

Abstract
SsgA-like proteins (SALPs) are a family of homologous cell division-related proteins that occur exclusively in morphologically complex actinomycetes. We show that SsgB, a subfamily of SALPs, is the archetypal SALP that is functionally conserved in all sporulating actinomycetes. Sporulation-specific cell division of Streptomyces coelicolor ssgB mutants is restored by introduction of distant ssgB orthologues from other actinomycetes. Interestingly, the number of septa (and spores) of the complemented null mutants is dictated by the specific ssgB orthologue that is expressed. The crystal structure of the SsgB from Thermobifida fusca was determined at 2.6 Å resolution and represents the first structure for this family. The structure revealed similarities to a class of eukaryotic “whirly” single-stranded DNA/RNA-binding proteins. However, the electro-negative surface of the SALPs suggests that neither SsgB nor any of the other SALPs are likely to interact with nucleotide substrates. Instead, we show that a conserved hydrophobic surface is likely to be important for SALP function and suggest that proteins are the likely binding partners.
The mechanisms governing the correct timing and localization of cell division is one of the most studied topics in cell biology. In unicellular bacteria like Escherichia coli, cell division occurs at the mid-cell position, away from the chromosomes (1–3). The key step in this process is the appropriate timing and localization of cell division protein FtsZ to the future septum site, followed by polymerization to the Z-ring and sequential recruitment of the divisome components (1, 4). One of the major advances in our understanding of the cell division process and, in particular, of the function of FtsZ came from elucidation of the three-dimensional structure of FtsZ, which showed striking similarity to the eukaryotic protein tubulin, despite very low sequence similarity (5). Such prokaryotic ancestry was later also revealed for the cytoskeletal proteins MreB and Mbl, which belong to the actin family (6, 7), and underscored the notion of generally conserved principles in cytokinesis.
Spore-forming Gram-positive Streptomyces bacteria are an important source of clinically useful antibiotics and anticancer agents (8). In these morphologically complex microorganisms, cell division is distinctly different from that in unicellular bacteria in several ways. For one, they are the only known organisms where FtsZ and MreB are both dispensable for growth (9, 10), which makes streptomycetes ideal for the study of cytokinesis. Streptomycetes have a complex life cycle that is mechanistically very similar to filamentous fungi, in producing a mycelium and propagating by sporulation (11, 12). During sporulation, the cell division machinery produces up to 100 septa simultaneously, spaced at around 1 μm, resulting in long chains of uniform and unigenomic spores (10, 13, 14). Besides the simultaneous production of multiple septa, cell division in mycelial actinomycetes also differs from that in other bacteria at the molecular level; actinomycetes lack orthologues of MinC and MinE for septum site localization (15, 16) as well as the nucleoid occlusion system Noc and Z-ring anchoring proteins, such as FtsA and ZipA (1, 6). Instead, several unique protein families have been identified that play a role in the control of cell division, including CrgA and the SsgA-like proteins (17–19). However, molecular details of their mode of action are so far lacking.
The SsgA (sporulation of Streptomyces griseus)-like proteins (SALPs)3 are small (around 130–140 residues), actinomycete-specific proteins, which control sporulation-related processes in streptomycetes (17, 20). Streptomyces coelicolor contains seven SALP paralogues (SsgA to SsgG). SsgA and SsgB are essential for sporulation of S. coelicolor (21, 22). SALPs are involved in the control of cell wall-related events, such as septum localization and synthesis, thickening of the spore wall, and autolytic spore separation (17, 20), and SsgA itself directly activates sporulation-specific cell division (22, 23). The morphological complexity of actinomycetes apparently correlates to the number of SALP homologues in each organism, with one paralogue in single spore-forming actinomycetes (e.g. Salinispora or Thermobifida) and up to seven in multispore formers (two in erythromycin producer Saccharopolyspora erythraea, 3–5 in Frankia, and 6–7 in Streptomyces) (17). Most SALPs can be assigned to three subfamilies (SsgA, SsgBG, and SsgDE) based on phylogenetic analysis (17). At present, there are no functional homologues for the SALPs, and structural information is lacking. To advance our understanding of how SALPs function at the molecular level and to provide a structural template for a unique protein family without obvious relatives in any other organism, we selected the single SALP homologue from the thermophilic soil bacterium Thermobifida fusca (a major degrader of plant cell walls used in waste remediation (24)) for detailed structural analysis by x-ray crystallography, as part of our structural genomics program.
In this work, we show that SsgB is most likely the archetypical SALP that occurs in morphologically complex actinomycetes, with an evolutionarily conserved function in the control of development. The three-dimensional structure of the SsgB orthologue from T. fusca was determined and revealed significant structural similarity to a eukaryotic family of ssDNA/gRNA-binding proteins. However, the structure and experimental data both suggest that SALPs probably interact with protein ligands through a hydrophobic region on their surface.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Culturing Conditions
E. coli K-12 strains JM109 (25) and ET12567 (26) were used for propagating plasmids and were grown and transformed using standard procedures (25). Transformants were selected in L broth containing 1% (w/v) glucose and the appropriate antibiotics. The Streptomyces strains used in this work are listed in supplemental Table S1.
S. coelicolor M145 was used for transformation and propagation of Streptomyces plasmids. Preparation of media for Streptomyces growth, protoplast preparation and transformation was done according to standard procedures (27). SFM medium was used to make spore suspensions; R2YE medium was used for regenerating protoplasts and, after the addition of the appropriate antibiotic, for selecting recombinants.
Plasmids and Constructs
The plasmids described in this work are summarized in supplemental Table S1.
General Cloning Vectors
pIJ2925 (28) is a pUC19-derived plasmid used for routine subcloning. For cloning in Streptomyces, we used the shuttle vectors pHJL401 (29), for which maintenance in streptomycetes occurs via the SCP2* ori (30) (5 copies/chromosome). Plasmid DNA was isolated from ET12567 prior to transformation to Streptomyces. For selection of plasmids in E. coli, ampicillin was used, and chloramphenicol was added for ET12567 transformants. For selection in S. coelicolor, we used thiostrepton for pHJL401 and derivatives.
Construction of pGWS271, pGWS294, and pGWS295
Fragments of ∼900 bp harboring the SALP-encoding genes and flanking regions from Salinispora tropica (designated ssgB(Stro)) or from Sacch. erythraea (ssgB(Sery) and SACE5535) (see supplemental Table S3) were amplified by PCR using Pfu polymerase (Stratagene, La Jolla, CA) and oligonucleotide pairs SalTr_BF/SalTr_BR, Sery_BF/Sery_BR or Sery_2F/Sery_2R (supplemental Table S2), respectively. Genomic DNA from S. tropica was a kind gift from Paul Jensen (University of California San Diego). PCR products were cloned as EcoRI/BamHI-digested (for ssgB(Sery)) or EcoRI-HindIII-digested (for ssgB(Stro) and for SACE5535) fragments into pHJL401 digested with the same enzymes, resulting in pGWS271 (ssgB(Stro)), pGWS294 (ssgB(Sery)), and pGWS295 (SACE5535) (S1). For the precise sequences of the inserts, see supplemental Fig. S1.
Construction of pGWS298, pGWS299, pGWS554, pGWS555, and pGWS556
To ensure proper expression, the open reading frames of the ssg orthologues from Kineococcus radiotolerans (ssgB(Krad)), Thermobifida fusca (ssgB(Tfus)), and Frankia sp. CcI3 (ssgB(Fran)) and a second SALP-encoding gene from Frankia sp. CcI3 (Francci3_3418) (see supplemental Table S3) were fused to the S. coelicolor ssgB promoter region (nucleotides −171 to −1 relative to the ATG start codon of ssgB (31)) by gene synthesis (Genscript Corp.). The exact sequences of the inserts of all five constructs are presented as supplemental Fig. S1. Sequences were designed so as to ensure that the promoter sequence was fused to the most likely start codon (nucleotide position 3147086 for ssgB(Krad), 2471763 for ssgB(Tfus), 1632970 for ssgB(Fran), and 4062696 for Francci3_3418 on their respective genomes), based on sequence homology with Streptomyces ssgB. EcoRI/HindIII-digested fragments were cloned into pHJL401 digested with the same enzymes, resulting in pGWS299, pGWS554, pGWS555, and pGWS556 (supplemental Table S1).
Microscopy
Phase-Contrast Light Microscopy
Strains were grown against coverslips inserted into solid SFM agar plates, as described previously (32). After 7 days, spore formation was assessed by phase-contrast light microscopy. For visualization, a Zeiss standard 25 phase-contrast light microscope was used.
Fluorescence Microscopy
DNA in spore chains was stained with a mixture of the membrane-impermeant fluorescent dye propidium iodine (PI) (1 μg/ml) (Sigma) and the membrane-permeant Syto-82 Orange (8 μm) (Invitrogen). Samples were analyzed with an Olympus BH2 fluorescence microscope using a 495-nm band pass filter and a 515-nm long pass filter and a 505-nm dichroic mirror in between. In this way, a single image can be made where DNA-bound PI emits a red-colored and DNA-bound Syto-82 emits a green-colored fluorescent signal.
Electron Microscopy
Morphological studies on surface-grown aerial hyphae and/or spores by cryo-scanning electron microscopy were performed using a JEOL JSM6700F scanning electron microscope, as described previously (21).
Protein Production and Crystallization
The ssgB gene from T. fusca YX-ER1 (GenBankTM, YP_290167.1; GI, 72162510; Swiss-Prot, Q47N25) was amplified by PCR from genomic DNA using PfuTurbo (Stratagene) and primers corresponding to the predicted 5′- and 3′-ends. The PCR product was cloned into plasmid pSpeedET, which encodes an expression and purification tag, followed by a tobacco etch virus protease cleavage site (MGSDKIHHHHHHENLYFQG) at the amino terminus of the full-length protein. The cloning junctions were confirmed by DNA sequencing. Protein expression was performed in a selenomethionine-containing medium using the E. coli strain GeneHogs (Invitrogen). At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/ml, and the cells were harvested. After one freeze/thaw cycle, the cells were homogenized in lysis buffer (50 mm HEPES, pH 8.0, 50 mm NaCl, 10 mm imidazole, 1 mm TCEP) and passed through a Microfluidizer (Microfluidics). The lysate was clarified by centrifugation at 32,500 × g for 30 min and loaded onto nickel-chelating resin (GE Healthcare) pre-equilibrated with lysis buffer. The resin was washed with wash buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 40 mm imidazole, 10% (v/v) glycerol, 1 mm TCEP), and the protein was eluted with elution buffer (20 mm HEPES, pH 8.0, 300 mm imidazole, 10% (v/v) glycerol, 1 mm TCEP). The eluate was buffer-exchanged with HEPES crystallization buffer (20 mm HEPES, pH 8.0, 200 mm NaCl, 40 mm imidazole, 1 mm TCEP) using a PD-10 column (GE Healthcare) and treated with 1 mg of tobacco etch virus protease per 15 mg of eluted protein. The digested eluate was passed over nickel-chelating resin (GE Healthcare) pre-equilibrated with HEPES crystallization buffer, and the resin was washed with the same buffer. The flow-through and wash fractions were combined and concentrated for crystallization assays to 14.4 mg/ml by centrifugal ultrafiltration (Millipore). SsgB(Tfus) was crystallized using the nanodroplet vapor diffusion method with standard Joint Center for Structural Genomics crystallization protocols (33). Screening for diffraction was carried out using the Stanford Automated Mounting system (34) at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA). The crystallization reagent contained 40% (v/v) 1,2-propanediol and 0.1 m acetate, pH 4.5. No additional cryoprotectant was added to the crystal. Diffraction images were initially indexed in tetragonal space group P4, and subsequent structure solution identified the space group as P41 (Table 1). The molecular weight and oligomeric state of SsgB(Tfus) were determined using a PROTEIN KW-803 column (Shodex) precalibrated with gel filtration standard (Bio-Rad). The mobile phase consisted of 20 mm HEPES, pH 7.5, 200 mm NaCl, and 0.5 mm TCEP.
TABLE 1.
Data collection and refinement statistics (Protein Data Bank code 3cm1)
| Parameter | Value | ||
|---|---|---|---|
| Space group | P41 | ||
| Unit cell |
a = b = 64.8 Å, c = 130.6 Å |
||
| λ1 MADSe | λ2 MADSe | λ3 MADSe | |
| Data collection | |||
| Wavelength (Å) | 0.9184 | 0.9793 | 0.9790 |
| Resolution range (Å) | 46.0–2.6 | 46.0–2.7 | 46.0–2.7 |
| No. of observations | 62,690 | 55,956 | 56,250 |
| No. of unique reflections | 16,527 | 14,741 | 14,382 |
| Completeness (%) | 99.5 (99.3)a | 99.6 (100)a | 99.6 (99.9)a |
| Mean I/σ(I) | 15.0 (2.0)a | 16.0 (2.7)a | 15.0 (2.3)a |
| Rsym on I (%)b | 4.6 (72)a | 4.4 (52)a | 4.9 (63)a |
| Model and refinement statistics | |||
| Resolution range (Å) | 46.0–2.6 | Data set used in refinement | λ1 MADSe |
| No. of reflections (total) | 16,493 | Cut-off criteria | |F| > 0 |
| No. of reflections (test) | 846 | Rcrystc | 0.23 |
| Completeness (% total) | 99.4 | Rfreed | 0.27 |
| Stereochemical parameters; restraints (r.m.s. observed) | |||
| Bond lengths (Å) | 0.011 | ||
| Bond angles (degrees) | 1.41 | ||
| Average isotropic B-value (Å2) | 88.7e | ||
| ESUf based on Rfree (Å) | 0.31 | ||
| Protein residues/atoms | 384/2,892 | ||
a Highest resolution shell in parentheses.
bRsym = Σ|Ii − 〈Ii〉|/Σ|Ii|, where Ii is the scaled intensity of the ith measurement, and 〈Ii〉 is the mean intensity for that reflection.
c Rcryst = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fc and Fo are the calculated and observed structure factor amplitudes, respectively.
d Rfree as for Rcryst, but for 5.0% of the total reflections chosen at random and omitted from refinement.
e This value represents the total B that includes TLS and residual B components. The estimated B-value based on the Wilson plot is 79 Å2.
f Estimated overall coordinate error (48).
Data Collection, Structure Solution, and Refinement
Multiwavelength anomalous diffraction (MAD) data were collected at Stanford Synchrotron Radiation Lightsource beamline 11-1. The data were collected at wavelengths corresponding to the high energy remote (λ1), inflection (λ2), and peak (λ3) of a selenium MAD experiment. The data sets were collected at 100 K using a MarCCD 325 detector (Rayonix). The data processing and structure solution were performed using an automatic structure solution pipeline developed at the Joint Center for Structural Genomics. The MAD data were integrated and reduced using XDS and then scaled with the program XSCALE (35). Selenium sites were located with SHELXD (36). Phase refinement and automatic model building were performed using autoSHARP (37) and RESOLVE (38). Model completion and refinement were performed with COOT (39) and REFMAC (40). CCP4 programs were used for data conversion and other calculations (41). Data and refinement statistics are summarized in Table 1. Independently, the structure was also solved by single-wavelength anomalous diffraction by the Midwest Center for Structural Genomics). Data were reduced, and the structure was solved at 2.75 Å with the HKL-3000 package (42) using data previously collected on the ID-19 beamline at the Advanced Photon Source. Refinement was performed in an analogous way, as described above. The refined model was similar to the one refined using the higher resolution data presented here. Analysis of the stereochemical quality of the model was accomplished using MolProbity (43). All molecular graphs were prepared with PyMOL (DeLano Scientific) unless specifically stated. The electrostatic potential in Fig. 4C was calculated using the program APBS (44).
FIGURE 4.

Crystal structure of SsgB(Tfus). A, monomer of SsgB from T. fusca (SsgB(Tfus)). Shown is a stereo ribbon diagram of the SsgB(Tfus) monomer color-coded from N terminus (blue) to C terminus (red). Helices α1–α3 and β-strands, β1–β8, are indicated. B, SsgB(Tfus) trimer. Each protomer is colored differently. Residue numbers at the ends of any gaps in the model are labeled. C, electrostatic surfaces of a SsgB(Tfus) monomer (left) and trimer (right). The color was scaled from −3 to +3 kT/e (blue, positive; red, negative electrostatic potential).
RESULTS
SsgB Functionality and Developmental Cell Division of Actinomycetes
The gene organization around the ssgB genes from Streptomyces species is similar to that of the genetic loci of the single ssg genes from T. fusca, Kineococcus radiotolerans, Nocardioides sp. JS614, Acidothermus cellulolyticus, Salinispora tropica and Salinispora arenicola and to one of the ssg genes from Frankia species alni, CcI3, and EAN1pec and from Sacch. erythraea (Fig. 1). Hence, gene synteny evidence suggests that the ssg genes have been derived from spread and/or duplication of an ancestral ssgB gene in actinomycetes (17). This gene synteny includes a gene for tRNAVal upstream of ssgB in all genera and at least three downstream-located tRNA genes (typically specifying tRNAGly, tRNACys, and tRNAVal) in all species except Salinispora. Furthermore, several conserved open reading frames are typically found in the vicinity of ssgB orthologues, including homologues of the S. coelicolor open reading frames SCO1531 for a threonine tRNA synthetase and SCO1540 for a conserved actinobacterial transmembrane protein (Pfam 09656) with three transmembrane domains (Fig. 1). As an exception, despite the gene synteny similarities, phylogenetic analysis places the SALP from A. cellulolyticus outside the SsgBG branch of the SALPs (17). Thus, it is unlikely that this protein functions as an SsgB orthologue in vivo. Studies on the horizontal transfer of pathogenicity islands in bacteria suggest that tRNA loci are common sites for the integration of foreign DNA sequences (45). Another striking feature of SsgB orthologues is that they are 99–100% conserved within the same species, which is the case in all streptomycetes (10 orthologues), in Frankia species (three orthologues), and in Salinispora (two orthologues). In all cases, no more than a single non-conservative amino acid change is afforded close to the C-terminal end, whereas at the nucleotide level, a much higher divergence is found.
FIGURE 1.

Genetic environment for ssgB loci in actinomycetes. Genes flanking Streptomyces ssgB that are conserved in the flanking regions of ssgB orthologues from other SALP-containing actinomycete genera are highlighted. Gene products for conserved genes are given in the box at the bottom.
To establish whether more divergent SsgB proteins from various actinomycetes have functional overlap with SsgB from S. coelicolor, the respective genes were introduced into the S. coelicolor ssgB null mutant GSB1 (21) to analyze their ability to restore sporulation. Low copy number plasmids based on pHJL401 were constructed, containing the ssgB orthologues from Frankia Cci3 (ssgB(Fran); pGWS555), K. radiotolerans (ssgB(Krad); pGWS299), Sacch. erythraea (ssgB(Sery); pGWS294), S. tropica (ssgB(Stro); pGWS271), or T. fusca (ssgB(Tfus); pGWS554). Additionally, we introduced the non-orthologous ssg genes from Sacch. erythraea (SACE5535) and from Frankia sp. CcI3 (Francci3_3418), which do not belong to the SsgBG subfamily (17). Details of the exact constructs are presented under “Experimental Procedures,” and the precise DNA sequences of the inserts and the predicted gene products are presented in supplemental Fig. S1. These constructs and the control plasmid (pHJL401 without insert) were individually introduced into the S. coelicolor ssgB mutant GSB1 (21) by transformation of protoplasts.
Mutants of S. coelicolor that produce aerial hyphae but fail to sporulate are called whi or white mutants, since they lack the gray spore pigment WhiE (46). Introduction of the control plasmid pHJL401 or the plasmid harboring Sacch. erythraea SACE5535 did not restore gray pigmentation. As a second important control, all plasmids were introduced into the ssgA mutant GSA3 (22), which is the only other SALP mutant of S. coelicolor with a non-sporulating phenotype. As expected, none of the constructs restored sporulation to GSA3 (data not shown). However, transformants carrying the expected ssgB orthologues ssgB(Fran), ssgB(Krad), ssgB(Sery), ssgB(Stro), or ssgB(Tfus) turned light gray after 5–6 days of incubation on SFM agar plates, indicating that sporulation had been restored to the S. coelicolor mutant and that these genes encoded functional homologues. Surprisingly, colonies expressing Francci3_3418 from Frankia Cci3 did have a gray appearance, despite gene synteny and phylogenetic analysis placing the gene product outside the SsgBG branch of the SALP phylogenetic tree.
For analysis by phase-contrast light microscopy, impression prints were made by pressing a microscope coverslip onto the surface of the solid-grown cultures of the transformants. The results largely corresponded to the degree of pigmentation. As expected, no spores were observed in the control strain containing pHJL401 or the construct expressing SACE5535 from Sacch. erythraea. Introduction of ssgB(Stro) or ssgB(Tfus) resulted in production, by the ssgB mutant, of many single spores and a few spore chains, whereas ssgB(Sery), ssgB(Krad), and ssgB(Fran) allowed formation of many spore chains, albeit with spores of irregular sizes and shapes (see the analysis by cryo-scanning electron microscopy below). Most surprising was the confirmation that transformants expressing the non-orthologous SALP Francci3_3418 from Frankia Cci3 sporulated abundantly. This exceptional result perhaps reflects the complex sporulation process in Frankia species, which may require two functional SsgB paralogues in a single organism.
Detailed Analysis of Transformants by Cryo-scanning Electron Microscopy
Detailed morphological analysis of surface-grown cultures of the various transformants and the parent M145 was performed by cryo-scanning electron microscopy. S. coelicolor M145 produced abundant and regular spore chains (Fig. 2A), whereas pHJL401 control transformants of the ssgB mutant produced the long non-coiling aerial hyphae typical of developmental mutants arrested in a very early stage of aerial growth (Fig. 2B). In agreement with LM results, aerial hyphae of ssgB mutant colonies expressing ssgB(Stro), ssgB(Sery), ssgB(Krad), ssgB(Tfus), or ssgB(Fran) produced a significant amount of spores (Fig. 2, C, D, and F–H). Expression of ssgB(Stro) resulted in the production of many single spores and few spore chains. Some variation in spore sizes was observed, although the majority of the spores had the characteristic oval shape (Fig. 2C). Colonies expressing ssgB(Sery), ssgB(Krad), ssgB(Tfus), or ssgB(Fran) produced predominantly spore chains. Spore-chain length and spore size varied significantly, and small (squashed) or very large (bloated) spores were found at a high frequency (Fig. 2, D and F–H). Colonies expressing SACE5535 produced only aseptate aerial hyphae (Fig. 2E), supporting the specific function of the ssgB orthologues. However, as was already observed by LM, colonies expressing Francci3_3418 (the second SALP from Frankia sp. CcI3) produced long spore chains (Fig. 2I).
FIGURE 2.

Analysis by cryo-scanning electron microscopy of 7-day-old surface-grown colonies. A, parental (wild type) strain S. coelicolor M145; B, S. coelicolor ssgB mutant GSB1 harboring pHJL401; C, GSB1 + pGWS271 (ssgB(Stro)); D, GSB1 + pGWS294 (ssgB(Sery)); E, GSB1 + pGWS295 (SACE5535); F, GSB1 + pGWS299 (ssgB(Krad)); G, GSB1 + pGWS554 (ssgB(Tfus)); H, GSB1 + pGWS555 (ssgB(Fran)); I, GSB1 + pGWS556 (Francci3_3418). White bars, 2.5 μm.
Spore Viability and Nucleoid Segregation
Considering the aberrant spore shapes and sizes, we analyzed nucleoid distribution in the spore chains of the transformants. DNA was visualized by simultaneous staining with PI and Syto-82 Orange (Syto). PI is a fluorescent dye that cannot penetrate the membrane of living cells and is used to stain DNA in dead or “leaky” cells, whereas the membrane-permeant Syto is used to stain DNA in living cells. Using a 515-nm long pass filter, DNA-bound PI emits a red-colored and DNA-bound Syto emits a green-colored fluorescent signal. In mature spore chains of the parental strain M145, well segregated chromosomes were observed (Fig. 3A). The DNA in these wild type spores was stained only by Syto, confirming that the cell wall of these spores was intact. In transformants of the ssgB null mutant containing any of the foreign ssgB genes, DNA was distributed over all spores (Fig. 3), suggesting proper DNA segregation and condensation, since defects in this process typically result in a high frequency of empty spores (20, 47). Even in spore chains consisting of both tiny and large spores, all compartments were fluorescently stained. Viability counts indicated that a number of spores in these transformants had a permeable (lysed) cell wall (supplemental Table S4). The lowest viability was found for transformants harboring SsgB(Stro), with around 35% dead spores, whereas close to 100% viability was found for transformants containing constructs expressing either SALP (SsgB or SsgB2/Francci3_3418) from Frankia.
FIGURE 3.

Analysis by phase-contrast and fluorescence microscopy. DNA was visualized by simultaneous staining with membrane-impermeant dye propidium iodine (red) and the membrane-permeant dye Syto-82 (green). A, wild type M145; B, S. coelicolor ssgB mutant GSB1 + pGWS271 (ssgB(Stro)); C, GSB1 + pGWS294 (ssgB(Sery)); D, GSB1 + pGWS299 (ssgB(Krad)); E, GSB1 + pGWS554 (ssgB(Tfus)); F, GSB1 + pGWS555 (ssgB(Fran)); G, GSB1 + pGWS556 (Francci3_3418).
Crystal Structure of the T. fusca SsgB Orthologue
The selenomethionine derivative of the full-length SsgB(Tfus) was expressed in E. coli with an N-terminal tobacco etch virus-cleavable His tag and purified by metal affinity chromatography. The crystal structure of SsgB(Tfus) was determined in space group P41 at 2.6 Å resolution using the MAD method. The structure was refined to Rcryst and Rfree of 23.0 and 27.0%, respectively. The model displays good geometry with 98.6% favorable main-chain torsion angles (one outlier) and 97.5% favorable side-chain rotamers according to MOLPROBITY (43). The mean residual error of the coordinates was estimated at 0.31 Å by an Rfree-based diffraction component precision index method (48). The final model contains one trimer (subunits A, B, and C; residues 1–137) per asymmetric unit. The β1-β2, β3-β4, and β6-β7 loops at the base of the trimer are highly mobile and were modeled only in promoter A, due to the lack of interpretable electron density in the other two subunits. The more ordered loops seen in promoter A are due to crystal contacts with a symmetry-related molecule. Data collection, model, and refinement statistics are summarized in Table 1.
SsgB(Tfus) is a single-domain protein consisting of eight β-strands (β1–β8) and three α-helices (α1–α3) arranged in an α + β fold (Fig. 4A). It contains two tandem repeats of a four-stranded β-sheet and an α-helix motif (β1–β4 + α1 and β5–β8 + α2) packed orthogonally. An additional helix α3 located at the C terminus interacts with another protomer in the trimer. Structural similarity searches with DALI (49) showed that the SsgB(Tfus) structure is most similar to mitochondrial RNA-binding protein 2 from Trypanosoma brucei (Protein Data Bank code 2gia, r.m.s. deviation of 3.6 Å for 107 aligned Cα, Z = 8.5) (50) and plant transcriptional regulator PBF-2 (Protein Data Bank code 1l3a, r.m.s. deviation of 4.4 Å for 107 aligned Cα atoms, Z = 6.4) (51), despite low sequence similarities (sequence identity <10%). Additionally, it has partial similarity to the Ral-binding domain of Exo84 (Protein Data Bank code 1zc3, r.m.s. deviation of 3.9 Å for 97 aligned Cα, Z = 5.8) (52), the N-terminal domain of coactivator-associated methyltransferase 1 (Protein Data Bank code 2oqb, r.m.s. deviation 3.4 Å for 83 aligned Cα, Z = 5.6) (53), and a “hypothetical” protein (Protein Data Bank code 2nvn, r.m.s. deviation 4.3 Å for 87 aligned Cα, Z = 5.5).
The SsgB(Tfus) trimer is bell-shaped, with dimensions of 71 × 67 × 47 Å (Fig. 4B), and is assembled primarily through α3 interaction with α1 and α2 of a neighboring protomer. The total buried surface area for the trimer is 4530 Å2 (1510 Å2/monomer). Interestingly, multimerization of PBF-2 and MRP1/2 also involves mostly helices, suggesting that the same strategy is followed for oligomer formation for other members of this fold family. SsgB(Tfus) is an acidic protein with a calculated isoelectric point of 4.5; the surface of SsgB(Tfus) has multiple electro negative patches throughout its surface for both monomer and trimer (Fig. 4C).
The structural similarity of SsgB(Tfus) to ssDNA-binding PBF-2 (51) and gRNA-binding proteins, MRP1 and MRP2 (50), is intriguing (Fig. 5). The positively charged concave surfaces, formed by the β-sheet, consisting of the first four strands in PBF-2 and MRPs, interact with ssDNA/gRNA. PBF-2 and MRP1-MRP2 both form “whirly” tetramers, such that ssDNA/gRNA can interact with extended surfaces of two promoters simultaneously. The core β-sheets of SsgB(Tfus) are highly similar to these proteins, especially to MRP1 and MRP2 (Fig. 5). Furthermore, SsgB(Tfus) also contains several equivalent positively charged residues (Arg29, Arg41, and His45), which in MRP1 are involved in binding phosphoryl groups of the gRNA backbone. However, SsgB(Tfus) lacks the concave-shaped surface (for nucleic acid interaction) due to differences in the mode of oligomerization (trimer versus tetramer) and in the conformation of the second half of β1 (Fig. 5).
FIGURE 5.

Comparison of SsgB(Tfus) with the plant transcriptional regulator PBF-2 (Protein Data Bank code 1l3a) and mitochondrial RNA-binding protein 2 MRP1-MRP2 (Protein Data Bank code 2gje). Ribbon representations of SsgB(Tfus), PBF-2, MRP1, and MRP2 are shown in the same superimposed orientation.
Analysis of Sequence Conservation and Mapping of Essential Residues
Although knowledge of their role in the developmental process and in cell division is accumulating, the precise molecular mode of action of SALPs is unknown. The crystal structure of SsgB(Tfus) provides a structural framework to understand mutational data (see below) and the evolution of SALPs.
We analyzed the sequence conservation of SsgBs presented in this study and two SsgAs (Fig. 6A). The hydrophobic core of the β-barrel is highly conserved. Conserved residues on the protein surface are predominantly clustered around the solvent-exposed edge of strand β4 (Fig. 6B). This strand interacts with the β5-β6 and β7-β8 loops. The first conserved cluster is close to Trp54 and consists of residues from the β4, β5-β6, and β7-β8 loops. The second cluster is centered on Tyr38 and also includes Tyr32, Asp36, Pro37, Ala39, Arg55, Glu123, and Asp130. This region is near the interface between helix α3 and the rest of the protein and also close to the trimer interface. Thus, maintenance of the proper orientation of α3 with respect to the rest of the monomer is important for SsgB(Tfus) function. Additionally, residues on helix α3 (Asp130 and Leu137) that are essential for interacting with neighboring promoter are also highly conserved, suggesting a critical role for α3 in oligomerization of SsgB(Tfus). The above observations also appear to be valid when all known homologous SALP sequences are considered, although the sequence diversity dramatically increases across species with only three residues strictly conserved, Trp54, Gly64, and Gly72. Thus, based on sequence conservation, we suggest that all SALPs are likely to adopt the same fold and function in a similar fashion.
FIGURE 6.

Mapping of sequence conservation and disruptive mutations of SsgA from Streptomyces coelicolor A3(2) (SsgA(Scoe)) onto SsgB(Tfus). A, sequence alignment between SsgB(Tfus) SsgB orthologues used in this study and two characterized SsgAs from S. coelicolor A3(2) and S. griseus. Secondary structures of SsgB(Tfus) are shown at the top. The disruptive mutants of SsgA(Scoe) are highlighted by stars in the bottom row. B, mapping of sequence conservation of sequences shown in A onto the structure of the SsgB(Tfus) monomer. Residues with a high degree of conservation are colored red. C, mapping of SsgA(Scoe) disruptive mutations onto the structure of the SsgB(Tfus) monomer. Residues that impact the function of SsgA(Scoe) are shown in orange. The surface representations of the disruptive mutations are shown on the right.
We previously constructed a large random mutant library of SsgA from S. coelicolor (SsgA(Scoe)), which shares 40% amino acid identity with SsgB(Tfus), and used it to map the functionally important residues (32). These data provided detailed insights into the location of functionally important residues, not only for SsgA but also (in light of the considerable number of highly conserved residues) for other SALPs, including SsgB. We mapped the functionally disruptive mutations (i.e. the functionally important residues) of SsgA(Scoe) onto the structure of SsgB(Tfus) (Fig. 6C). These residues correlate well with those that are most conserved among SALPs (Fig. 6B). Most of the disruptive mutations are located inside the hydrophobic core (Fig. 6C), confirming that the integrity of the SALP structure is important for their function. Several important residues map to either α3 (Leu137 SsgB(Tfus) or Leu134 SsgA(Scoe)) or the interface between α3 and the rest of the protein (Tyr38 and Arg58 of SsgB(Tfus); Tyr35 and Arg55 of SsgA(Scoe)), supporting the importance of α3 in SALP function. Arg55 and Glu123 form a buried salt bridge inside the protein. Several hydrophobic residues (such as Trp54, Leu91, Ala98, and Phe100), whose mutations are disruptive to SsgA function, map to a conserved surface patch (Fig. 6, B and C). Two other reported disruptive mutations that are not associated with this conserved surface, Leu85 and Leu96 to Pro mutations in SsgA(Scoe) (corresponding to Asn88 and Arg99, respectively, in SsgB(Tfus)), are likely to affect the stability of the protein, since they would hinder main-chain hydrogen bonding across the β-sheet in which they are located. Therefore, the current available experimental data are consistent with the notion that the conserved solvent-exposed residues are important for SALP function.
Oligomerization of SsgB and Potential Binding Site
Unlike PBF-2 and MRP1-MRP2, which form homo- or hetero-tetramers, SsgB(Tfus) was crystallized as a trimer. It is currently not entirely clear if this represents the physiologically relevant oligomeric state for SsgB. Size exclusion chromatography indicated that SsgB(Tfus) exists mainly as a monomer in solution. It is, therefore, possible that an equilibrium exists between monomeric and trimeric forms. At the same time, we have extensive evidence of SALP multimerization both in vitro and in vivo, including formation of trimers by S. coelicolor SsgE in vivo, formation of larger complexes by purified SsgA and SsgF (size exclusion chromatography), and focal localization of SsgA and SsgB as visualized by fluorescence microscopy. Purified SsgF forms very stable dimers and trimers, as identified by LC-MS (17, 32).4
In the context of the trimer, a portion of the conserved surfaces discussed above are exposed to solvent at the trimer base, whereas others are buried around the central channel of the trimer (Fig. 7). The long strands of β1 and β2 of SsgB(Tfus) are tilted toward β8 and form a barrel-like shape (Fig. 4A). A valley is created by the surrounding loops β3-β4, β1-β2, β7-β8, and β5-β6 and is lined by highly conserved hydrophobic residues Leu16, Val18, Val24, Leu26, Phe44, Leu46, Ala98, Val52, Trp54, Leu91, Pro94, and Phe100 that emanate mainly from either end of these loops (Fig. 7). The three patches, one from each monomer, are close to each other in the trimer near the 3-fold axis (Fig. 7). Two nearby loops (β5-β6 and β7-β8) are also probably important for SALPs. The β5-β6 loop has a highly conserved sequence motif 70GXGDV74 across all SALPs. Interestingly, Asp73 in this loop is close to the 3-fold axis and not readily accessible to solvent. The β7-β8 loop is also highly conserved among SsgBs, with a sequence motif 91LSSPXGXA98. SsgAs have a different sequence motif (LQVGX(D/E)XA), raising the possibility that the β7-β8 loop may be one of the structural determinants that differentiate SsgBs from SsgA and other SALPs.
FIGURE 7.

Hydrophobic surfaces of SsgB(Tfus) trimer for potential intermolecular recognition. A, mapping of disruptive mutations of SsgA(Scoe) onto the SsgB(Tfus) trimer, viewed down the 3-fold, non-crystallographic axis. Disruptive mutations are colored orange. The β-hairpin of a symmetry-related protomer B is shown in blue. B, hydrophobic base of SsgB(Tfus) trimer (white, hydrophobic; green, hydrophilic).
DISCUSSION
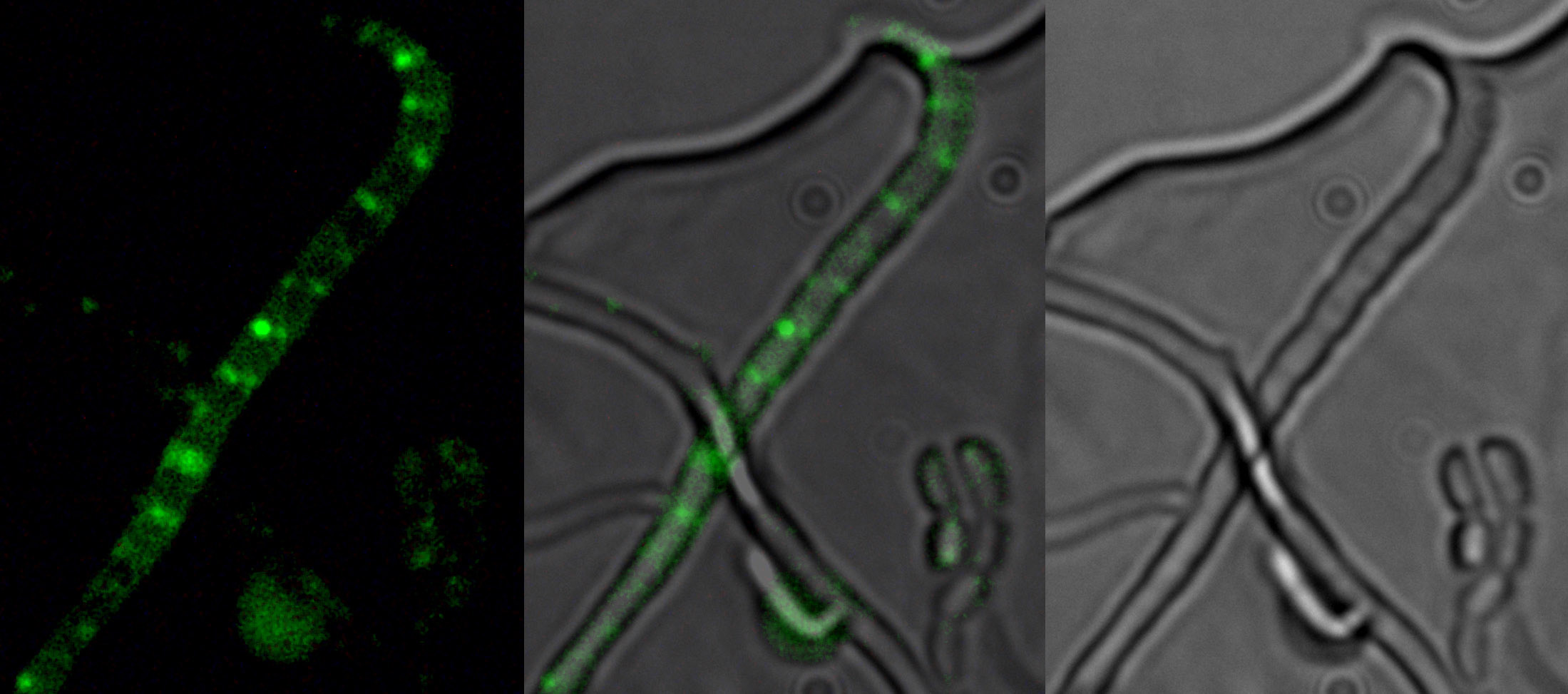
SsgA-like proteins form a unique family of cell division proteins in morphologically complex actinomycetes, which have no overt sequence similarity to any other characterized proteins. In Streptomyces, the well studied SsgA and SsgB proteins have previously been shown to play an important role in the control of sporulation-specific cell division. However, several important issues concerning SALP function have not yet been resolved. In particular, why are multiple SALPs found in actinomycetes that produce multiple spores, and how do they act at the molecular level? Also, what are the interacting partners of the SALPs? In this work, we show that SsgB is the archetypal SALP that is functionally conserved in all sporulating actinomycetes and plays a role in septum site localization. This functional relationship of SALPs to cell division is further substantiated by our recent discovery that SsgB localizes to the sporulation septa, strongly suggesting that SsgB is, in fact, a component of the divisome (supplemental Fig. S2).5
The three-dimensional structure of the archetypal SsgB protein is an important step forward in obtaining molecular insights into the function of SALPs. Toward this goal, we determined the crystal structure of the SsgB orthologue from T. fusca at 2.6 Å resolution, thus providing the first structural description of an SALP family member. The structure reveals structural similarities to a class of eukaryotic “whirly” ssDNA/RNA-binding proteins. The ssgB gene is surrounded by structural RNA-related genes (Fig. 1). These gene correlations coupled with structural similarity to RNA/DNA-binding proteins raise the possibility that SsgB could bind nucleic acids. However, experimental and structural evidence strongly argue against this notion. DNA binding studies using purified SsgA and SsgF with radiolabeled genomic DNA did not identify relevant nucleic acid binding (17).5 Furthermore, SsgB localizes exclusively to the sporulation septa, making it less likely that SALPs function primarily through binding of nucleic acids. Structurally, the electro-negative surface of the SsgB(Tfus) is not favorable for interaction with the electro-negative backbone of nucleic acids (Fig. 4C), whereas PBF-2 and MRP1/2 both have large positive electrostatic potentials to support ssDNA/single-stranded RNA binding. Homology modeling studies have indicated that other SALPs also possess highly electro-negative surfaces.
SsgB is essential for sporulation and determines colony size in S. coelicolor, as shown by the strictly non-sporulating and extremely large colonies that result from ssgB mutations. So far, it is unclear whether these two functionalities are the consequence of single or separate activities. ssgB orthologues from Frankia sp. Cci3, S. erythraea, S. tropica, K. radiotolerans, and T. fusca were all able to restore the capacity to initiate sporulation-specific cell division to ssgB mutants of S. coelicolor but not to ssgA mutants, strongly suggesting conserved functionality between ssgB orthologues. The large colony phenotype of the ssgB mutant was not complemented by the above orthologues, and therefore, the determinants for sporulation and for cessation of aerial growth may be separate activities of SsgB, perhaps through interacting with different cellular partners.
Interestingly, introduction of the non-orthologous SALP Francci3_3418 from Frankia Cci3 allowed production of viable spores by S. coelicolor ssgB mutants to similar levels as the ssgB orthologue (Francci3_1359) from the same species. The observation that, besides the likely SsgB orthologue Francci3_1359, at least one other Frankia SALP (Francci3_3418) has SsgB-like activity and suggests possible differences between sporulation processes in the actinomycetes, Streptomyces and Frankia.
Although the intrinsic ability to sporulate was restored by ssgB orthologues, significant anomalies were observed in spore and spore-chain morphologies (Fig. 2). The spore-chain lengths also varied considerably and correlated with the particular ssgB orthologue that was introduced; for example, expression of ssgB(Stro) resulted in the production of many single spores and very few (short) spore chains. Thus, spore-chain length may be yet another functionality determined by SsgB. DNA segregation and condensations were not significantly affected (Fig. 3). These data suggest that, although the intrinsic capacity to initiate and complete cell division had been restored to the S. coelicolor ssgB mutant GSB1, correct septum site localization was strongly perturbed. Precise positioning of the septum is primarily determined by localization of the Z-ring, which consists of polymeric rings of FtsZ (1). Variable spore sizes, as a result of incorrect septum placement, suggest that SsgB is possibly required to ensure correct localization of the Z-rings during sporulation. Indeed, as shown in supplemental Fig. S2, SsgB localizes to the divisome in sporogenic aerial hyphae.
Thus, our experiments provide evidence that the ability to initiate septum formation has been conserved over evolution in actinomycete SsgB orthologues. However, this complementation is imperfect, as illustrated by misplacement of the septa and variable and aberrant spore sizes. Since MinD and FtsZ are well conserved between E. coli and actinomycetes, one might expect a certain degree of similarity between these systems. The SsgB structure is not similar to other known cell division proteins in E. coli, such as MinE and ZipA. Interestingly, analogous to SsgB, the FtsZ binding domain of ZipA, which anchors FtsZ protofilaments to the membrane during invagination of the septum, also has a fold that is similar to many RNA-binding proteins (54). The C terminus of FtsZ interacts with the β-sheet surface of ZipA through hydrophobic contacts. It is possible that SsgB makes use of the conserved hydrophobic surface centered on Trp54 to interact with protein partners, such as FtsZ. This proposed binding site in protomer A is in contact with a symmetry-related β-hairpin at the N terminus of promoter B (residues 1–10), suggesting a possible mode of protein-protein recognition (Fig. 7A).
One of the remaining mysteries is how SALPs utilize a single conserved fold to carry out complex molecular functions. The SsgB(Tfus) structure suggests a few possibilities. First, the properties of the conserved hydrophobic surface could be altered by residues at the perimeter that are more variable. Second, we observed that the fold of SALPs can support different oligomeric states through similar contacts made through the α-helices. For example, PBF-2 is a homotetramer. Geometrically feasible models of dimers or tetramers of SsgB can be assembled by computer modeling (not shown). Therefore, it is plausible that different SALPs may function in different oligomeric states. Third, as seen in the example of the MRP1-MRP2 heterocomplex, the fold adopted by SALPs could also support formation of heterocomplexes between different types of SALPs, since they have similar structures. We have previously shown that SsgC may antagonize the activity of SsgA, since SsgA mutants have a similar phenotype to strains overexpressing SsgC, and vice versa (20). However, at least with the bacterial two-hybrid system (55), we have so far only been able to demonstrate interaction between the same SALPs (e.g. SsgA-SsgA or SsgB-SsgB),5 although the two-vector system makes analysis of simultaneous interactions between three different proteins (such as SsgABC trimers) impossible.
In conclusion, our studies highlight SsgB as an important control protein for sporulation-specific cell division in morphologically complex actinomycetes, as illustrated by the clear functional relationship between SsgB and spore-chain length and localization of SsgB exclusively to the septa in Streptomyces. The crystal structure of this archetypal SALP protein reveals structural similarity to whirly ssDNA/guide RNA-binding proteins that are found in mitochondria or in plants, although interaction of SsgB with nucleic acids is unlikely. In contrast, we identified a conserved surface for protein-protein interaction. We are currently investigating the role of SsgB in septum-site localization in sporulating actinomycetes and are conducting an extensive search for interaction partners for the different SALP proteins. These combined structural and functional results have shed new light on the function of this interesting protein family in the control of sporulation-specific cell division.
Supplementary Material
Acknowledgments
Genomic DNA from S. tropica was a kind gift from Paul Jensen (University of California San Diego, La Jolla, CA). Genomic DNA from the protease-deficient ER1 strain derived from T. fusca YX was a gift from David B. Wilson (Cornell University). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and the National Institutes of Health (National Center for Research Resources, Biomedical Technology Program, and NIGMS).
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grants U54 GM074898 and U54 GM074942 (to the Protein Structure Initiative, which supported the structural genomics project) and Netherlands Council for Scientific Research ECHO Grant 700.54.002 (to G. P. v. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 3cm1) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
G. P. van Wezel, unpublished data.
E. de Waal and G. P. van Wezel, unpublished data.
- SALP
- SsgA-like family of proteins
- MAD
- multiwavelength anomalous diffraction
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride
- ssDNA
- single-stranded DNA
- gRNA
- guide RNA
- PI
- propidium iodine
- Syto
- Syto-82 Orange.
REFERENCES
- 1.Errington J., Daniel R. A., Scheffers D. J. (2003) Microbiol. Mol. Biol. Rev. 67, 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goehring N. W., Beckwith J. (2005) Curr. Biol. 15, R514–R526 [DOI] [PubMed] [Google Scholar]
- 3.Rothfield L., Taghbalout A., Shih Y. L. (2005) Nat. Rev. Microbiol. 3, 959–968 [DOI] [PubMed] [Google Scholar]
- 4.Bramhill D. (1997) Annu. Rev. Cell Dev. Biol. 13, 395–424 [DOI] [PubMed] [Google Scholar]
- 5.Löwe J., Amos L. A. (1998) Nature 391, 203–206 [DOI] [PubMed] [Google Scholar]
- 6.Löwe J., van den Ent F., Amos L. A. (2004) Annu. Rev. Biophys. Biomol. Struct. 33, 177–198 [DOI] [PubMed] [Google Scholar]
- 7.van den Ent F., Amos L. A., Löwe J. (2001) Nature 413, 39–44 [DOI] [PubMed] [Google Scholar]
- 8.Hopwood D. A. (2007) Streptomyces in Nature and Medicine: the Antibiotic Makers, Oxford University Press, New York [Google Scholar]
- 9.Mazza P., Noens E. E., Schirner K., Grantcharova N., Mommaas A. M., Koerten H. K., Muth G., Flärdh K., van Wezel G. P., Wohlleben W. (2006) Mol. Microbiol. 60, 838–852 [DOI] [PubMed] [Google Scholar]
- 10.McCormick J. R., Su E. P., Driks A., Losick R. (1994) Mol. Microbiol. 14, 243–254 [DOI] [PubMed] [Google Scholar]
- 11.Elliot M. A., Buttner M. J., Nodwell J. R. (2008) in Myxobacteria: Multicellularity and Differentiation ( Whitworth D. E. ed) pp. 419–438, American Society for Microbiology Press, Washington, D. C. [Google Scholar]
- 12.Chater K. F. (2001) Curr. Opin. Microbiol. 4, 667–673 [DOI] [PubMed] [Google Scholar]
- 13.Flärdh K., Leibovitz E., Buttner M. J., Chater K. F. (2000) Mol. Microbiol. 38, 737–749 [DOI] [PubMed] [Google Scholar]
- 14.Wildermuth H., Hopwood D. A. (1970) J. Gen. Microbiol. 60, 51–59 [DOI] [PubMed] [Google Scholar]
- 15.Marston A. L., Thomaides H. B., Edwards D. H., Sharpe M. E., Errington J. (1998) Genes Dev. 12, 3419–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Autret S., Errington J. (2001) Dev. Cell 1, 10–11 [DOI] [PubMed] [Google Scholar]
- 17.Traag B. A., van Wezel G. P. (2008) Antonie Leeuwenhoek 94, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Del Sol R., Mullins J. G., Grantcharova N., Flärdh K., Dyson P. (2006) J. Bacteriol. 188, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chater K. F., Chandra G. (2006) FEMS Microbiol. Rev. 30, 651–672 [DOI] [PubMed] [Google Scholar]
- 20.Noens E. E., Mersinias V., Traag B. A., Smith C. P., Koerten H. K., van Wezel G. P. (2005) Mol. Microbiol. 58, 929–944 [DOI] [PubMed] [Google Scholar]
- 21.Keijser B. J., Noens E. E., Kraal B., Koerten H. K., van Wezel G. P. (2003) FEMS Microbiol. Lett. 225, 59–67 [DOI] [PubMed] [Google Scholar]
- 22.van Wezel G. P., van der Meulen J., Kawamoto S., Luiten R. G., Koerten H. K., Kraal B. (2000) J. Bacteriol. 182, 5653–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawamoto S., Watanabe H., Hesketh A., Ensign J. C., Ochi K. (1997) Microbiology 143, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 24.Lykidis A., Mavromatis K., Ivanova N., Anderson I., Land M., DiBartolo G., Martinez M., Lapidus A., Lucas S., Copeland A., Richardson P., Wilson D. B., Kyrpides N. (2007) J. Bacteriol. 189, 2477–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26.MacNeil D. J., Gewain K. M., Ruby C. L., Dezeny G., Gibbons P. H., MacNeil T. (1992) Gene 111, 61–68 [DOI] [PubMed] [Google Scholar]
- 27.Kieser T., Bibb M. J., Buttner M. J., Chater K. F., Hopwood D. A. (2000) Practical Streptomyces Genetics, the John Innes Foundation, Norwich, UK [Google Scholar]
- 28.Janssen G. R., Bibb M. J. (1993) Gene 124, 133–134 [DOI] [PubMed] [Google Scholar]
- 29.Larson J. L., Hershberger C. L. (1986) Plasmid 15, 199–209 [DOI] [PubMed] [Google Scholar]
- 30.Lydiate D. J., Malpartida F., Hopwood D. A. (1985) Gene 35, 223–235 [DOI] [PubMed] [Google Scholar]
- 31.Kormanec J., Sevcikova B. (2002) Mol. Genet. Genomics 267, 536–543 [DOI] [PubMed] [Google Scholar]
- 32.Noens E. E., Mersinias V., Willemse J., Traag B. A., Laing E., Chater K. F., Smith C. P., Koerten H. K., van Wezel G. P. (2007) Mol. Microbiol. 64, 1244–1259 [DOI] [PubMed] [Google Scholar]
- 33.Santarsiero B. D., Yegian D. T., Lee C. C., Spraggon G., Gu J., Scheibe D., Uber D. C., Cornell E. W., Nordmeyer R. A., Kolbe W. F., Jin J., Jones A. L., Jaklevic J. M., Schultz P. G., Stevens R. C. (2002) J. Appl. Crystallogr. 35, 278–281 [Google Scholar]
- 34.Cohen A. E., Ellis P. J., Miller M. D., Deacon A. M., Phizackerley R. P. (2002) J. Appl. Crystallogr. 35, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 36.Schneider T. R., Sheldrick G. M. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 37.Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 38.Terwilliger T. C., Berendzen J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40.Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 41.CCP4 ( 1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 42.Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 43.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ochman H., Lawrence J. G., Groisman E. A. (2000) Nature 405, 299–304 [DOI] [PubMed] [Google Scholar]
- 46.Chater K. F. (1972) J. Gen. Microbiol. 72, 9–28 [DOI] [PubMed] [Google Scholar]
- 47.Jakimowicz D., Brzostek A., Rumijowska-Galewicz A., Zydek P., Dołzbłasz A., Smulczyk-Krawczyszyn A., Zimniak T., Wojtasz L., Zawilak-Pawlik A., Kois A., Dziadek J., Zakrzewska-Czerwińnska J. (2007) Microbiology 153, 4050–4060 [DOI] [PubMed] [Google Scholar]
- 48.Cruickshank D. W. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 583–601 [DOI] [PubMed] [Google Scholar]
- 49.Holm L., Sander C. (1995) Trends Biochem. Sci. 20, 478–480 [DOI] [PubMed] [Google Scholar]
- 50.Schumacher M. A., Karamooz E., Zíková A., Trantírek L., Lukes J. (2006) Cell 126, 701–711 [DOI] [PubMed] [Google Scholar]
- 51.Desveaux D., Allard J., Brisson N., Sygusch J. (2002) Nat. Struct. Biol. 9, 512–517 [DOI] [PubMed] [Google Scholar]
- 52.Jin R., Junutula J. R., Matern H. T., Ervin K. E., Scheller R. H., Brunger A. T. (2005) EMBO J. 24, 2064–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Troffer-Charlier N., Cura V., Hassenboehler P., Moras D., Cavarelli J. (2007) EMBO J. 26, 4391–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mosyak L., Zhang Y., Glasfeld E., Haney S., Stahl M., Seehra J., Somers W. S. (2000) EMBO J. 19, 3179–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karimova G., Ullmann A., Ladant D. (2000) Methods Enzymol. 328, 59–73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}