

Abstract
α-Hemolysin (HlyA) is an exotoxin secreted by some pathogenic strains of Escherichia coli that causes lysis of several mammalian cells, including erythrocytes of different species. HlyA is synthesized as a protoxin, pro-HlyA, which is activated by acylation at two internal lysines Lys-563 and Lys-689. It has been proposed that pore formation is the mechanism of cytolytic activity for this toxin, as shown in experiments with whole cells, planar lipid membranes, and liposomes, but these experiments have yielded conflicting results about the structure of the pore. In this study, HlyA cysteine replacement mutant proteins of amino acids have been labeled with Alexa-488 and Alexa-546. Fluorescence resonance energy transfer measurements, employing labeled toxin bound to sheep ghost erythrocytes, have demonstrated that HlyA oligomerizes on erythrocyte membranes. As the cytotoxic activity is absolutely dependent on acylation, we have studied the role of acylation in the oligomerization, demonstrating that fatty acids are essential in this process. On the other hand, fluorescence resonance energy transfer and the hemolytic activity decrease when the erythrocyte ghosts are cholesterol-depleted, hence indicating the role of membrane microdomains in the clustering of HlyA. Simultaneously, HlyA was found in detergent-resistant membranes. Pro-HlyA has also been found in detergent-resistant membranes, thus demonstrating that the importance of acyl chains in toxin oligomerization is the promotion of protein-protein interaction. These results change the concept of the main role assigned to acyl chain in the targeting of proteins to membrane microdomains.
Escherichia coli α-hemolysin, HlyA,4 is an exotoxin that elicits a number of responses from mammalian target cells and also alters the membrane permeability of host cells, causing lysis and death (1, 2). Synthesis, maturation, and secretion of E. coli HlyA are determined by the hlyCABD operon (3). The gene A product is a 110-kDa polypeptide corresponding to protoxin (Pro-HlyA), which is matured in bacterial cytosol to the active form (HlyA) by HlyC-directed acylation. This post-translational modification involves a covalent amide linkage of fatty acids at two internal lysine residues (Lys-563 and Lys-689) for activation (4). HlyA activated in vivo consists of a heterogeneous family of up to nine different covalent structures (two acylation sites and three possible modifying groups in each site, C14:0 (68%), C15:0 (26%) and C17:0 (6%) (5)). Although these fatty acids are not required for the binding of the toxin to membranes, they are essential for the hemolytic process, inducing a molten globule conformation and promoting the irreversibility of the binding (6, 7).
It has been proposed that pore formation is the mechanism of cytolytic activity for this toxin, as shown in experiments with whole cells, planar lipid membranes, and liposomes. However, these experiments have yielded conflicting results. Although a group of researchers is in favor of a monomer as the active species of the toxin in membranes, other groups postulate that an oligomerization process is involved. Based on experiments with lipid bilayers, Menestrina et al. (8) have suggested that one single HlyA molecule is responsible for the formation of the channel. HlyA has also been recovered from deoxycholate-solubilized erythrocyte membranes as a monomer, indicating either that oligomerization is not required for pore formation or that oligomers are dissociated in the detergent (1).
On the other hand, Benz et al. (9) have found that small variations of toxin concentration have had a considerable effect on the specific membrane conductance. An increase in HlyA concentration, by a factor of 5, results in about 40–100-fold higher membrane conductance. This means that several HlyA molecules could be involved in channel formation (9). Besides, they have found that the active channel-forming oligomer and inactive monomer are in an association-dissociation equilibrium (10). In addition, the complementation of inactive deleted mutant proteins of HlyA with the corresponding wild type toxin produces hemolytic activity, suggesting that two or more toxin molecules aggregate before pore formation (11). All of the evidence suggests the formation of an oligomer.
Experiments employing erythrocytes and model membranes have shown that the lesion created by HlyA is perhaps a more complicated event than the creation of a simple, static protein-lined pore. We have recently found that addition of nanomolar concentrations of toxin to planar lipid membranes have resulted in a decrease in membrane lifetime up to 3 orders of magnitude in a voltage-dependent manner, a typical behavior of proteolipidic pores (12). Moayeri and Welch (13) have previously demonstrated that osmotic protection of erythrocytes by sugars of different sizes is a function of toxin concentration and assay time. It appears that HlyA induces heterogeneous erythrocyte lesions that increase in size over time and that the rate of the putative growth in the size of HlyA-mediated lesions is temperature-dependent (13).
On the other hand, it has been recognized that a variety of pathogens and toxins interacts with microdomains in the plasma membrane. These microdomains are enriched in cholesterol and sphingolipids and probably exist in a liquid-ordered phase, in which lipid acyl chains are extended and ordered (14). Many proteins are targeted to these membrane microdomains by their favorable association with ordered lipids. Interestingly, these proteins are linked to saturated acyl chains, which partition well into these domains (15).
In this context, and in view of the fact that acyl chains covalently bound to proteins are determinant of specific protein-protein interactions, this research presents a study of HlyA oligomerization on sheep erythrocytes, as well as the implication of fatty acids and cholesterol-enriched microdomains in this process.
EXPERIMENTAL PROCEDURES
Proteins and Mutant Proteins
HlyA is a protein of 1023 amino acids devoid of cysteine. HlyA and Pro-HlyA were purified from culture filtrates of E. coli strains WAM 1824 (16) and WAM 783, respectively (17). The HlyA cysteine mutant protein K344C (HlyA K344C) was purified from the E. coli strain WAM 2205 (18), which was kindly provided by Dr. R. A. Welch, University of Wisconsin, Madison. Pro-HlyA cysteine mutant protein (Pro-HlyA K344C) was obtained by site-directed mutagenesis of plasmid (pSF4000ΔBamH1) extracted from WAM 783, using the QuickChange method (Stratagene, Cedar Creek, TX). The sequence for the coding strands of mismatch primer was as follows: 5′-CCATCGTATCCAAGGCATTTGAATCGTTGTGAATACTCC-3′; The mismatch codon is underlined. The mutation has been confirmed by DNA sequencing at Macrogen (Seoul, Korea). The mutated plasmid was transformed into E. coli BL-21 for further purification of Pro-HlyA K344C.
Protein Purification
Cultures of the corresponding E. coli strain were grown to late log phase in Luria-Bertani medium to an absorbance at 600 nm (A600) of 0.8–1.0. Cells were pelleted, and the supernatant was concentrated and partially purified by precipitation with 20% cold ethanol. The precipitate containing the protein was collected by centrifugation (1 h, 14,500 × g in a Sorvall centrifuge, rotor SSA 34) and then resuspended in 20 mm Tris, pH 7.4, and 150 mm NaCl (TC buffer). SDS-PAGE analysis of this preparation showed a main band at 110 kDa corresponding to more than 90% of the total protein. Proteins of lower molecular mass were removed by dialysis (membrane cutoff, 30 kDa). The protein was stored at −70 °C in 20 mm Tris, pH 7.4, 150 mm NaCl, and 6 m guanidine hydrochloride (TCGn). Proteins were first dialyzed in TC (1:100 v/v) before each experiment at 4 °C for 4 h.
The purification of Pro-HlyA and Pro-HlyA K344C by the same process employed for HlyA gave low yield, so it was purified from inclusion bodies as described by Sanchez Magraner et al. (19).
Measurements of Intrinsic Fluorescence
Protein intrinsic fluorescence spectra were recorded on a SLM 4800 Aminco spectrofluorometer. The excitation wavelength was 295 nm to minimize tyrosine emission, and the emission spectra were recorded in the 310–370 nm wavelength range. Slit width was 4 nm for both excitation and emission.
Labeling of Mutant Proteins with Fluorescence Probes
HlyA and Pro-HlyA cysteine mutant proteins in degassed TCGn at pH 7.4 were incubated with sulfhydryl-specific probes at room temperature for 1 h in the presence of 0.4 mm tris(2-carboxyethyl)phosphine hydrochloride. The probes (dissolved in the same buffer) were added in small volumes while stirring in the dark. Alexa-488 was added to a final molar ratio of 30:1 probe to protein. A molar excess of 5:1 probe to protein and 0.1% sodium cholate were used for the specific labeling with Alexa-546. Labeled proteins were separated from unbound probe by elution through PD-10 columns (GE Healthcare) and dialysis overnight into TC buffer. HlyA was reacted alongside the mutant proteins to detect nonspecific labeling. The efficiency of labeling was determined from the molar extinction coefficients for Alexa-488, ϵ492 = 67,100 cm−1 m−1, and for Alexa-546, ϵ554 = 90,300 cm−1 m−1 (specifications from Molecular Probes) and ϵ280 = 73,960 cm−1 m−1 for HlyA. All mutant proteins were specifically labeled (molar ratio 0.9–1.1 probe to protein). The nonspecific labeling detected with the wild type toxin HlyA was <10%. The hemolytic activity of the HlyA K344C mutant protein was not detectably decreased by labeling.
Hemolytic Assays
The hemolysis was determined by measuring the decrease in turbidity (scattered light at 595 nm) of a standardized sheep erythrocyte suspension. Proteins were serially diluted in TC buffer containing 10 mm CaCl2 on a 96-well microtiter plate. One hundred μl of the diluted suspensions were mixed with 100 μl of standardized sheep erythrocytes.
The standardization of the sheep erythrocytes was done just before the assay. The erythrocytes were washed in 0.9% NaCl and then diluted to 12.5 μl in 1 ml of distilled water to give a reading of 0.6 absorbance unit at 412 nm (20).
The plate was then incubated at 37 °C for 30 min, and the absorbance at 595 nm was measured at a Multimode Detector DTX 880 Beckman Coulter.
The hemolysis percentage was calculated as shown in Equation 1,
where ODc is the optical density of control erythrocytes; ODx is the optical density of erythrocytes treated with different concentrations of toxin, and ODTx is the optical density of erythrocytes after Triton X-100 addition.
The hemolytic activity is defined as the dilution of HlyA preparation producing 50% lysis of the erythrocyte suspension. Specific activity is calculated as the hemolytic activity (in hemolytic units/ml) of the respective protein divided by its concentration (mg/ml).
Cholesterol Depletion
Cholesterol extraction from sheep erythrocytes was performed by two methods, one using small unilamellar vesicles made of egg phosphatidylcholine (SUV-PC) (21) and the other employing a 3 mm methyl-β-cyclodextrin (CD) solution (22).
Sheep erythrocytes (10% hematocrit) were incubated with 1 μmol of SUV-PC at 37 °C. After 3 h, the cells were sedimented and washed five times with TC buffer.
For the cholesterol extraction using CD (Fluka), 1 ml of sheep erythrocytes was incubated with 10 ml of 3 mm CD in TC buffer at 37 °C for 30 min and washed three times with TC buffer. Half of the cells were used for experiments, and the other half was used for cholesterol quantification (Colestat, Weiner).
Hemolysis Kinetics
Hemolysis kinetics was determined by measuring the decrease in turbidity of a standardized sheep erythrocyte suspension (control and cholesterol-depleted) exposed to HlyA as a function of time at 37 °C. A series of hemolytic reactions were set up. The highest concentration of HlyA studied (0.2 μm) corresponded to the minimal amount of toxin that would produce 100% of hemolysis in 96-well plate dilution assays. The absorbance at 595 nm was measured at a Multimode Detector DTX 880 from Beckman Coulter. Initial rate of hemolysis was obtained from the linear portion of the kinetic curve (absorbance at 595 nm versus time).
Ghost Erythrocyte Preparation
1.5 milliliters of packed sheep erythrocytes (control and cholesterol-depleted) were washed with TC buffer and osmotically lysed in 10 mm Tris-HCl, pH 7.4 buffer, at 4 °C for 30 min. The membranes were pelleted by centrifugation (10 min at 14,500 × g) and washed until the supernatant remained clear. The membranes were finally resuspended in 3 ml of TC buffer. The concentration of these samples was 0.51 ± 0.06 mg of phospholipid/ml (n = 3).
Binding of Labeled Proteins to Ghost Erythrocytes
Twenty micrograms of protein was incubated with 75 μl of sheep ghost erythrocytes at 37 °C for 60 min in a TC buffer containing 10 mm CaCl2. The resulting final reaction volume was 1 ml. Membranes were pelleted by centrifugation (10 min, 14,500 × g, 4 °C) and washed three times with TC buffer to separate unbound protein. Binding percentage of protein to membranes was calculated as shown in Equation 2,
where FT is the maximum fluorescence of the corresponding labeled protein before its interaction with membranes, and sup is the fluorescence measured in the supernatant after the three washes. A blank of buffer and membranes was subtracted from all fluorescence measurements.
For proteins labeled with Alexa-488, the excitation wavelength was 480 nm, and emission fluorescence was measured at 520 nm, and proteins labeled with Alexa-546 were excited at 530 nm, and emission was measured at 570 nm.
Fluorescence Resonance Energy Transfer (FRET) Measurements
Fluorescence resonance energy transfer is a distance-dependent interaction between the electronic excited states of two fluorescent molecules, in which excitation energy is transferred from a donor molecule (D) to an acceptor molecule (A), without emission of a photon. The efficiency of energy transfer (E) is related to R0, the Förster radius, and to R, the distance between donor and acceptor, as shown by the Equation 3,
R0 represents the distance where the transfer is 50% efficient (23).
When donor and acceptor probes in FRET experiments are chemically different, FRET can be detected by the following: (a) the increase in the fluorescence intensity of the acceptor; (b) the decrease in the fluorescence intensity of the donor; or (c) the decrease in the fluorescence lifetime of the donor. For our experiments, Alexa-488 (D) and Alexa-546 (A) were chosen as donor and acceptor, respectively, and method a was selected for the determination of FRET efficiencies. For each HlyA mutant protein tested, three samples were measured differing in the contents of the labeled variants (D and A) and in the corresponding unlabeled mutant protein: (a) D/unlabeled mutant protein, (b) D/A, and (c) unlabeled mutant protein/A. The molar ratio between donor and acceptor was 1:1.
Determination of the Enhancement of the Acceptor Fluorescence Emission
FRET was calculated from spectral data as described by Gohlke et al. (24). This approach allows for the calculation of E from three fluorescence spectra. The fluorescence emission spectrum of ghost erythrocytes containing D/A, FD/A (480, λem) (excited at 480 nm), is fitted to the weighted sum of two spectral components as follows: (a) a standard emission spectrum of ghost erythrocytes labeled only with donor, FD(480, λem), and (b) a spectrum of ghost erythrocytes containing D/A, FD/A (530, λem) (excited at 530 nm where only the acceptor absorbs), as shown in Equation 4,
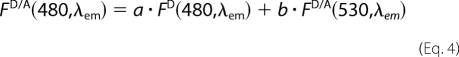 |
The coefficients a and b are the fitted fractional contributions of the two spectral components; b is the acceptor fluorescence signal due to FRET from the donor, normalized by FD/A(530 nm, λem). E was obtained as shown in Equation 5, using the b value obtained from the fitting of the spectra using Sigmaplot 8.0 (Jandel Scientific, San Rafael, CA).
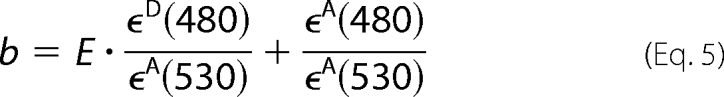 |
ϵD and ϵA are the molar absorption coefficients of D and A at the given wavelengths, respectively; ϵD(480 nm)/ϵA(530 nm) and ϵA(480 nm)/ϵA(530 nm) were determined from the absorption spectra of D and A in singly labeled preparations (D/HlyA and HlyA/A). The absorption spectrum of membranes with unlabeled toxin was subtracted in all the samples.
For FRET measurements, mutant proteins labeled with both fluorophores (ranging from 3 to 50 μg of total protein) were added to 75 μl of ghost erythrocyte suspension. The condition for the binding of the proteins to ghost membranes was the one previously described. Steady-state fluorescence spectra were recorded at room temperature on an SLM 4800 Aminco spectrofluorometer. Alexa-488 was excited at 480 nm, and emission was recorded between 470 and 600 nm. Direct excitation of Alexa-546 was achieved at 530 nm, and the emission was recorded between 540 and 600 nm.
FRET Kinetics
For kinetics studies, ghost erythrocytes were mixed with either donor plus acceptor or acceptor plus unlabeled HlyA K344C using a stopped flow (RX 2000, Applied Photophysics Ltd.) adapted to the SLM 4800 Aminco spectrofluorometer. Assays were performed as follows: 5 μg of total toxin per 100 μg of phospholipids (erythrocytes membranes). The excitation monochromator was set at 480 nm, and the emission monochromator was set at 570 nm with 8 nm slit widths. Alexa-546 emission was measured at a rate of 25 samples/s for 240 s at 37 °C. The curves represent the average of three independent experiments obtained from five replicates each.
Isolation of Detergent-resistant Membranes (DRMs) by Sucrose Gradient Centrifugation
We adapted the procedure from that of Quinn and co-workers (25). One volume of protein-bound ghost erythrocytes was resuspended in 4 volumes of TNE buffer (10 mm Tris, 200 mm NaCl, 1 mm EDTA, pH 7.4) containing 1% (v/v) Triton X-100 at 4 °C and dispersed by 10 passages through a 21-gauge needle. The mixture was incubated on ice for 20–30 min. The lysate was diluted with an equal volume of 80% (v/v) sucrose solution in TNE buffer. One ml of this suspension was overlayered by 2.3 ml of 35% sucrose solution in TNE buffer, followed by 1 ml of a 5% sucrose solution in TNE buffer. The samples were centrifuged at 190,000 × g in a Beckman SW60 Ti rotor at 4 °C for 18 h. Twelve 0.33-ml fractions were collected from the top of each tube. Pelleted material recovered from the bottom of the tube was named “P” fraction, and the material layered at the 5–35% sucrose interface was designated as DRMs (fractions 3–5). All fractions were stored frozen at −20 °C prior to further analysis.
Analysis of Sucrose Density Gradient Fractions
Immunoblotting Analysis
Fifteen microliters from each of the 13 sucrose fractions were separated by 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane by the Towbin et al. method (26). Blots were blocked with 3% skim milk in PBST buffer (140 mm NaCl, 2.7 mm KCl, 1.5 mm KH2PO4, 8.1 mm Na2HPO4, and 0.05% Tween, pH 7.4) at room temperature for 2 h. They were then incubated with a solution containing a polyclonal rabbit anti-hemolysin antibody (1:500) or anti-Flotillin-1 antibody (Santa Cruz Biotechnology, Inc.) (1:1000) in 3% skim milk/PBST at 4 °C overnight, washed with PBST buffer, and finally reacted with peroxidase-conjugated anti-rabbit IgG antibody (Sigma) (1:1000) in PBST buffer with 3% skim milk at room temperature for 2 h. After incubation and washing as stated above, blots were developed by chemiluminescence.
Lipid Analysis
Total lipids of sucrose density gradient fractions were extracted by procedure of Floch et al. (27). Lipids were recovered from the original chloroform extract and subjected to analysis by TLC on high performance TLC plates (Whatman), developed with a two solvent system consisting first of chloroform/methanol/acetic acid/water (25:18.75:1.75:1 by volume) and then with hexane/diethyl ether/acetic acid (40:10:1 by volume). Lipid spots were detected by 5% sulfuric acid in ethanol.
RESULTS
Mutant Protein Characterization
HlyA does not contain Cys residues in its sequences; therefore, Lys-344 was replaced by Cys (HlyA K344C). The same point mutation was introduced in the unacylated protein (Pro-HlyA K344C). Prior to the FRET experiments, the hemolytic activity of the mutant proteins has been measured and compared with the wild type toxin (HlyA). Fig. 1A shows that mutation located in the insertion region of the toxin into membranes (28) does not affect the hemolytic activity of the toxin. As expected, Pro-HlyA K344C is hemolytically inactive as Pro-HlyA, because the fatty acids covalently bound are lacking in both proteins.
FIGURE 1.

Characterization of HlyA and Pro-HlyA cysteine mutants. A, specific hemolytic activity of the different cysteine mutant proteins (n = 3) compared with the wild type toxin. HU, hemolytic units. B, emission spectra of Trp of wild type (WT) HlyA (solid black line), HlyA K344C (light gray dashed line), Pro-HlyA K344C (dark gray dashed line), and Pro-HlyA (black dotted line). The amount of toxin measured was of 10 g/ml, λexc was 295 nm.
As HlyA contains hydrophobic domains in its sequence, it presents a great tendency to aggregate. The aggregation percentage of each protein in solution was measured. Briefly, 20 μg/ml of protein in TC buffer was centrifuged at 110,000 × g in a Beckman 70.1 rotor at 4 °C for 2 h. The amount of proteins in the pellet (aggregates) and supernatants was quantified by the Bradford procedure. The aggregation percentage obtained was 21.6 ± 1.6 and 15.9 ± 0.48% for HlyA K344C and Pro-HlyA K344C, respectively. If we compare these values with the aggregation percentage of the wild type protein (27.2 ± 5%), the HlyA mutant protein does not present any difference with HlyA. Instead, Pro-HlyA has less of a tendency to aggregate, probably because of its compact structure (6).
HlyA contains four tryptophan residues that can serve as intrinsic fluorophores. They are located at positions 431, 479, 578, and 913. Fig. 1B shows that maximum emission does not change because of the presence of mutation, indicating that the Cys introduced does not induce important conformational changes in either HlyA or Pro-HlyA. The blue shift of Pro-HlyA indicates that the Trp residues are located in a more hydrophobic environment than in the acylated protein as published before (7).
FRET Experiments to Determine HlyA Oligomerization
Resonance energy transfer is a photochemical process whereby one fluorescent molecule or fluorophore, the “donor,” excited by an initial photon of light spontaneously transfers its energy to another molecule, the “acceptor,” by a nonradioactive dipole-dipole interaction (29, 30).
The distance over which energy can be transferred depends on the spectral characteristics of the fluorophores, but it is generally in the 10–100-Å range. Hence, FRET can be used for measuring structure (31), conformational changes (32), and interactions between molecules (33). This has been the aim of our experiment, where labeled mutant proteins of HlyA have been used to study the oligomerization of the toxin on erythrocytes membranes. To carry out this study, two populations of HlyA K344C mutant proteins, one labeled with donor (Alexa-488) and the other with acceptor fluorophores (Alexa-546), have been bound to sheep ghost erythrocytes.
The hemolytic activity of the labeled proteins has been the same as that of the unlabeled ones, indicating that the presence of fluorophores does not change the active conformation of the toxin. On the other hand, the binding percentage of the mutant proteins labeled with both fluorophores to ghost erythrocytes has been calculated as described under “Experimental Procedures.” The mean binding percentage of three independent experiments has been 23.47 ± 3.61 and 15.36 ± 5.63% for HlyA K344C Alexa-546 (HlyA A) and HlyA K344C Alexa-488 (HlyA D), respectively. The difference is not significant, demonstrating that fluorophores do not change the binding properties of the toxin. These values give us the certainty that the ratio of D:A added in the following experiments is maintained when the toxin is bound to membranes.
The fluorescence emission spectrum of a mixture containing HlyA D and HlyA A in solution at a molar ratio of 1:1 was then recorded. Fig. 2A shows that the emission fluorescence of that mixture corresponds to the fluorescence emission of donor; otherwise, when ghost erythrocytes are added, an enhancement of acceptor fluorescence is observed. This result demonstrates that the small aggregation of the toxin in solution (21.6 ± 1.6%, as described before) is not detected by FRET, but when ghost erythrocytes are added some molecules of the protein get in close proximity.
FIGURE 2.

FRET, a method used to study oligomerization. A, fluorescence spectra of a mixture of HlyA K344C D and HlyA K344C A in the absence (dark circle) and presence (light gray circle) of ghost erythrocytes. The excitation wavelength was set at 480 nm and emission was scanned from 500 to 620 nm. The initial amounts of protein and ghost erythrocytes phospholipids used were 20 and 37.5 μg, respectively. The molar ratio was 1:1 donor to acceptor. Blanks of buffer, membranes, and acceptor fluorescence excited at 480 nm were subtracted from all spectra. B, FRET measurement calculated as the enhancement of acceptor fluorescence at different lipid/protein molar ratio. Lipids concentration was 0.05 mm. This graph represents an example of four experiments. C, example of the three spectra measured at 1.2 × 109 lipid/protein ratio. Fluorescence emission spectrum of ghost erythrocytes containing D/A, FD/A(480, λem) (excited at 480 nm) (●), emission spectrum of HlyA labeled only with donor, bound to ghost erythrocytes FD(480, λem) (inverted gray triangle), spectrum of ghost erythrocytes containing D/A, FD/A(530, λem) (excited at 530 nm where only the acceptor absorbs) (■), and emission spectrum of HlyA labeled only with acceptor, bound to ghost erythrocytes FA(480, λem) (light gray circle).
FRET, calculated as the enhancement of acceptor fluorescence, has been measured as a function of lipid/protein ratio. Fig. 2B shows that FRET decreases with the enhancement of the lipid/protein ratio until it reaches a constant value, indicating that FRET can be measured, even when the amount of protein is very small. This means that at least two molecules of HlyA are close enough to transfer fluorescence energy, forming an oligomer when the toxin is bound to a membrane. An example of the spectra employed to calculate FRET is shown in Fig. 2C.
Implication of Fatty Acids Covalently Bound to HlyA in the Oligomerization Processes
Taking into account that the two fatty acids covalently bound to the toxin induce a molten globule conformation, exposing intrinsic disordered regions that may promote protein-protein interactions, we have designed the mutant protein Pro-HlyA K344C. This protein is the inactive form of HlyA (without both acyl chains) with mutation introduced in the insertion region. Fig. 3 shows that its analogous mutant protein in the active protein (HlyA K344C) presents FRET, so construction of this mutant protein ensures that the distance and orientation of the fluorophores are adequate for FRET occurrence. However, this Pro-HlyA mutant protein does not present FRET. It is important to mention that the binding percentage of Pro-HlyA and HlyA to ghost erythrocytes is 24.8 ± 6.09 and 23.47 ± 3.61%, respectively. This is consistent with what has been published by Soloaga et al. (45). This result supports that the absence of FRET in the Pro-HlyA mutant protein is because of the fact that oligomerization does not occur, thus indicating the implication of fatty acids in this process.
FIGURE 3.

Oligomerization of HlyA in ghost erythrocytes. FRET calculated for HlyA K344C and Pro-HlyA K344C was bound to ghost erythrocytes. The lipid/protein ratio was 109. FRET was calculated as mentioned before under “Experimental Procedures” (n = 4). Inset, example of spectra measured for Pro-HlyA K344C. Fluorescence emission spectrum of ghost erythrocytes containing D/A, FD/A(480, λem) (excited at 480 nm) (●), emission spectrum of ghost erythrocytes labeled only with donor, FD(480, λem) (light gray circle), and spectrum of ghost erythrocytes containing D/A, FD/A(530, λem) (excited at 530 nm where only the acceptor absorbs) (inverted gray triangle).
Role of Cholesterol-enriched Microdomains in the Action Mechanism of HlyA
Knowing that HlyA has to be acylated to be active and that some acylated proteins interact with membrane microdomains, we have studied their role in the hemolytic process. For this purpose, and taking into account that these microdomains are enriched in cholesterol and sphingolipids, the hemolytic activity of the toxin on sheep erythrocytes has been compared with the activity on erythrocytes cholesterol-depleted by SUV-PC. This method has been used because cholesterol efflux with CD led to spontaneous hemolysis. The cholesterol content of the membranes was decreased by approximately 30%. When hemolysis kinetics was measured in both control and cholesterol-depleted erythrocytes, the latter were less sensitive to toxin. As shown in Fig. 4A, the initial hemolysis rate is lower for cholesterol-depleted erythrocytes than for the one obtained with control erythrocytes for all toxin amounts tested. This result indicates that cholesterol-enriched microdomains facilitate the hemolytic process. Moreover, Fig. 4B shows that t50 values (time to reach 50% of hemolysis) are longer for cholesterol-depleted erythrocytes than for control ones.
FIGURE 4.

Hemolysis of HlyA employing cholesterol-depleted erythrocytes. A, initial hemolytic rate (−ΔDO/Δt) as a function of HlyA concentration (μm), employing control erythrocytes (●) and cholesterol-depleted erythrocytes (▼). Initial rate of hemolysis was obtained from the linear portion of kinetic curve (scattered light at 595 nm versus time; data not shown). Cholesterol was extracted from erythrocytes with egg SUV-PC (n = 3). B, plot of t50 versus reciprocal of toxin concentration, employing control erythrocytes (●) and cholesterol-depleted erythrocytes (▼). t50 was calculated from the kinetic curve as the time required to reach half of the initial absorbance at 595 nm.
To determine whether the decrease of the hemolytic rate observed in the cholesterol-depleted erythrocytes is caused by the impairment of toxin oligomerization, we repeated the FRET experiments using sheep ghost erythrocytes cholesterol-depleted by CD. In this case, the cholesterol content of the membrane was also decreased by approximately 30%. The binding percentage of HlyA to ghost cholesterol-depleted erythrocytes is 17.44 ± 1.46%. By comparing this value with the binding percentage of the protein to control ghost erythrocytes (23.47 ± 3.61%), it is possible to conclude that the changes produced in the membrane by the extraction of cholesterol do not significantly modify the binding percentage of the toxin to membranes.
FRETs of HlyA K344C bound to control and cholesterol-depleted ghosts are shown in Fig. 5. As shown in Fig. 5, CD treatment led to a decrease of 75% of FRET compared with control. This result suggests that cholesterol-enriched microdomains play an important role in the oligomerization process.
FIGURE 5.

Oligomerization of HlyA on cholesterol-depleted ghost erythrocytes. FRET measurement of HlyA K344C using control (−CD) and cholesterol-depleted (+CD) sheep ghost erythrocytes is shown. In this case, the cholesterol depletion was done with 3 mm CD treatment. Lipid concentration was 0.05 mm, and the lipid/protein ratio was 109. FRET was calculated as mentioned before under “Experimental Procedures” (n = 4). Inset, example of spectra measured for HlyA K344C bound to cholesterol-depleted ghost erythrocytes. Fluorescence emission spectrum of ghost erythrocytes containing D/A, FD/A(480, λem) (excited at 480 nm) (●), emission spectrum of ghost erythrocytes labeled only with donor, FD(480, λem) (gray circle), and spectrum of ghost erythrocytes containing D/A, FD/A(530, λem) (excited at 530 nm where only the acceptor absorbs) (inverted gray triangle).
FRET kinetics has been performed to obtain a more detailed study of the effect of cholesterol-enriched microdomains in the oligomerization process. Fig. 6 shows the enhancement of acceptor fluorescence for 4 min after mixing labeled HlyA K344C (acceptor and donor fluorophores) with control and cholesterol-depleted ghost erythrocytes. The curves in Fig. 6 show biphasic behavior of the enhancement of fluorescence with time, indicating that two phases take place during the oligomerization process. A first fast phase of ∼20 s, independent of the cholesterol content of the membrane, as the enhancement of acceptor fluorescence presents the same exponential increase for control as for cholesterol-depleted erythrocytes ghosts. On the other hand, the period between the two phases is cholesterol content-dependent because the second enhancement of acceptor fluorescence occurs 50 s later in cholesterol-depleted ghost (110 s) than in control ones (60 s).
FIGURE 6.

FRET kinetics. Measurement of acceptor fluorescence at 570 nm as a function of time of a mixed composed by HlyA K344C labeled with donor and acceptor plus control erythrocytes (black line) and cholesterol-depleted erythrocytes (light gray line) are shown. Measurement of a mixture of unlabeled and labeled with acceptor HlyA K344C with control erythrocytes (dark gray line) was done as FRET negative control. Assays were performed at a ratio of 5 μg of total toxin per 100 μg of phospholipids (erythrocytes membranes). The excitation monochromator was set at 480 nm, and the emission monochromator was set at 570 nm. Alexa-546 emission was measured at a rate of 25 samples/s during 240 s, at 37 °C. The curves represent the average value of three independent experiments obtained from five replicates each.
The acceptor fluorescence of a mixture composed of unlabeled and acceptor-labeled HlyA K344C with ghost erythrocytes was also measured. This mixture was excited at 480 nm, the excitation wavelength of the donor. As seen in Fig. 6 the enhancement of fluorescence is not as abrupt as in other cases. This demonstrates that the first phase of oligomerization described before corresponds to the enhancement of acceptor fluorescence because of FRET and not to an increase in the quantum yield of the fluorophore by the insertion of the toxin into membranes. Finally, we can hypothesize that the oligomerization process occurs in two phases as follows: a first fast phase where small oligomers are formed, and a second phase where these oligomers coalesce in a large pore. This second phase is cholesterol-dependent. This means that cholesterol-enriched microdomains favor the fusion of oligomers.
Interaction of HlyA and Pro-HlyA with DRMs
It is important to remember that the terms “membrane microdomains” and “DRMs” should not be used as synonyms because they have different origins and conceptual meanings (34). However, a very much used approach in the current literature to investigate the interaction between a protein and membrane microdomains is the DRM technique. This technique takes profit from the selective solubilization of different lipids occurring when a biomembrane is submitted to the action of a nonionic detergent such as Triton X-100. In this paper, this technique has been used to compare the affinity of HlyA and Pro-HlyA to DRMs. For this purpose, ghost erythrocytes were incubated with purified HlyA or Pro-HlyA and DRMs were separated by sucrose gradient ultracentrifugation as described under “Experimental Procedures.” DRMs fractions have been defined by their enrichment in cholesterol, sphingomyelin, and Flotillin-1, a specific protein marker of erythrocyte microdomains (35).
Fig. 7A shows the immunoblot analysis of these fractions, revealing that most of the ghost-associated HlyA has been detected in low density fractions (lanes 3 and 4). Control experiments were performed with free HlyA, i.e. in the absence of ghost erythrocytes. As expected, after ultracentrifugation, the whole toxin was found at the bottom of the gradient (Fig. 7B, lanes 12 and P). Hence, the ability of HlyA to reach the top of the gradient depends on the presence of cell lysate, indicating that the flotation of the toxin to the lighter fractions is not because of the presence of detergent and the centrifugation procedure. The low density fractions that contained HlyA were enriched in cholesterol, sphingomyelin (Fig. 7C), and Flotillin-1 (Fig. 7D), typical characteristics of DRMs.
FIGURE 7.

Interaction of HlyA with DRMs. A, 30 μg of HlyA was incubated with 100 μl of ghost erythrocytes for 30 min at 37 °C. Cells were lysed with 1% Triton X-100, and insoluble cell components were then separated by sucrose density gradient centrifugation. The gradient fractions were analyzed by immunoblotting with anti-HlyA antibodies. HlyA was present in fractions 3 and 4 (A, lanes 3 and 4). B, when sucrose density gradient centrifugation was applied to free HlyA, it appeared at the bottom of the gradient (B, lanes 12 and P). C, enrichment of cholesterol (Ch) and sphingomyelin (SM) in DRM was performed by high performance TLC analysis. Total lipids of sucrose density gradient fractions were extracted by the procedure of Folch. Lipids were recovered from the original chloroform extract and subjected to analysis on high performance TLC plates, developed with a two solvent system. Because Triton X-100 in the bottom fractions (lanes 7–12) interferes with the separation of lipids on TLC plates, the distribution pattern of cholesterol and sphingomyelin in these fractions was not shown. D, gradient fractions were also analyzed by immunoblotting with anti-Flotillin-1 antibodies. Flotillin-1 appears mainly in fractions 3 and 4 (D, lanes 3 and 4). E, Pro-HlyA was incubated in the same conditions as HlyA. The gradient fractions were analyzed by immunoblotting with anti-HlyA antibodies. Pro-HlyA co-localizes with HlyA (E, lanes 3 and 4).
The sheep erythrocytes were then cholesterol-depleted with CD, and ghosts were prepared and incubated with HlyA. Finally, DRMs were obtained as described before. The immunoblot analysis has revealed that HlyA in low density fractions completely disappears (Fig. 8A, lanes 3 and 4), and it appears mainly at the bottom of the gradient (Fig. 8A, lanes 12 and P). Flotillin-1 was exclusively located at the bottom of the gradient after the treatment with CD, representing a positive control for the disruption of DRM (Fig. 8B, lanes 12 and P).
FIGURE 8.

Interaction of HlyA with cholesterol-depleted erythrocytes. Thirty micrograms of HlyA was incubated with 100 μl of cholesterol-depleted ghost erythrocytes for 30 min at 37 °C. Cells were lysed with 1% Triton X-100, and insoluble cell components were then separated by sucrose density gradient centrifugation. The gradient fractions were analyzed by immunoblotting with anti-HlyA (A) and anti-Flotillin-1 (B) antibodies.
To test if fatty acids covalently bound to protein are responsible for toxin association with DRMs, Pro-HlyA has been incubated with ghost erythrocytes, and DRMs have been obtained. Interestingly, Pro-HlyA co-localizes with HlyA and Flotillin-1 in low density fractions of the gradient (Fig. 7E, lanes 3 and 4). Also, control experiments have been done with Pro-HlyA in the absence of ghost membrane, and it was found at the bottom of the gradient (data not shown) as HlyA, after Triton X-100 treatment and ultracentrifugation.
To sum up, the comparison between control and cholesterol-depleted erythrocytes in FRET experiments, kinetic assays, and DRM technique confirms that cholesterol-enriched microdomains facilitate the hemolytic process of HlyA. In addition, the fact that Pro-HlyA is found in DRMs indicates that the main role assigned to saturated acyl chain to target proteins to these microdomains is not valid for HlyA.
DISCUSSION
There is a substantial debate about how HlyA creates lesions in target cell membranes. Some studies have hinted that oligomerization can occur, although others have suggested that there is neither oligomerization nor a single oligomer size. Both linear and nonlinear dependences of membrane conductance have been reported, leading to confusion about whether the pore formation depends on toxin oligomerization. However, in those previous studies, oligomerization has been indirectly examined.
Our report shows, by FRET experiments, that an oligomer is involved in the hemolytic mechanism of HlyA. FRET can be used to study the distribution of molecules in membranes because the average spacing between molecules of interest will depend primarily on their lateral distribution. Molecules may be within FRET distance either because they are clustered or because they are randomly distributed at such high surface densities that there is a fraction of randomly distributed labeled molecules in FRET proximity. The last effect has been avoided in our experiments by using a high lipid/protein molar ratio (109), to ensure that the observed FRET corresponds to oligomerization of the toxin on the erythrocyte surface (Fig. 2B).
On the other hand, the absence of FRET in the mutant protein, Pro-HlyA K344C (Fig. 3), confirms the participation of the fatty acids covalently bound in the oligomerization process. At first glance, this absence of FRET may be attributed to a reduced binding of the mutant protein to ghost erythrocytes, but this was discarded because the percentage of binding to membranes of both proteins is similar, as shown under “Results.” It is important to mention that fatty acids are essential for the hemolytic activity (Fig. 1) and, considering that the fatty acids are needed for oligomerization, we can state that oligomerization is necessary for hemolysis. We feel tempted to propose that the presence of fatty acids covalently bound to the protein leads to the exposure of regions that are implicated in protein-protein interactions. In addition, an important role of acylation in the oligomerization process to form hemolytic pores has been proposed for adenylate cyclase toxin from Bordetella pertussis, another RTX toxin (36).
Finally, if we consider that pores formed by HlyA are sensitive to proteases on the cis side of the planar lipid membranes (8), it is possible to conclude that the part of the toxin remaining external to the membrane is involved in protein-protein interaction responsible for oligomerization, and thus in pore formation. The kinetics of HlyA-induced hemolysis is also consistent with multimeric pore formation. Simulations indicate that the escape time of a small molecule from a vesicle through a pore is on the order of 10 ms (37), which is about 50,000-fold faster than the half-time of hemolysis shown in Fig. 4B. This suggests that the half-times represent the time required to form pores.
As seen in Fig. 4A, the hemolysis rate of cholesterol-depleted erythrocytes is lower than the hemolysis rate of control erythrocytes at each HlyA concentration tested, suggesting the participation of cholesterol-enriched microdomains in the oligomerization process. For cholesterol-depleted erythrocytes, at low toxin concentration, the kinetics of hemolysis seems to be more complex, suggesting that toxin diffusion in membranes is the limiting step. This was confirmed with the results of FRET kinetics. The biphasic behavior of FRET seen in Fig. 6 suggests the first formation of small oligomers, followed by their assembly to form multimeric structures. The concentration of the small oligomers is favored by the cholesterol-enriched microdomains, diminishing the diffusion time in the membrane. The number of HlyA molecules that associate to form the pore is uncertain; however, it is not unreasonable to assume that several molecules could oligomerize to form a pore. An extension of this reasoning suggests that at high doses progressive oligomerization of HlyA leads to the fusion of the pore and rapid destruction of the cell membrane with little time for activation of the central apoptotic pathway. On the other hand, at lower concentration, pores would be smaller and fewer in number so that cells, although injured, survive long enough so that apoptosis can be observed (38).
These results can explain why toxin association with erythrocytes at 0–2 °C is characterized as a prelytic state, and following a shift to 23 °C, after a lag period, lysis begins (16). In conclusion, the fusion of oligomers may be the rate-limiting step in pore formation, and the integrity of the cholesterol-enriched microdomains is necessary for the local concentration of HlyA-induced hemolysis. This is in agreement with Moayeri and Welch (13), who observed that the degree of osmotic protection of erythrocytes afforded by protectants of varying sizes depends on the amount of toxin applied and the duration of the assay. They suggest that HlyA creates a lesion with a very small initial size that increases in apparent diameter over time. Consequently, the larger the oligomer, the bigger the pore size becomes.
In this study, we provide direct biochemical evidence indicating that HlyA associates with DRMs. When ghost erythrocytes were incubated with purified HlyA and DRMs were separated by sucrose gradient ultracentrifugation, the immunoblot analysis revealed that most of the ghost-associated HlyA was associated with DRMs (Fig. 7A, lanes 3 and 4). The data suggest that the binding of HlyA to erythrocyte membranes is mediated by membrane microdomains that serve as concentration platforms for the toxin oligomerization as we have demonstrated previously (Fig. 6). The fact that Pro-HlyA co-localizes with HlyA and flotillin in DRMs (Fig. 7E, lanes 3 and 4) emphasizes our hypothesis that the main role of the saturated acyl chain covalently bound to HlyA is the participation in the oligomerization process, and not the targeting to cholesterol-enriched membranes.
A key feature of cholesterol-enriched microdomains is the tight packing of lipid acyl chains in the liquid-ordered phase, in which lipid acyl chains are extended and ordered (14). Because of the difficulty in packing membrane-spanning helices into the ordered lipid environment, some proteins are linked to saturated acyl chains, which partition well into them (39). However, Shogomori et al. (40) have found that acylation does not measurably enhance microdomain association, and they have concluded that the acylated linker for activation of T cell transmembrane domain has low inherent cholesterol-enriched microdomain affinity. It is possible to conclude that acylation is not sufficient for the targeting of any transmembrane protein, and a second mechanism, such as protein-protein interactions for microdomains association, is required (41, 42).
In this context, our results show that there is no difference in DRMs association between acylated and unacylated HlyA. However, FRET experiments clearly show the importance of the acyl chain covalently bound to toxin in the oligomerization is essential for the lytic process (Fig. 3).
Recent results we have published are consistent with a molten globular form of the acylated protein. Moreover, we have proposed that the fatty acids covalently bound to the protein expose intrinsically disordered regions present in the toxin sequence that may be involved in different steps of the action mechanism of the toxin (6). The intrinsic lack of structure can confer functional advantages on the protein, including the ability to bind to several different targets or, among toxin monomers, to form the active oligomer responsible for the hemolytic action of the toxin. In addition, we have found that the presence of two acyl chains in HlyA confers this protein with the property of irreversible binding to membranes, which is essential for the lytic process to take place (7).
The formation of oligomers by HlyA is consistent with the results of lipid bilayer experiments with asolectin membranes. In those experiments, we have found that HlyA increases membrane conductance by many orders of magnitude in a concentration-dependent fashion. An amplitude histogram of HlyA pores demonstrates a significant dependence of pore size on toxin concentration, indicating a possible participation of a variable number of monomers in the structure of a pore.5
In this way, HlyA pore behavior differs from the fixed oligomeric stoichiometric seen in other membrane-inserting toxins such as staphylococcal α-hemolysin (43). Rather, HlyA oligomerization appears to be more similar to that of the diphtheria toxin, which can form pores and oligomers of various size (44).
To conclude, we propose that fatty acids covalently bound to HlyA and membrane microdomains are implicated in the hemolysis process. Fatty acids are essential because they expose intrinsic disorder regions that enhance protein-protein interaction to form the oligomer, and the membrane microdomains act as platforms that concentrate the toxin in this oligomerization process. Unfortunately, pore size cannot be used to estimate the monomer number, because the HlyA pore is partly bordered by lipids (12).
Acknowledgments
We thank Prof. Nelba Lema for revising the English grammar and Dr. Ben Corry for helpful comments concerning FRET.
This work was supported by grants from the Comisión de Investigaciones Científicas de la Provincia de Buenos Aires, Agencia Nacional de Promoción Científica Grant BID 1728/OC-AR 26228, and PIP CONICET 5742, Argentina.
V. Herlax and L. Bakás, unpublished results.
- HlyA
- α-hemolysin
- FRET
- fluorescence resonance energy transfer
- DRM
- detergent-resistant membrane
- CD
- methyl-β-cyclodextrin
- SUV-PC
- small unilamellar vesicles made of egg phosphatidylcholine.
REFERENCES
- 1.Bhakdi S., Mackman N., Nicaud J. M., Holland I. B. (1986) Infect. Immun. 52, 63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gadeberg O. V., Orskov I. (1984) Infect. Immun. 45, 255–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch R. A. (2001) Curr. Top. Microbiol. Immunol. 257, 85–111 [DOI] [PubMed] [Google Scholar]
- 4.Stanley P., Koronakis V., Hughes C. (1998) Microbiol. Mol. Biol. Rev. 62, 309–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim K. B., Walker C. R., Guo L., Pellett S., Shabanowitz J., Hunt D. F., Hewlett E. L., Ludwig A., Goebel W., Welch R. A., Hackett M. (2000) J. Biol. Chem. 275, 36698–36702 [DOI] [PubMed] [Google Scholar]
- 6.Herlax V., Bakas L. (2007) Biochemistry 46, 5177–5184 [DOI] [PubMed] [Google Scholar]
- 7.Herlax V., Bakás L. (2003) Chem. Phys. Lipids 122, 185–190 [DOI] [PubMed] [Google Scholar]
- 8.Menestrina G., Mackman N., Holland I. B., Bhakdi S. (1987) Biochim. Biophys. Acta 905, 109–117 [DOI] [PubMed] [Google Scholar]
- 9.Benz R., Döbereiner A., Ludwig A., Goebel W. (1992) FEMS Microbiol. Immunol. 5, 55–62 [DOI] [PubMed] [Google Scholar]
- 10.Benz R., Schmid A., Wagner W., Goebel W. (1989) Infect. Immun. 57, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludwig A., Benz R., Goebel W. (1993) Mol. Gen. Genet. 241, 89–96 [DOI] [PubMed] [Google Scholar]
- 12.Bakás L., Chanturiya A., Herlax V., Zimmerberg J. (2006) Biophys. J. 91, 3748–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moayeri M., Welch R. A. (1994) Infect. Immun. 62, 4124–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown D. A., London E. (1998) J. Membr. Biol. 164, 103–114 [DOI] [PubMed] [Google Scholar]
- 15.Pike L. J. (2003) J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 16.Moayeri M., Welch R. A. (1997) Infect. Immun. 65, 2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boehm D. F., Welch R. A., Snyder I. S. (1990) Infect. Immun. 58, 1959–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pellett S., Welch R. A. (1996) Infect. Immun. 64, 3081–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez-Magraner L., Cortajarena A. L., Goñi F. M., Ostolaza H. (2006) J. Biol. Chem. 281, 5461–5467 [DOI] [PubMed] [Google Scholar]
- 20.Soloaga A., Ramírez J. M., Goñi F. M. (1998) Biochemistry 37, 6387–6393 [DOI] [PubMed] [Google Scholar]
- 21.Giraud F., M'Zali H., Chailley B., Mazet F. (1984) Biochim. Biophys. Acta 778, 191–200 [DOI] [PubMed] [Google Scholar]
- 22.Kilsdonk E. P., Yancey P. G., Stoudt G. W., Bangerter F. W., Johnson W. J., Phillips M. C., Rothblat G. H. (1995) J. Biol. Chem. 270, 17250–17256 [DOI] [PubMed] [Google Scholar]
- 23.Lackowicz J. (1984) Principle of Fluorescence Spectroscopy, pp. 305–337, Plenum Publishing Corp., New York [Google Scholar]
- 24.Gohlke C., Murchie A. I., Lilley D. M., Clegg R. M. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 11660–11664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koumanov K. S., Tessier C., Momchilova A. B., Rainteau D., Wolf C., Quinn P. J. (2005) Arch Biochem. Biophys. 434, 150–158 [DOI] [PubMed] [Google Scholar]
- 26.Towbin H., Staehelin T., Gordon J. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folch J., Lees M., Sloane Stanley G. H. (1957) J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 28.Hyland C., Vuillard L., Hughes C., Koronakis V. (2001) J. Bacteriol. 183, 5364–5370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster T. (1948) Ann. Phys. 2, 55–75 [Google Scholar]
- 30.Stryer L. (1978) Annu. Rev. Biochem. 47, 819–846 [DOI] [PubMed] [Google Scholar]
- 31.Lakowicz J. R., Gryczynski I., Wiczk W., Laczko G., Prendergast F. C., Johnson M. L. (1990) Biophys. Chem. 36, 99–115 [DOI] [PubMed] [Google Scholar]
- 32.Heyduk T. (2002) Curr. Opin. Biotechnol. 13, 292–296 [DOI] [PubMed] [Google Scholar]
- 33.Parsons M., Vojnovic B., Ameer-Beg S. (2004) Biochem. Soc. Trans. 32, 431–433 [DOI] [PubMed] [Google Scholar]
- 34.Lichtenberg D., Goñi F. M., Heerklotz H. (2005) Trends Biochem. Sci. 30, 430–436 [DOI] [PubMed] [Google Scholar]
- 35.Salzer U., Prohaska R. (2001) Blood 97, 1141–1143 [DOI] [PubMed] [Google Scholar]
- 36.Hackett M., Walker C. B., Guo L., Gray M. C., Van Cuyk S., Ullmann A., Shabanowitz J., Hunt D. F., Hewlett E. L., Sebo P. (1995) J. Biol. Chem. 270, 20250–20253 [DOI] [PubMed] [Google Scholar]
- 37.Parente R. A., Nir S., Szoka F. C., Jr. (1990) Biochemistry 29, 8720–8728 [DOI] [PubMed] [Google Scholar]
- 38.Lally E. T., Hill R. B., Kieba I. R., Korostoff J. (1999) Trends Microbiol. 7, 356–361 [DOI] [PubMed] [Google Scholar]
- 39.de Planque M. R., Killian J. A. (2003) Mol. Membr. Biol. 20, 271- 284 [DOI] [PubMed] [Google Scholar]
- 40.Shogomori H., Hammond A. T., Ostermeyer-Fay A. G., Barr D. J., Feigenson G. W., London E., Brown D. A. (2005) J. Biol. Chem. 280, 18931–18942 [DOI] [PubMed] [Google Scholar]
- 41.Fragoso R., Ren D., Zhang X., Su M. W., Burakoff S. J., Jin Y. J. (2003) J. Immunol. 170, 913–921 [DOI] [PubMed] [Google Scholar]
- 42.Cherukuri A., Shoham T., Sohn H. W., Levy S., Brooks S., Carter R., Pierce S. K. (2004) J. Immunol. 172, 370–380 [DOI] [PubMed] [Google Scholar]
- 43.Song L., Hobaugh M. R., Shustak C., Cheley S., Bayley H., Gouaux J. E. (1996) Science 274, 1859–1866 [DOI] [PubMed] [Google Scholar]
- 44.Sharpe J. C., London E. (1999) J. Membr. Biol. 171, 209–221 [DOI] [PubMed] [Google Scholar]
- 45.Soloaga A., Ostolaza H., Goñi F. M., de la Cruz F. (1996) Eur. J. Biochem. 238, 418–422 [DOI] [PubMed] [Google Scholar]