGL3 basic together with 50 pmol of siRNA against Bmal1 (n = 6) or Bmal2 (n = 6). Co-transfection of siRNA with control siRNA (n = 6) had no effect on the cellular circadian rhythm. siRNA against luciferase (n = 4) reduced the luciferase activity (in yellow) down to 2–5% level of the maximal signals observed in cells transfected with control siRNA. Even in this case, we observed an extremely low level of the circadian oscillation of the bioluminescence signals.
In the knockdown experiments, we noticed that the peak level of the bioluminescence signals from the Bmal2-suppressed cells (red curves in Fig. 3, B–G) was consistently higher (1.3–2.5-fold) than that of the Bmal1-suppressed cells (blue curves) within a single set of experiments (p < 0.05, n = 6). This observation further emphasizes the unique mode of BMAL2 action that is distinguishable from that of BMAL1 action in the clockwork. Here we recognized that the profiles of the bioluminescence signals recorded from the BMAL2-suppressed cells (red curves in Fig. 3, B–G) resemble those of PER2-overexpressing cells (Fig. 2, H and I) in terms of not only the temporal profile of the bioluminescence signals but also the increased maximal intensity. The maximal levels of the bioluminescence signals observed in PER2-overexpressing cells were 1.2-1.8-fold higher than that in the control cells (p < 0.05, n = 3; Fig. 2, compare G with H). The similarity in the waveform between the BMAL2-suppressed cells and PER2-overexpressing cells implies an intimate functional linkage between BMAL2 and PER2 in the network of circadian feedback loops.
BMAL2 Is Less Sensitive than BMAL1 to Repression by CRYs but More Sensitive to PER2
A potential difference between BMAL1 and BMAL2 was examined by comparing their sensitivities to CRY-mediated negative regulation of the E-box-dependent transactivation in the presence of CLOCK (Fig. 4). In HEK293 cells, transfection of the same amount of BMAL1 or BMAL2 expression plasmid yielded largely diverged levels in expression of the two BMAL proteins (supplemental Fig. 4). Accordingly, we evaluated the inhibitory effects of CRYs on BMAL1 or BMAL2 under two controlled conditions; BMAL1 and BMAL2 were expressed either at similar protein levels or at protein levels delivering comparable transcriptional activities.
FIGURE 4.

CRY1 and CRY2 repress transactivation of the mPer1 promoter by BMAL1-CLOCK more effectively than that by BMAL2-CLOCK. A, HEK293 cells were co-transfected with combinations of constant amounts of FLAG-BMAL1, FLAG-BMAL2, and/or CLOCK expression plasmids (25, 250, and 250 ng, respectively) and various amounts of CRY1-HA or CRY2-HA expression plasmid (0, 0.4, 2, 10, 50, and 250 ng). The expression levels of FLAG- or HA-tagged proteins of BMAL1, BMAL2, CRY1, and CRY2 were examined by immunoblot analysis with anti-FLAG and anti-HA antibodies. The asterisks indicate nonspecific bands. Relative luciferase activities were shown by mean fold increases from the value of the control sample that was co-transfected with the 2.1-kb mPer1 promoter plasmid and pcDNA3.1/V5-His empty vector, and plotted against the amount of CRY1-HA (B) or CRY2-HA expression plasmid (C). D and E, similarly, HEK293 cells were co-transfected with combinations of constant amounts of FLAG-BMAL1, FLAG-BMAL2, and/or CLOCK expression plasmids (4, 50, and 250 ng, respectively) with various amounts of CRY1-HA or CRY2-HA expression plasmid (0, 0.4, 2, 10, 50, and 250 ng). The total amount of the transfected plasmids was adjusted to 1.0 μg by the addition of empty vector. Data are means ± S.E. from three independent experiments (Student's t test, #, p < 0.05; ##, p < 0.01; ###, p < 0.001).
For the first condition, we managed to set the two BMAL levels similar to each other by transfecting the cells with either 25 ng of FLAG-BMAL1 expression plasmid or 250 ng of FLAG-BMAL2 expression plasmid (Fig. 4A and supplemental Fig. 5). With these fixed combinations of co-expressed CLOCK and BMALs, co-transfection of increasing amounts of CRY1 expression plasmid dose-dependently inhibited the transactivation (Fig. 4B). A comparison of the doses of CRY1 expression plasmid inducing the half semi-maximal inhibition demonstrates that the transactivation by BMAL1-CLOCK was more sensitive to the CRY1 action than that by BMAL2-CLOCK (Fig. 4B). A similar or more evident difference between the two BMALs was observed when CRY1 was replaced by CRY2 (Fig. 4C). In the second condition, comparable levels of transactivation of the mPer1 promoter were achieved by transfecting the cells with either 4 ng of FLAG-BMAL1 or 50 ng of FLAG-BMAL2 expression plasmid. Under this condition, it was again demonstrated that BMAL1-CLOCK was more sensitive than BMAL2-CLOCK to the inhibitory action by CRY2 (Fig. 4E), and CRY1 also showed a similar tendency toward the two BMALs (Fig. 4D). The results obtained in these two sets of transcription assays suggest that BMAL2 serves as a less sensitive target of CRY2-mediated negative regulation when compared with BMAL1. The divergent sensitivities to the inhibitory action of CRY2 were not because of the difference in protein abundance between the two BMALs (Fig. 4C) but were attributable to their molecular properties.
Based on the implication for functional linkage between BMAL2 and PER2 (as described earlier for Figs. 2 and 3), we examined BMAL2-mediated transactivation for sensitivity to negative regulation by PER2. In contrast to the dull inhibitory effect of CRY2, transfection of increasing amounts of PER2 expression plasmid sharply inhibited BMAL2-CLOCK-dependent transactivation, whereas it had a modest effect on BMAL1-dependent activation (Fig. 5B) under the condition of the two BMALs being expressed at equivalent protein levels (Fig. 5A). A typical difference between the two BMALs was observed at a lower dose of co-transfected Per2 plasmid (10 ng), which caused ∼30% repression of the transcription mediated by BMAL2-CLOCK, whereas the same dose caused no inhibition on BMAL1-CLOCK-dependent transactivation (Fig. 5B). In a parallel experiment, BMAL1 and BMAL2 levels were adjusted to deliver comparable transactivation, and we again observed a remarkably higher sensitivity of BMAL2 than BMAL1 to PER2-mediated suppression of transactivation (Fig. 5C).
FIGURE 5.

PER2 represses transactivation of the mPer1 promoter by BMAL2-CLOCK more effectively than that by BMAL1-CLOCK. A, HEK293 cells were co-transfected with combinations of constant amounts of FLAG-BMAL1, FLAG-BMAL2, and CLOCK expression plasmids (25, 250, and 250 ng, respectively) and various amounts of PER2-HA expression plasmid (0, 10, 50, and 250 ng). The expressed levels of FLAG- or HA-tagged proteins of BMAL1, BMAL2, and PER2 were examined by immunoblot analysis with anti-FLAG and anti-HA antibodies. The asterisks indicate nonspecific bands. Relative luciferase activities were shown by mean fold increases from the value of the control sample that was co-transfected with 2.1-kb mPer1 promoter plasmid and pcDNA3.1/V5-His empty vector and plotted against the amount of PER2-HA expression plasmid (B). C, similarly, HEK293 cells were co-transfected with combinations of constant amounts of FLAG-BMAL1, FLAG-BMAL2, and/or CLOCK expression plasmids (4, 50, and 250 ng, respectively) and various amounts of PER2-HA expression plasmid (0, 10, 50, and 250 ng). The total amount of the transfected plasmids was adjusted to 1.0 μg by the addition of empty vector. Data are means ± S.E. from three independent experiments (Student's t test, #, p < 0.05; ##, p < 0.01; ###, p < 0.001).
Physical Interaction of PER2 with BMAL2 Is Stronger than That with BMAL1
PER2 is known to interact with BMAL1 (42, 43), whereas the intimate functional linkage between PER2 and BMAL2 (Fig. 5) suggested that PER2 may associate with BMAL2 more efficiently than with BMAL1. We performed co-immunoprecipitation assay in HEK293 cells, in which PER2-HA was co-expressed with either FLAG-BMAL1 or FLAG-BMAL2. For semi-quantitative analysis, the two BMALs were expressed at protein levels similar to each other. Then we found that FLAG-BMAL2 associated with PER2-HA was 3.6-fold larger in amount than FLAG-BMAL1 with PER2-HA (Fig. 6), in the presence of comparable amounts of PER2 in the precipitates (data not shown). These results strongly suggest that PER2 has a greater affinity for BMAL2 than for BMAL1, and this property of BMAL2 appears to support its higher sensitivity to PER2 action when compared with BMAL1-mediated transactivation.
FIGURE 6.
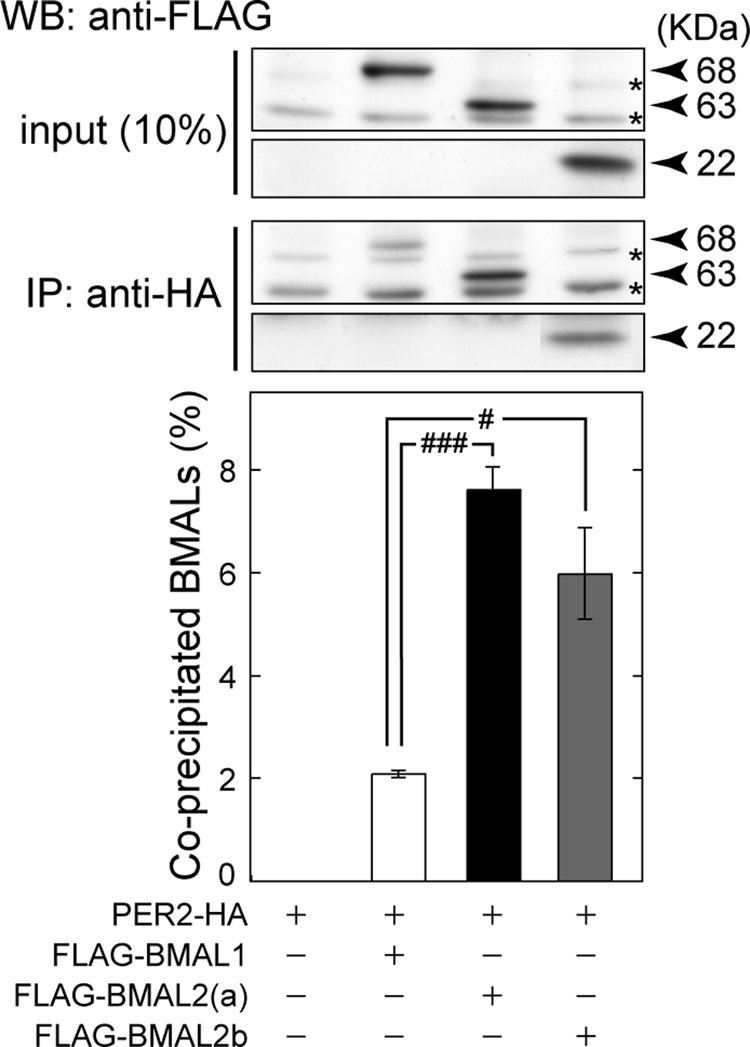
Co-immunoprecipitation of BMAL with PER2 protein. HEK293 cells were transfected with PER2-HA expression plasmids (250 ng) together with FLAG-tagged BMAL1 (75 ng), BMAL2a (750 ng), or BMAL2b (750 ng) expression plasmids to yield the same protein levels among the three BMALs. The total amount of the transfected plasmids was adjusted to 1.0 μg by the addition of empty vector. PER2-HA protein in each cell lysate was immunoprecipitated (IP) with anti-HA antibody and was subjected to immunoblotting with anti-FLAG antibody. WB, Western blot. The asterisks indicate nonspecific bands. The band intensities of the precipitated FLAG immunoreactivities were quantified, and the value relative to the total amount of FLAG-BMALs was shown. Data are means ± S.E. from three independent experiments (Student's t test, #, p < 0.05; ###, p < 0.001).
In addition to full-length BMALs, the short variant BMAL2b was tested for its ability to interact with PER2. FLAG-BMAL2b also associated with PER2 (Fig. 6), and the amount of precipitated BMAL2b was significantly higher than that of BMAL1 and comparable with that of BMAL2a. This observation falls into line with the previous finding by Langmesser et al. (43) that PER2 can interact with a truncated form of BMAL1 (amino acids 1–278 of mBMAL1b isoform) lacking the C-terminal half (including PAS-B domain). Consistently, the bHLH and PAS-A domains of BMAL2 share 60 and 73% amino acid identities with those of BMAL1, respectively. Our observation with BMAL2b narrows down the PER2-interacting domain of BMAL2a to amino acids 1–198.
BMAL2-CLOCK Exhibits a Distinct Preference for E/E′-box-containing Sequence
To address the functional difference between BMAL1 and BMAL2 in the circadian oscillatory system, we then examined BMAL2-CLOCK-dependent transcriptional regulation via E/E′-box of potential target genes by performing transcription assays using reporter constructs containing a short stretch of various E/E′-box elements with their flanking sequences clipped from the clock or clock-controlled genes (Fig. 7, A–J, solid symbols). The transactivation mediated by BMAL2 and CLOCK was compared with that achieved by BMAL1 and CLOCK (Fig. 7, A–J, open symbols). In all the cases with the 10 constructs, the reporter activities were significantly up-regulated by increasing the amounts of the two BMALs in combination with CLOCK expression, suggesting that these E/E′-box elements serve as the target of the two BMALs. Importantly, the ratio of BMAL2-CLOCK-mediated transactivation to BMAL1-CLOCK-mediated one varied remarkably among the E/E′-box-containing sequences (Fig. 7K), even though the transcription levels activated by BMAL2-CLOCK were consistently lower than that activated by BMAL1-CLOCK in HEK293 cells (Fig. 7, A–J, see “Discussion”). In the transcription assays with mCry1 E′-box, mDec2 E′-box, and hPAI-1 E-box, the ratios were also less than 0.5 (0.44 ± 0.03, 0.44 ± 0.02, and 0.46 ± 0.02, respectively). In Hep3B cells, it was reported that the M34 element (described as M34 E-box in this paper)-mediated transactivation achieved by BMAL2-CLOCK was higher than that achieved by BMAL1-CLOCK (31), whereas in our experiments the transcription level achieved by BMAL2-CLOCK was 0.78-fold that caused by BMAL1-CLOCK via M34 E-box in HEK293 cells (Fig. 7J). In the transcription assays using mRev-erbα E-box1, mRev-erbα E-box2, and mDec1 E-box constructs, the ratios of BMAL2-CLOCK over BMAL1-CLOCK transactivation were comparable with or greater than that of M34 E-box (0.71 ± 0.04, 0.82 ± 0.02, and 0.70 ± 0.02, respectively). The ratios of BMAL2-CLOCK over BMAL1-CLOCK activation of mRev-erbβ E-box1, mRev-erbβ E-box2, and mDbp E-box were also greater than 0.5 (0.58 ± 0.03, 0.60 ± 0.02, and 0.61 ± 0.02, respectively). Together, our data demonstrate similar but obviously distinct roles for BMAL1 and BMAL2 in the molecular oscillatory system.
DISCUSSION
This study aimed at clarifying any functional difference between BMAL1 and BMAL2 in the circadian clockwork. In real time reporter systems of the cellular rhythms, the bioluminescence rhythm in NIH3T3 cells was abolished reproducibly by siRNA-mediated Bmal1 suppression (Fig. 3), and this result is compatible with the complete loss of circadian rhythmicity of wheel-running activities in Bmal1-deficient mice (38). Our observation confirms the essential role of BMAL1 for circadian rhythm generation. On the other hand, because of the lack of information about Bmal2-deficient mice, it is of great importance to evaluate the effect of Bmal2 knockdown on perturbation of the circadian clockwork in NIH3T3 cells (Fig. 3). It should be emphasized that RNAi-mediated suppression of Bmal2 abolished the cellular bioluminescence rhythms, albeit with exceptions (see below). The observations together indicate nonredundant essential roles of the two BMALs in the cellular clockwork.
As a possible mechanism for the nonredundancy, one may speculate that BMAL1 and BMAL2 are included in a single multimeric protein complex responsible for the E-box-dependent transactivation. Then the two BMALs could both be indispensable for the circadian oscillation of the molecular clock. This idea, however, disagrees readily with the efficient transactivation of the Per promoters even in the presence of one BMAL with CLOCK in HEK293 cells (Fig. 1). The observed nonredundancy can be explained by an alternative mechanism, in which the two BMALs contribute independently to the clockwork. As suggested from the studies on various E/E′-boxes (Fig. 7), BMAL1 and BMAL2 may activate their preferential sets of downstream clock-controlled genes, which possibly include those encoding unique regulators of core components of the molecular clock. If we postulate that the target genes of BMAL1 are more central to the core oscillatory mechanism than those of BMAL2, then we can easily understand the basis for the different sensitivities of the cellular clock to Bmal1 and Bmal2 knockdown (Fig. 3). Importantly, CRY2 inhibited transactivation of the Per1 promoter mediated by BMAL1-CLOCK more efficiently than that by BMAL2-CLOCK (Fig. 4), whereas PER2 showed a stronger inhibitory effect on BMAL2-CLOCK than on BMAL1-CLOCK (Fig. 5). Thus, in HEK293 cells, BMAL1- and BMAL2-induced transactivation of the Per1 promoter appears to be regulated differently by these negative regulators. The preferential inhibition by PER2 and CRY2 toward the two BMALs supports a model that BMAL1-CLOCK and BMAL2-CLOCK may play distinguishable roles, both of which are essential for maintaining normal oscillation of the cellular clock (Fig. 7L).
In the model, we postulated that distinct regulatory mechanisms are operative on each E/E′-box sequence. Some clock(-controlled) genes are mainly regulated by BMAL2-CLOCK and PER2, whereas others are regulated by BMAL1-CLOCK and CRYs (Fig. 7L). Indeed, the ratios of transactivation mediated by BMAL2-CLOCK to that by BMAL1-CLOCK were different from each other among the E-box-containing sequences (Fig. 7, A–K). Particularly, BMAL2-CLOCK-mediated transactivation through CACGTT-type E′-box sequences, mCry1 E′-box and mDec2 E′-box, were both far less effective (∼45%, Fig. 7, A, B and K) than that mediated by BMAL1-CLOCK, suggesting minor contribution of BMAL2 to the E′-box-dependent transactivation. These observations are likely indicative of a possible functional difference between BMAL1 and BMAL2 in regulation of E/E′-box-dependent circadian gene transcription. Also, the transactivation ratio of BMAL2-CLOCK to BMAL1-CLOCK appears strongly dependent on the cell types. In our transcription assays using HEK293 cells, the maximal level of transactivation of 2.1-kb mPer1 promoter or 1.6-kb mPer2 promoter mediated by BMAL1-CLOCK was both 1.6-fold higher than that achieved by BMAL2-CLOCK (Fig. 1, A and B). In bovine aortic endothelial cells, on the other hand, BMAL2-CLOCK transactivated the 2.0-kb mPer1 promoter more effectively than BMAL1-CLOCK did (37). In the endothelial cells, the transcription level of the PAI-1 promoter activated by BMAL2-CLOCK was remarkably higher than that by BMAL1-CLOCK when either of the two BMAL proteins was expressed at a level comparable with each other in the transcription assay. In contrast, activation of hPAI-1 E-box by BMAL1-CLOCK was 2-fold stronger than that by BMAL2-CLOCK in HEK293 cells (Fig. 7, C and K). It is probable that BMAL1 and BMAL2 not only bind to various promoters with different affinities but also transactivate them by recruiting different sets of co-activators in a manner depending on the cell types and the promoter sequences. The complexity of the functions of BMALs has been further highlighted by the presence of multiple isoforms (for each BMAL) that show different transactivation abilities (36). In this study, we found that BMAL2b, an isoform that is truncated largely at the C-terminal part of full-length BMAL2, showed no transactivation ability with CLOCK but weakly inhibited BMAL1-CLOCK-mediated transactivation (supplemental Fig. 1). It is possible to speculate that BMAL-dependent transcriptional regulation may be fine-tuned by combinatorial actions of the isoforms.
We propose a model for the roles of BMAL1 and BMAL2 (Fig. 7L), in which the lower degree of the rhythm disturbance in Bmal2-suppressed cells than in Bmal1-suppressed cells (Fig. 3) may be ascribed to a difference in relative importance of the clock regulators that are driven uniquely by BMAL1 or BMAL2. In our transcription assays, BMAL2-CLOCK weakly transactivated the E′-box sequence found in the promoter regions of core clock genes such as mCry1. The lower efficiency in transactivation mediated by BMAL2 for the core clock genes is compatible with the fact that BMAL2 cannot compensate for BMAL1 function in Bmal1-deficient mice (38). It is notable that BMAL2-CLOCK caused relatively efficient activation of the E-box-containing sequences derived from mRev-erbα/β, mDbp, and mDec1 (Fig. 7, E–K), suggesting that BMAL2 plays a more important role in a regulatory loop than that in the core loop. Bmal2 might be an important gene as well for clock resetting, like Dec1 (29).
Interestingly, the Bmal1 promoter activity was up-regulated more efficiently by RNAi-mediated BMAL2 suppression than by BMAL1 suppression (Fig. 3), suggesting that BMAL2-CLOCK activates the inhibitory pathway of Bmal1 expression. This pathway is thought to involve REV-ERBα/β acting as BMAL2 downstream regulators for several reasons. (i) REV-ERBα/β negatively regulate Bmal1 expression through RORE sequences found in Bmal1 promoter (9). (ii) Promoter regions of Rev-erbα/β possess E-box sequences that are important for circadian oscillation (10). (iii) BMAL2 shows efficient transactivation through E-box-containing sequences that are found in promoter region of Rev-erbα/β genes (Fig. 7, E–H). These lines of evidence raise a putative transcriptional circuit from BMAL2 to BMAL1 through REV-ERBα/β. As discussed above, BMAL2 is a preferable target of negative regulation by PER2, and therefore it is possible that the expression of REV-ERBα/β may be inhibited by PER2 as well as RNAi-mediated suppression of BMAL2. In fact, overexpression of PER2 causes up-regulation of the Bmal1 promoter-mediated bioluminescence (Fig. 2, H and I) as seen in suppression of BMAL2 (Fig. 3). Conversely, Bmal1 expression is down-regulated in Per2-deficient mice (39), in which E-box-dependent transactivation of Rev-erbα/β is likely up-regulated to inhibit the Bmal1 promoter activity. Detailed analysis of Rev-erbα/β promoters should provide a concrete illustration of this hypothetical circuit.
It has been considered that PER2 serves as the negative regulator for E-box-dependent transactivation, although it shows a weaker inhibition of the transactivation when compared with CRYs (30). This study provides experimental evidence supporting a more potent role of PER2 in regulation of BMAL2-CLOCK-mediated transactivation. Our model of the preferable regulation mechanism shared by PER2-BMAL2 and CRY-BMAL1 (Fig. 7L) reemphasizes the physiological importance of BMAL2 and PER2, because the model assumes their roles that do not fully overlap with BMAL1 and CRY proteins, respectively. To understand the clock oscillation mechanism to which BMAL2 contributes, it should be important to determine the binding specificities and preferences of the two BMALs to nucleotide sequences encompassing E/E′-box elements. A genome-wide analysis would also reveal temporal and spatial variations of the molecular structures among the negative complexes formed on a variety of E-box and E-box-like elements.
Supplementary Material
Acknowledgments
We thank Nobuhiro Kurabayashi for helpful discussions.
This work was supported in part by grants-in-aid for scientific research and by Global COE Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5.
- bHLH
- basic helix-loop-helix
- PAS
- Period-Aryl hydrocarbon receptor nuclear translocator-Single minded
- HA
- hemagglutinin
- siRNA
- small interfering RNA
- RNAi
- RNA interference.
REFERENCES
- 1.Pittendrigh C. S. (1993) Annu. Rev. Physiol. 55, 16–54 [DOI] [PubMed] [Google Scholar]
- 2.Reppert S. M., Weaver D. R. (2002) Nature 418, 935–941 [DOI] [PubMed] [Google Scholar]
- 3.Yamazaki S., Numano R., Abe M., Hida A., Takahashi R., Ueda M., Block G. D., Sakaki Y., Menaker M., Tei H. (2000) Science 288, 682–685 [DOI] [PubMed] [Google Scholar]
- 4.Balsalobre A., Damiola F., Schibler U. (1998) Cell 93, 929–937 [DOI] [PubMed] [Google Scholar]
- 5.King D. P., Takahashi J. S. (2000) Annu. Rev. Neurosci. 23, 713–742 [DOI] [PubMed] [Google Scholar]
- 6.Gekakis N., Staknis D., Nguyen H. B., Davis F. C., Wilsbacher L. D., King D. P., Takahashi J. S., Weitz C. J. (1998) Science 280, 1564–1569 [DOI] [PubMed] [Google Scholar]
- 7.Yoo S. H., Ko C. H., Lowrey P. L., Buhr E. D., Song E. J., Chang S., Yoo O. J., Yamazaki S., Lee C., Takahashi J. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maemura K., de la Monte S. M., Chin M. T., Layne M. D., Hsieh C. M., Yet S. F., Perrella M. A., Lee M. E. (2000) J. Biol. Chem. 275, 36847–36851 [DOI] [PubMed] [Google Scholar]
- 9.Preitner N., Damiola F., Lopez-Molina L., Zakany J., Duboule D., Albrecht U., Schibler U. (2002) Cell 110, 251–260 [DOI] [PubMed] [Google Scholar]
- 10.Ueda H. R., Hayashi S., Chen W., Sano M., Machida M., Shigeyoshi Y., Iino M., Hashimoto S. (2005) Nat. Genet. 37, 187–192 [DOI] [PubMed] [Google Scholar]
- 11.Sun Z. S., Albrecht U., Zhuchenko O., Bailey J., Eichele G., Lee C. C. (1997) Cell 90, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 12.Tei H., Okamura H., Shigeyoshi Y., Fukuhara C., Ozawa R., Hirose M., Sakaki Y. (1997) Nature 389, 512–516 [DOI] [PubMed] [Google Scholar]
- 13.Albrecht U., Sun Z. S., Eichele G., Lee C. C. (1997) Cell 91, 1055–1064 [DOI] [PubMed] [Google Scholar]
- 14.Shearman L. P., Zylka M. J., Weaver D. R., Kolakowski L. F., Jr., Reppert S. M. (1997) Neuron 19, 1261–1269 [DOI] [PubMed] [Google Scholar]
- 15.Takumi T., Matsubara C., Shigeyoshi Y., Taguchi K., Yagita K., Maebayashi Y., Sakakida Y., Okumura K., Takashima N., Okamura H. (1998) Genes Cells 3, 167–176 [DOI] [PubMed] [Google Scholar]
- 16.Takumi T., Taguchi K., Miyake S., Sakakida Y., Takashima N., Matsubara C., Maebayashi Y., Okumura K., Takekida S., Yamamoto S., Yagita K., Yan L., Young M. W., Okamura H. (1998) EMBO J. 17, 4753–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zylka M. J., Shearman L. P., Weaver D. R., Reppert S. M. (1998) Neuron 20, 1103–1110 [DOI] [PubMed] [Google Scholar]
- 18.Kume K., Zylka M. J., Sriram S., Shearman L. P., Weaver D. R., Jin X., Maywood E. S., Hastings M. H., Reppert S. M. (1999) Cell 98, 193–205 [DOI] [PubMed] [Google Scholar]
- 19.van der Horst G. T., Muijtjens M., Kobayashi K., Takano R., Kanno S., Takao M., de Wit J., Verkerk A., Eker A. P., van Leenen D., Buijs R., Bootsma D., Hoeijmakers J. H., Yasui A. (1999) Nature 398, 627–630 [DOI] [PubMed] [Google Scholar]
- 20.Akashi M., Tsuchiya Y., Yoshino T., Nishida E. (2002) Mol. Cell. Biol. 22, 1693–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagita K., Tamanini F., Yasuda M., Hoeijmakers J. H., van der Horst G. T., Okamura H. (2002) EMBO J. 21, 1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takano A., Isojima Y., Nagai K. (2004) J. Biol. Chem. 279, 32578–32585 [DOI] [PubMed] [Google Scholar]
- 23.Loop S., Katzer M., Pieler T. (2005) EMBO Rep. 6, 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee C., Etchegaray J. P., Cagampang F. R., Loudon A. S., Reppert S. M. (2001) Cell 107, 855–867 [DOI] [PubMed] [Google Scholar]
- 25.Lee C., Weaver D. R., Reppert S. M. (2004) Mol. Cell. Biol. 24, 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunlap J. C. (1999) Cell 96, 271–290 [DOI] [PubMed] [Google Scholar]
- 27.Bae K., Jin X., Maywood E. S., Hastings M. H., Reppert S. M., Weaver D. R. (2001) Neuron 30, 525–536 [DOI] [PubMed] [Google Scholar]
- 28.Zheng B., Albrecht U., Kaasik K., Sage M., Lu W., Vaishnav S., Li Q., Sun Z. S., Eichele G., Bradley A., Lee C. C. (2001) Cell 105, 683–694 [DOI] [PubMed] [Google Scholar]
- 29.Kon N., Hirota T., Kawamoto T., Kato Y., Tsubota T., Fukada Y. (2008) Nat. Cell Biol. 10, 1463–1469 [DOI] [PubMed] [Google Scholar]
- 30.Hirayama J., Sassone-Corsi P. (2005) Curr. Opin. Genet. Dev. 15, 548–556 [DOI] [PubMed] [Google Scholar]
- 31.Hogenesch J. B., Gu Y. Z., Moran S. M., Shimomura K., Radcliffe L. A., Takahashi J. S., Bradfield C. A. (2000) J. Neurosci. 20, RC83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cermakian N., Whitmore D., Foulkes N. S., Sassone-Corsi P. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 4339–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda M., Yu W., Hirai M., Ebisawa T., Honma S., Yoshimura K., Honma K. I., Nomura M. (2000) Biochem. Biophys. Res. Commun. 275, 493–502 [DOI] [PubMed] [Google Scholar]
- 34.Okano T., Yamamoto K., Okano K., Hirota T., Kasahara T., Sasaki M., Takanaka Y., Fukada Y. (2001) Genes Cells 6, 825–836 [DOI] [PubMed] [Google Scholar]
- 35.Okano T., Sasaki M., Fukada Y. (2001) Neurosci. Lett. 300, 111–114 [DOI] [PubMed] [Google Scholar]
- 36.Dardente H., Fortier E. E., Martineau V., Cermakian N. (2007) Biochem. J. 402, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schoenhard J. A., Smith L. H., Painter C. A., Eren M., Johnson C. H., Vaughan D. E. (2003) J. Mol. Cell. Cardiol. 35, 473–481 [DOI] [PubMed] [Google Scholar]
- 38.Shearman L. P., Sriram S., Weaver D. R., Maywood E. S., Chaves I., Zheng B., Kume K., Lee C. C., van der Horst G. T., Hastings M. H., Reppert S. M. (2000) Science 288, 1013–1019 [DOI] [PubMed] [Google Scholar]
- 39.Bunger M. K., Wilsbacher L. D., Moran S. M., Clendenin C., Radcliffe L. A., Hogenesch J. B., Simon M. C., Takahashi J. S., Bradfield C. A. (2000) Cell 103, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Travnickova-Bendova Z., Cermakian N., Reppert S. M., Sassone-Corsi P. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7728–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reick M., Garcia J. A., Dudley C., McKnight S. L. (2001) Science 293, 506–509 [DOI] [PubMed] [Google Scholar]
- 42.Kiyohara Y. B., Tagao S., Tamanini F., Morita A., Sugisawa Y., Yasuda M., Yamanaka I., Ueda H. R., van der Horst G. T., Kondo T., Yagita K. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10074–10079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmesser S., Tallone T., Bordon A., Rusconi S., Albrecht U. (2008) BMC Mol. Biol. 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.