

Abstract
Filamins are actin filament cross-linking proteins composed of an N-terminal actin-binding domain and 24 immunoglobulin-like domains (IgFLNs). Filamins interact with numerous proteins, including the cytoplasmic domains of plasma membrane signaling and cell adhesion receptors. Thereby filamins mechanically and functionally link the cell membrane to the cytoskeleton. Most of the interactions have been mapped to the C-terminal IgFLNs 16–24. Similarly, as with the previously known compact domain pair of IgFLNa20–21, the two-domain fragments IgFLNa16–17 and IgFLNa18–19 were more compact in small angle x-ray scattering analysis than would be expected for two independent domains. Solution state NMR structures revealed that the domain packing in IgFLNa18–19 resembles the structure of IgFLNa20–21. In both domain pairs the integrin-binding site is masked, although the details of the domain-domain interaction are partly distinct. The structure of IgFLNa16–17 revealed a new domain packing mode where the adhesion receptor binding site of domain 17 is not masked. Sequence comparison suggests that similar packing of three tandem filamin domain pairs is present throughout the animal kingdom, and we propose that this packing is involved in the regulation of filamin interactions through a mechanosensor mechanism.
Actin cytoskeleton is a dynamic network that is involved in many fundamental cellular processes such as cell differentiation, morphology, endocytosis, exocytosis, cytokinesis, and cell movement. These events are regulated by proteins that interact with monomeric and filamentous actin. Filamins are actin filament-binding and cross-linking proteins. Filamin A and filamin B are both ubiquitously expressed, and their mutations in human patients cause developmental abnormalities in brain, cartilage, bones, and epithelial tissues (1). Filamin C is muscle-specific, and mutations thereof cause myofibrillar myopathy (2). Mice with targeted deletion of any of the filamin genes die either during development or soon after birth (3–6). These phenotypes are thought to reflect the roles of filamins as scaffolds of signaling pathways required for cell differentiation, regulators of cell migration, and stabilizers of cytoskeleton and cell membranes (1, 7).
Filamins bind to actin filaments mainly via their N-terminal actin-binding domains and interact with other proteins via the 24 filamin type immunoglobulin-like domains (IgFLN),3 also called filamin repeats (8). Especially the C-terminal IgFLNs 16–24 contain several protein-protein interaction sites (1). Our previous structural studies have revealed that many proteins interact with filamins by forming an additional β-strand next to strand C of an individual IgFLN. The platelet von Willebrand factor receptor, glycoprotein (GP) Ibα, interacts in this way with IgFLNa17 (9). The integrin family adhesion receptor β subunits interact with IgFLNa21 and to a lesser extent with IgFLNa19 (10, 11). Furthermore, some signaling proteins use a similar interaction mode: the adaptor protein migfilin interacts with IgFLNa21 (12), and the Rho family GTPase-activating protein FilGAP interacts with IgFLNa23 (13, 14).
Although structural details are known from many filamin interactions, it is not completely clear how these interactions are regulated. In some cases the regulation involves competition between multiple binding partners (10, 11). Alternative splicing (15), proteolysis of filamin (16–18), and ligand phosphorylation (11) also contribute to the regulation. Recently, it has become apparent that conformational changes in filamins may also be involved. For instance, actomyosin contraction exposes hidden cysteine residues in filamins (19). This opens the possibility that forces transmitted through actin filament may open up binding sites, and filamin may thus be involved in mechanosensor signaling.
We have recently found a structural mechanism by which mechanical forces could regulate interactions at the C-terminal part of filamin. Our recent crystal structure revealed that IgFLNa20 forms a compact pair with IgFLNa21, and in this pair the N-terminal part of IgFLNa20 masks the integrin-binding site on IgFLNa21 (15). It is possible that this masking could be released by mechanical forces. Four lines of evidence led us to hypothesize that in addition to the IgFLNa20–21 pair, other similar domain pairs could exist at the C terminus of filamin: (i) the overall structure of the C-terminal part (IgFLNs 16–24) of filamin is relatively more compact than the N-terminal part of the molecule (IgFLNs 1–15) (8); (ii) the N-terminal sequences of even-numbered domains 16, 18, and 20 differ from other IgFLNs (20) (sequence alignment is shown in supplemental Fig. S1); (iii) in single-domain solution NMR structures of IgFLNc16, IgFLNb16, 18, and 20, the N-terminal part is not folded with the rest of the domain; and (iv) according to biochemical experiments, IgFLNa18 masks integrin binding to IgFLNa19 (15). We report here small angle x-ray scattering (SAXS) analysis showing that IgFLNa16–17 and 18–19 have overall dimensions very similar to those of the previously known domain pair IgFLNa20–21. The IgFLNa22–23 construct was much more elongated, which is indicative for two independently folded noninteracting domains. Further, the atomic structures solved with NMR spectroscopy show that IgFLNa18–19 forms a pair similar to IgFLNa20–21, but the details of the interaction and orientation of the domains differ. On the other hand, IgFLNa16–17 forms an entirely novel type of domain pair. Sequence comparisons predict that these three interdependent domain pairs are conserved from nematodes to vertebrates, suggesting that the arrangement has special regulatory functions.
EXPERIMENTAL PROCEDURES
Recombinant Proteins
The IgFLNa12–13 (amino acids 1353–1542), IgFLNa16–17 (amino acids 1772–1956), IgFLNa18–19 (amino acids 1954–2141), IgFLNa20–21 (amino acids 2141–2329), and IgFLNa22–23 (amino acids 2330–2522) fragments were generated by polymerase chain reaction and cloned into the modified pGEX vector (GE Healthcare). The inserts were checked by DNA sequencing. Glutathione S-transferase fusion proteins then were produced in Escherichia coli BL21 Gold cells and purified with glutathione-Sepharose 4 Fast Flow (GE Healthcare) according to the manufacturer's instructions. Glutathione S-transferase was cleaved by TEV protease at 4 °C for 16 h. The buffer was changed in a HiPrep 26/10 desalting column (GE Healthcare) to 100 mm NaCl, 1 mm dithiothreitol, 20 mm Tris, pH 8, and glutathione S-transferase was removed in a glutathione-Sepharose 4 Fast Flow column. The further protein purifications were performed by gel filtration in a HiLoad 26/60 Superdex 75 column (GE Healthcare), and finally the proteins were concentrated with Centriprep YM-3000 or YM-10000 (Millipore).
Small Angle X-ray Scattering and Data Processing
Synchrotron radiation x-ray scattering data were collected on the EMBL X33 beamline at the DORIS III storage ring, DESY, Hamburg (21). Solutions of FLNa fragments IgFLNa12–13, IgFLNa16–17, IgFLNa18–19, IgFLNa20–21, and IgFLNa22–23 in 100 mm NaCl, 10 mm dithiothreitol, 20 mm Tris, pH 8.0, were adjusted to concentrations of 2.8–9.6, 2.3–9.9, 1.9–7.8, 3.7–9.7, and 3.3–10.0 mg/ml, respectively. The MAR345 image plate at sample-detector distance 2.7 m and wavelength λ = 0.15 nm, covering the momentum transfer range 0.12 < s < 4.9 nm−1 (s = 4π sin(θ)/λ, where 2θ is the scattering angle) was used. The data were averaged after normalization to the intensity of the incident beam, the scattering of the buffer was subtracted, and the difference data were extrapolated to zero solute concentration following standard procedures. All of the data manipulations were performed using the program package PRIMUS (22).
The forward scattering I(0) and the radius of gyration (Rg) were evaluated using the Guinier approximation (23), assuming that at very small angles (s < 1.3/Rg) the intensity is represented as I(s) = I(0) exp(−1/3(Rgs)2). These parameters were also computed from the entire scattering patterns using the program GNOM (24), providing the distance distribution functions p(r) and the maximum particle dimensions Dmax. The molecular mass of the solute was evaluated by comparison of the forward scattering with that from reference solutions of bovine serum albumin (molecular mass, 66 kDa). The excluded volume of the hydrated particle (the Porod volume) was computed as follows (25).
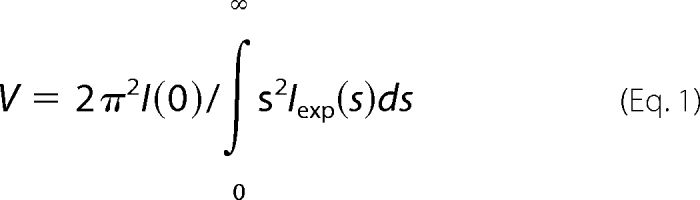 |
Prior to this analysis, an appropriate constant was subtracted from each data point to force the s−4 decay of the intensity at higher angles following Porod's law (25) for homogeneous particles. This “shape scattering” curve was further used to generate low resolution ab initio models of fragments IgFLNa12–13, 16–17, 18–19, 20–21, and 22–23 by the program DAMMIN (26) and DAMMIF (27), which represent the protein by an assembly of densely packed beads. Simulated annealing was employed to build a compact interconnected configuration of beads inside a sphere with the diameter Dmax that fits the experimental data Iexp(s) to minimize the discrepancy,
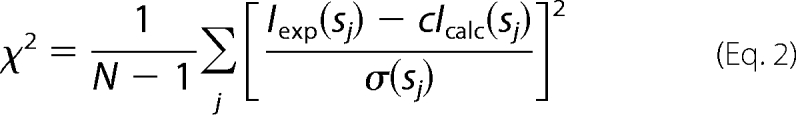 |
where N is the number of experimental points, c is a scaling factor, and Icalc(sj) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively.
The program GASBOR (28) was used to create ab initio models of proteins consisting of dummy residues instead of beads. In this program a simulated annealing protocol is employed to construct a model with a protein-like distribution of beads that provides the best fit to the experimental data. For the ab initio analyses, multiple runs were performed to verify the stability of the solution. The results from 10 separate runs of DAMMIN, DAMMIF, and GASBOR were averaged to determine common structural features using the program DAMAVER (29).
The calculated parameters for IgFLNa20–21 were estimated based on the crystal structure of IgFLNa19–21 (Protein Data Bank code 2j3s) (15) and for IgFLNa16–17 and FLNa18–19 based on the NMR structures using the program CRYSOL (30).
Structure Determination of IgFLNa16–17 and IgFLNa18–19
NMR sample preparation and conditions, spectroscopic details, and chemical shift assignment have been described earlier (31). For structure determination three-dimensional 13C- and 15N-edited NOE spectroscopy-heteronuclear single-quantum coherence spectra were recorded on a Varian INOVA 800-MHz spectrometer equipped with 5-mm z-gradient triple resonance probehead at 30 °C. Spectrum acquisition and processing was done using VNMRJ 2.1 and VNMR 6.1C software (Varian Inc.). Sparky 3.110 was used for spectrum analysis (Goddard TD, Kneller DG. University of California, San Francisco, CA). Dihedral angle constraints for χ and ψ angles were extracted from chemical shift data using TALOS software (32). The dihedral angle restraints were parameterized as (TALOS prediction ± 2 S.D.). Structure calculation was done using the automatic NOE assignment and torsion angle dynamics mode of CYANA 2.1 (33). Based on the lowest target function, 10% of calculated structures were chosen for further refinement. Molecular dynamics refinement of the final structures was done using a generalized Born implicit solvent model in AMBER 8.0 (38). Quality control of the structure families was done with WHAT CHECK (34) and PROCHECK-NMR (35) programs. Domain-domain interaction interfaces were analyzed on a ProtorP server (36). Experimental procedures of relaxation rate and heteronuclear NOE determination and peptide titrations can be found in the supplemental materials.
Binding Assays
To compare the binding of IgFLNa16–17 and IgFLNa17 fragments to GPIbα, the synthetic GPIbα peptide containing residues 556–577 (EZBiolab Inc., Westfield, IN) was coupled to an N-hydroxysuccinimide activated Sepharose 4 Fast Flow (GE Healthcare) at 4 °C according to the manufacturer's instructions. Purified IgFLNa16–17 and IgFLNa17 were incubated for 1 h at 23 °C with 20 μl of the peptide-Sepharose in 1% Triton X-100, 150 mm NaCl, 20 mm Tris, pH 7.4. The Sepharose was centrifuged at 15,000 × g for 1 min and washed twice with 300 μl of the binding buffer. The proteins were eluted with 10 μl of SDS electrophoresis sample buffer and run on a SDS-polyacrylamide gel.
RESULTS
Low Resolution Analysis Reveals Three Compact Domain Pairs
To study the presence of compact domain pairs in the C-terminal part of filamin A, we expressed and purified two-domain fragments and analyzed them by SAXS. The location of the studied fragments in filamin is shown in Fig. 1. IgFLNa12–13 was used as a control because no domain pair formation was expected in this area. All of the constructs behaved well in SAXS, and neither aggregation nor dimerization was observed. The experimental SAXS curves from the constructs are shown in Fig. 2, and the overall parameters computed from the data are presented in Table 1. The experimental values for radius of gyration (Rg) and maximal dimension (Dmax) from IgFLNa16–17, 18–19, and 20–21 were similar to the theoretical values calculated from the crystallographic structure of the IgFLNa20–21 pair (15) (Table 1). However, the values of Rg and Dmax for the IgFLNa12–13 and 22–23 were significantly higher, pointing to more extended structures. The long-tailed shape of distance distribution function p(r) for IgFLNa12–13 and 22–23 (Fig. 2, insets) was also consistent with elongated shapes.
FIGURE 1.

A diagram of filamin A domains and protein constructs used in this study. Filamin A is a dimer. Each polypeptide contains an N-terminal actin-binding domain (ABD) and IgFLNa 1–24. Filamin A dimerizes through the IgFLNa24. The constructs used in this study are marked in the diagram. Blue, IgFLNa12–13; magenta, IgFLNa16–17; yellow, IgFLNa18–19; cyan, IgFLNa20–21; green, IgFLNa22–23.
FIGURE 2.

Experimental SAXS data of two-domain fragments and fits of ab initio models. The x-ray scattering data from IgFLNa16–17, 18–19 and 22–23 (A) and IgFLNa12–13 and 22–23 (B) are displayed as dots. The scattering from typical ab initio models computed by DAMMIN/GASBOR is displayed as full lines. The plots display the logarithm of the scattering intensity as a function of momentum transfers, and successive curves are displaced down by one logarithmic unit for clarity. The distance distribution functions are presented in the insets.
TABLE 1.
Summary of SAXS measurements
C, concentration; Rg, radius of gyration; Dmax, maximum size of the particle, Vp, excluded volume of the hydrated particle, MMexp experimental molecular mass of the solute, and χab, discrepancy factors between the experimental scattering curves and those calculated from ab initio models.
| FLNa12–13 |
FLNa16–17 |
FLNa18–19 |
FLNa20–21 |
FLNa22–23 |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Observed | Observed | Calculated | Observed | Calculated | Observed | Calculated | Observed | ||
| C (mg/ml) | 2.8/9.6 | 2.3/9.9 | 1.9/7.8 | 3.7/9.3 | 3.3/10.0 | ||||
| Rg (nm) | 2.39 ± 0.01 | 1.93 ± 0.03 | 1.81 | 2.11 ± 0.04 | 2.11 | 1.91 ± 0.03 | 1.97 | 2.77 ± 0.05 | |
| Dmax (nm) | 8.6 ± 0.5 | 6.0 ± 0.5 | 6.3 | 6.5 ± 0.5 | 7.6 | 6.1 ± 0.5 | 6.3 | 9.0 ± 0.5 | |
| Vp (nm3) | 25 ± 4 | 33 ± 4 | 27 | 34 ± 4 | 33 | 31 ± 4 | 29 | 32 ± 4 | |
| MMexp (kDa) | 14 ± 4 | 22 ± 4 | 18.7 | 21 ± 3 | 20.4 | 16.8 ± 4 | 18.4 | 24 ± 4 | |
| χab | 1.45 | 1.69 | 1.87 | 1.37 | 1.78 | ||||
Typical low resolution shapes of IgFLNa16–17 and 18–19 reconstructed ab initio by DAMMIN (26) and GASBOR (28) (Fig. 3) provided good fits to the experimental data with the discrepancy factors of χ = 1.69–1.87 (curves are shown in Fig. 2) and displayed similar overall dimensions with IgFLNa20–21 (Fig. 3). The shape of IgFLNa18–19 had more pronounced features of two domains, whereas the envelope of IgFLNa16–17 was a bit more compact. Typical ab initio low resolution models of IgFLNa12–13 and 22–23 revealed that the shapes of these tandem domains significantly differ from those of IgFLNa16–17, 18–19, and 20–21. The shape of IgFLNa12–13 and 22–23 is elongated, suggesting a conventional head-to-tail arrangement of the domains (Fig. 3). In conclusion, our SAXS analysis suggests that IgFLNa16–17 and 18–19 form interacting domain pairs similar to those of IgFLNa20–21. IgFLNa22–23, on the other hand, does not appear to form such a pair.
FIGURE 3.

Ab initio SAXS models of two-domain fragments. The ab initio envelopes obtained by DAMMIN and GASBOR of IgFLNa12–13, 16–17, 18–19, 20–21, and 22–23 as determined from solution scattering experiment. The two views are rotated with respect to each other by a 90° rotation about the vertical axis. All ab initio envelopes are presented in the same scale. The dimensions of IgFLNa16–17, 18–19, and 20–21 are 6.0, 6.5, and 6.5 nm (vertical), 3 nm (horizontal, upper), and 3.5 nm (horizontal, lower), whereas the corresponding dimensions of IgFLNa12–13 and 22–23 are 9, 3.0, and 3.5 nm.
Structure of IgFLNa18–19 Resembles That of IgFLNa20–21
To study the atomic details of the IgFLNa18–19 and IgFLNa16–17 domain pairs, NMR spectroscopy was employed. The chemical shift assignments have been published elsewhere (31). Chemical shift mapping between single domains IgFLNa18 and 19 and the two-domain construct IgFLN18–19 showed that the largest changes were located at β-strands C and D as well as at the EF loop of IgFLNa19 (supplemental Fig. S3). These changes confirmed that the two domains indeed interact with each other in solution and that the interaction resembles the one found previously for IgFLNa20–21 (15). The structure of IgFLNa18–19 was solved with solution state NMR spectroscopy. Details of the structure calculations are described in supplemental materials, and structure quality indicators are presented in Table 2. Altogether, 2930 NOE-derived distance restraints and 235 chemical shift-based dihedral angle restraints were used in the structure calculation. The mutual orientation of the domains was based on 76 interdomain distance restraints, of which 42 are located between β-strand A of domain 18 and the CD face of domain 19 (see supplemental Fig. S2B for graphical representation of the interdomain distance restraints). Average backbone RMSD from the mean structure for the double-domain (residues 1960–2135) was ∼1 Å. The coordinate precision of single domains was better; average backbone RMSD from the mean structure was 0.8 Å for IgFLNa18 (residues 1960–2045) and 0.3 Å for IgFLNa19 (residues 2046–2135). There was some fluctuation in the mutual orientation of the domains in the structure ensemble (Fig. 4A). However, relaxation properties of the two domains were similar (supplemental Fig. S7), suggesting that the domains tumble in solution as a single unit and do not substantially wobble relative to each other. The fluctuation in domain orientation in the structure ensemble is most probably due to the relatively low number of NOE restraints between the main bodies of domain 18 and domain 19. Over 90% of the residues reside in the most favored regions of the Ramachandran plot. The IgFLNa18–19 structure family conforms well to the SAXS data (Table 1).
TABLE 2.
NMR structure statistics
| FLNa16–17 | FLNa18–19 | |
|---|---|---|
| Amino acids | 1772–1956 | 1954–2141 |
| Number of structures | 40 | 20 |
| Structure restraints | ||
| Total distance restraints | 3439 | 2930 |
| Short range |i − j| ≤ 1 | 1655 | 1483 |
| Medium range, 1 < |i − j| < 5 | 317 | 331 |
| Long range, |i − j| ≥ 5 | 1467 | 1116 |
| Interdomain | 99 | 76 |
| Distance restraints/residue | 18.5 | 15.3 |
| ϕ and ψ dihedral angle restraints | 202 | 235 |
| Violation statistics | ||
| Maximum NOE restraint violation (Å) | 0.22 | 0.22 |
| Number of NOE violations > 0.10 Å (n ± S.D.) | 8.2 ± 2.6 | 2.2 ± 1.1 |
| Maximum ϕ/ψ dihedral angle violation (°) | 7.6 | 14.2 |
| Number of ϕ/ψ dihedral angle violations > 5° (n ± S.D.) | 0.13 ± 0.33 | 0.15 ± 0.37 |
| Energies | ||
| Average restraint violation energy (kcal/mol ± S.D.) | 29.24 ± 1.30 | 16.23 ± 1.74 |
| Average AMBER energy (kcal/mol ± S.D.) | −4640.48 ± 13.01 | −5440.69 ± 16.61 |
| RMSD from ideal covalent geometry | ||
| Bond lengths (Å ± S.D.) | 0.0098 ± 0.0001 | 0.0097 ± 0.0002 |
| Bond angles (° ± S.D.) | 2.18 ± 0.01 | 2.22 ± 0.01 |
| Average coordinate RMSD from the mean structure (Å ± S.D.) | Residues 1787–1954 | Residues 1960–2135 |
| Backbone atoms | 0.48 ± 0.08 | 1.01 ± 0.32 |
| Heavy atoms | 0.85 ± 0.07 | 1.31 ± 0.29 |
| Ramachandran map regions (%) | ||
| Residues in most favored regions | 89.1 | 91.7 |
| Additionally allowed regions | 9.8 | 7.5 |
| Generously allowed regions | 0.9 | 0.4 |
| Disallowed regions | 0.2 | 0.4 |
FIGURE 4.

Solution Structure of IgFLNa18–19. IgFLNa18 (residues 1954–2045) is colored in blue, and IgFLNa19 (residues 2046–2141) is in red. A, superimposed backbone traces of the structure ensemble (20 structures) demonstrating the coordinate precision. Superimposition was done for residues 1960–2135. B, ribbon presentation showing the secondary structure elements (only residues 1958–2137 are shown). Structure visualizations A and B were generated with MOLMOL (37). C, detailed view of IgFLNa18 β-strand A bound to CD face of IgFLNa19. The most important residues for the domain interaction are labeled, and hydrogen bonds are indicated with dashed lines. D, detailed view of Tyr2077 of IgFLNa19 (red) and the hydrophopic residues of IgFLNa 18 in the domain interface. Note that the orientation of the structure is different from in other illustrations. C and D were created with PyMOL (DeLano Scientific LLC, Palo Alto, CA). E, sequence alignment of the BC loop of human IgFLNa19 containing Tyr2077 and the corresponding positions in other species.
The structure of the IgFLNa18-19 domain pair reveals that IgFLNa19 is folded as a conventional Ig domain, but IgFLNa18 does not constitute a complete Ig-fold (Fig. 4B). The first β-strand of IgFLNa18 is not folded as part of domain 18 but is instead bound to the CD face of IgFLNa19. In addition to the interaction between β-strand A of IgFLNa18 and β-strand C of IgFLNa19, also some hydrophobic contacts (particularly Leu1963 with Ile2092 and Thr2094) contribute to the interaction (Fig. 4C). A hydrogen bond is formed between the Ser2961 hydroxyl group and, depending on substructure of the structure family, either amide hydrogen or carbonyl oxygen of Val2090. The absence of β-strand A leaves the hydrophobic core of domain 18 partly exposed. This hydrophobic core anchors domain 18 orthogonally to the N-terminal end of IgFLNa19. A closer look at the structure shows that the side chain of Tyr2077 is pointing outwards from the BC loop of IgFLNa19 and sticks into the hydrophobic core of domain 18 (Fig. 4D). Many hydrophobic core residues of IgFLNa18 (Ile1971, Phe2011, Pro2013, Val2037, and Ile2039) interact with the aromatic side chain of Tyr2077. Also, residues of the AB loop of IgFLNa18 (e.g. Ala1969) and of the domain linker contribute to the domain interface. Total domain-domain interaction surface area between IgFLNa18 and 19 is 980 Å2. If the β-strand interaction is neglected, the interaction surface is considerably smaller: only 380 Å2. Overall structure of IgFLNa18–19 is similar to that of IgFLNa20–21 (15). In both structures, β-strand A of the even-numbered domain is masking the integrin-binding site on the odd-numbered domain. In our NMR titrations we could not detect binding of the integrin β7 peptide to IgFLNa18–19, whereas under equivalent experimental conditions the peptide clearly bound to isolated IgFLNa19 (supplemental Fig. S4). Although the overall fold of IgFLNa18–19 and IgFLNa20–21 are similar, there are several differences in the detailed interaction mode of the domains. These differences will be discussed later.
Structure of IgFLNa16–17 Reveals New Filamin Domain-Domain Interaction Mode
Based on chemical shift comparison between IgFLNa16–17 and IgFLNa17, it was predicted that the domain-domain interaction mode would be different from IgFLNa18–19 and IgFLNa20–21 and that domain 16 mainly interacts with the AG face of domain 17 (31). The structure calculation of IgFLNa16–17 was based on 3439 NOE-derived distance restraints and 202 dihedral angle constraints (Table 2). Ninety-nine interdomain distance restraints were found, and all of them were located between β-strands A and G of IgFLNa17 and B and G of IgFLNa16 (see supplemental Fig. S2A for graphical presentation of the interdomain distance restraints). Average backbone RMSD from the mean structure was ∼0.5 Å for the whole double domain (residues 1787–1954). The coordinate precision of the double domain was almost as good as for the individual domains: backbone RMSDs for IgFLNa16 (residues 1787–1865) and IgFLNa17 (residues 1866–1954) were 0.4 and 0.3 Å, respectively. The 99 interdomain distance restraints were sufficient to define the mutual orientation of the domains of IgFLNa16–17 in good precision. Based on the abundance of NOE distance restraints, high overall coordinate precision, and NMR relaxation analysis (see supplemental Fig. S6), the two domains are tightly bound to each other. Nearly all residues of the structure family reside on the favored areas of the Ramachandran plot. The structure also fits well with the SAXS data (Table 1).
As in IgFLNa18 and 20, the first predicted β strand of IgFLNa16 does not fold as in conventional filamin-type Ig domains. The residues 1772–1785 corresponding to β-strand A of IgFLNa16 lack long range distance restraints. The random coil conformations of these residues are well illustrated in Fig. 5A. The domain-domain interaction of IgFLNa16–17 turned out to be very different from the other two known IgFLN pairs. The two Ig domains are stacked on to each other so that the β-sheets are approximately parallel (Fig. 5B). As the first β strand of Ig domain 16 is unfolded, the hydrophobic core of the domain is exposed, and it binds tightly to the AG face of domain 17 (Fig. 5B). Interaction surface area between the two domains is 720 Å2. The domain-domain interaction is mostly based on hydrophobic interactions. Particularly the side chains of His1877 and Thr1876 from IgFLNa17 are located close to the exposed hydrophopic core of IgFLNa16 (Phe1791, Leu1793, Ile1795, Leu1856, and Phe1858) (Fig. 5C).
FIGURE 5.

Solution structure of IgFLNa16–17. IgFLNa16 (residues 1772–1865) is colored in green, and IgFLNa17 (residues 1866–1956) is in gray. A, superimposed backbone traces of the structure ensemble (40 structures) demonstrating the coordinate precision. Superimposition was done for residues 1787–1862 and 1867–1954. The first 13 residues of IgFLNa16 corresponding to strand A of the Ig-fold are random-coiled. B, ribbon presentation showing the secondary structure elements (only residues 1785–1956 are shown). Structure visualizations A and B were generated with MOLMOL (37). C, detailed view of the domain interface looking through IgFLNa16. The most important residues for the domain interaction are depicted with stick models and labeled. The picture was created with PyMOL (DeLano Scientific).
The structure of IgFLNa16–17 uncovers a completely new domain-domain interaction mode of IgFLNs. It also shows that the CD face of IgFLNa17 that interacts with the cytoplasmic tail of GPIbα (9) is not masked in the two-domain pair like the integrin-binding surfaces of IgFLNa19 and 20 (12). To verify this, we compared the binding of the solid phase-coupled GPIbα peptide to IgFLNa17 or 16–17. The data show that the binding of the one- and two-domain constructs is indistinguishable with the EC50 values of ∼10–20 μm (Fig. 6). The interaction of IgFLNa16–17 with GPIbα peptide was also verified with NMR spectroscopy (supplemental Fig. S5). GPIbα peptide binds to the CD face of domain 17 in the IgFLNa16–17 construct as it does with the isolated IgFLNa17. These data are consistent with the structure where IgFLNa16 interacts solely with the AG face of IgFLNa17 and does not interfere with the GPIbα interaction.
FIGURE 6.

Binding of GPIb peptide to single-domain and two-domain fragments. Binding of IgFLNa17 (closed squares) and IgFLNa16–17 (open circles) to GPIbα peptide as determined in a pull-down assay. The results from three independent experiments are shown so that the bound fraction is normalized to the value obtained with 200 μm protein concentration. The solid line shows the theoretical binding curve for 15 μm equilibrium dissociation constant (Kd).
DISCUSSION
We have here presented solution NMR structures of two new filamin domain pairs. The SAXS results showed that IgFLNa16–17 and 18–19 form tightly interacting domain pairs, whereas domains 22–23 do not exhibit such domain packing. IgFLNa16–17 seemed to be even more compactly packed than IgFLNa18–19. The structure of IgFLNa18–19 shares the overall folding pattern of IgFLNa domain pair 20–21 (15), but the details of domain-domain interaction are distinct. The structure of IgFLNa16–17 revealed completely new packing of Ig domains.
The folding pattern of IgFLNa18–19 closely resembles the one found previously for domain pair IgFLNa20–21 (15). The first β-strand of IgFLNa18 extends the CGF β sheet of domain 19 similarly to that of the IgFLNa20–21 pair (Fig. 7A). The main part of domain 18 lies on top of domain 19 (Fig. 7), interacting mainly with the BC loop of domain 19, again similarly as in the IgFLNa20–21 pair. However, there are notable differences in the relative domain orientations. Superimposition of the domains 19 and 21 (Cα RMSD = ∼1 Å) brings the even domains to the same position relative to the respective uneven domain, but in a different orientation. The angle between the longitudinal axes of the paired domains is approximately the same (90°), but the longitudinal axes of the even domains point to different directions (Fig. 7B). The most notable difference is the relative rotation of the even domains along their longitudinal axis. In consequence, the connecting loops between the two domains take completely different paths in the two pairs. The AB loop of IgFLNa18 is positioned close to domain 19 and is strongly involved in the domain interaction interface. Also the linker between IgFLNa18 and 19 is located close to the domain interface (Fig. 7A). On the contrary, in the 20–21 pair the AB loop of IgFLNa20 and the domain linker are exposed to solvent. Both domains 18 and 20 interact with the BC loop of their partners, but the mode of interaction is quite different. Although the G strand of IgFLNa20 makes a short β-strand interaction with IgFLNa21, in IgFLNa19 there is a single Tyr residue (Tyr2077) in the BC loop that is deeply buried in the hydrophobic core of domain 18 (Fig. 4D). In the 18–19 pair, the β strand G of 18 is outside from the interaction surface. In FLNa21 the position corresponding to Tyr2077 of FLNa19 is occupied by Ala2268. In conclusion, it seems that despite a similar overall arrangement, the interaction details of these IgFLNa pairs are not conserved.
FIGURE 7.

Comparison between the structures of IgFLNa18–19 and IgFLNa20–21 and conservation of domain arrangement. A and B, Ig domain 19 of IgFLNa18–19 structure was superimposed on Ig domain 21 of IgFLNa19–21 structure (Protein Data Bank code 2j3s) (15) using Cα atoms (pairwise RMDS, ∼1 Å). Ig domains are colored as follows: blue, IgFLNa18; red, IgFLNa19; yellow, IgFLNa20; cyan, Ig FLNa21. Two views from different perspectives are shown in A and B. Locations of the strands and loops mentioned under “Discussion” are indicated. The picture was created using PyMOL (DeLano Scientific). C, position of the three domain pairs in vertebrate filamins and predicted position of the homologous segments in D. melanogaster Cherio (Cher) and C. elegans Y66HB1.3–5 (constructed from predicted transcripts of three genes Y66HB1.3, Y66HB1.5, and Y66HB1.2 in WormBase Release WS201).
Based on similar overall dimensions in SAXS measurements, the structure of IgFLNa16–17 was expected to resemble the structures of the other two filamin domain pairs, IgFLNa18–19 and 20–21. The first sign of significant structural difference came from NMR resonance assignments (31). Comparison of the assignments of IgFLNa16–17 to the ones of isolated IgFLNa17 (9) showed that there were practically no chemical shift changes at the CD face that is mainly involved in the domain-domain interactions of IgFLNa18–19 and 20–21. On the contrary, the largest differences were found at the AG face. In line with this, NOE restraints established a strong domain-domain interaction with domain 16 at the AG face of domain 17 in the IgFLNa16–17 pair. There are several hydrophobic and aromatic residues at the AG surface of IgFLNa17, much more than in the corresponding parts of domains 19 and 21 (supplemental Fig. S1). As the hydrophobic core of the domain 16 is exposed because of unfolded strand A, there are strong prerequisites for this kind of domain-domain interaction to occur. To the best of our knowledge, this kind of Ig domain-domain interaction has not been presented before.
The interaction of IgFLNa17 with GPIbα has been characterized in detail by Nakamura et al. (9). The structure of domain 17 in the IgFLNa16–17 pair is very similar to the structure of isolated domain 17 (pairwise Cα RMSD of residues 1868–1954 is 1.1 Å), and because the domain-domain interaction takes place at the opposite face of domain 17, the interaction site at the C-strand is free. Our biochemical experiments (Fig. 6) and NMR titrations (supplemental Fig. S5) confirmed that the presence of IgFLNa16 does not interfere with GPIbα binding to IgFLNa17.
In conclusion, the three filamin A tandem Ig domain pairs characterized so far have surprised us with new structural features. There are two common denominators of the three domain pairs IgFLNa16–17, IgFLNa18–19, and IgFLNa20–21: (i) the two domains interact tightly with each other and form relatively compact structures, and (ii) the β-strand A of the even-numbered domains is not folded with its own domain but either is unstructured (IgFLNa16) or folds together with the following domain (IgFLNa18-19 and IgFLNa20–21). Incomplete folding of strand A of domains 16, 18, and 20 is also seen in single-domain structures from FLNb (Protein Data Bank codes 2EE9, 2DMC, 2DLG, and 2E9I) and FLNc (Protein Data Bank code 2D7N), implying that similar domain pairing is present also in these filamin isoforms. Furthermore, sequence comparisons show that similar segments of six IgFLNs where the first, third, and fifth domains have atypical A strand sequences can be found throughout the animal kingdom including Drosophila melanogaster (gene Cherio) and Caenorhabditis elegans (GenBankTM accession number Y66H1B) (supplemental Fig. S1). Remarkably, the residue corresponding to the key interaction sites between domains 18 and 19 (Tyr2077 in FLNa; Fig. 4E) as well as between domains 16–17 (marked in supplemental Fig. S1) are conserved. Interestingly, when this arrangement of three tandem domain pairs is found, it is located in the same position relative to the C terminus of the polypeptide independently on the total number of IgFLNs in the polypeptide (Fig. 7C). We suggest that this specific arrangement of three tandem domain pairs close to the C terminus of filamin is evolutionally conserved because it has specific functions in regulating protein binding to filamins. Most probably the interaction sites can be opened via mechanical stretching of the protein by actimyosin contraction, which would fit well in the role of filamin in mechanosensor signaling (19).
Supplementary Material
Acknowledgments
We thank Arja Mansikkaviita for excellent technical assistance. AMBER MD calculations were run on a computer cluster of The Finnish IT Center for Science. The 1H-15N heteronuclear single-quantum coherence resonance assignments of IgFLNa19 were kindly provided by Dr. Pengju Jiang and Dr. Iain Campbell (University of Oxford).
This work was supported by funds from the Finnish National Graduate School in Informational and Structural Biology (to S. R. and O. K. H.), Academy of Finland Grant 114713 (to J. Y.), European Union Design Study SAXIER Contract 011934 (to D. I. S. and P. V. K.), and European Union Framework Program 6 Contract RII3/CT/2004/506008.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7, supplemental Methods, and supplemental references.
The atomic coordinates and structure factors (codes 2k7p and 2k7q) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- IgFLN
- filamin immunoglobulin-like domain
- GP
- glycoprotein
- NOE
- nuclear Overhauser enhancement
- RMSD
- root mean square deviation
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1.Feng Y., Walsh C. A. (2004) Nat. Cell Biol. 6, 1034–1038 [DOI] [PubMed] [Google Scholar]
- 2.Vorgerd M., van der Ven P. F., Bruchertseifer V., Löwe T., Kley R. A., Schröder R., Lochmüller H., Himmel M., Koehler K., Fürst D. O., Huebner A. (2005) Am. J. Hum. Genet. 77, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalkilic I., Schienda J., Thompson T. G., Kunkel L. M. (2006) Mol. Cell. Biol. 26, 6522–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng Y., Chen M. H., Moskowitz I. P., Mendonza A. M., Vidali L., Nakamura F., Kwiatkowski D. J., Walsh C. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19836–19841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X., Tian F., Sandzén J., Cao R., Flaberg E., Szekely L., Cao Y., Ohlsson C., Bergo M. O., Borén J., Akyürek L. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3919–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrington-Rock C., Kirilova V., Dillard-Telm L., Borowsky A. D., Chalk S., Rock M. J., Cohn D. H., Krakow D. (2008) Hum. Mol. Genet. 17, 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popowicz G. M., Schleicher M., Noegel A. A., Holak T. A. (2006) Trends Biochem. Sci. 31, 411–419 [DOI] [PubMed] [Google Scholar]
- 8.Nakamura F., Osborn T. M., Hartemink C. A., Hartwig J. H., Stossel T. P. (2007) J. Cell Biol. 179, 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura F., Pudas R., Heikkinen O., Permi P., Kilpeläinen I., Munday A. D., Hartwig J. H., Stossel T. P., Ylänne J. (2006) Blood 107, 1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiema T., Lad Y., Jiang P., Oxley C. L., Baldassarre M., Wegener K. L., Campbell I. D., Ylänne J., Calderwood D. A. (2006) Mol. Cell 21, 337–347 [DOI] [PubMed] [Google Scholar]
- 11.Takala H., Nurminen E., Nurmi S. M., Aatonen M., Strandin T., Takatalo M., Kiema T., Gahmberg C. G., Ylänne J., Fagerholm S. C. (2008) Blood 112, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 12.Lad Y., Jiang P., Ruskamo S., Harburger D. S., Ylänne J., Campbell I. D., Calderwood D. A. (2008) J. Biol. Chem. 283, 35154–35163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohta Y., Hartwig J. H., Stossel T. P. (2006) Nat. Cell Biol. 8, 803–814 [DOI] [PubMed] [Google Scholar]
- 14.Nakamura F., Heikkinen O., Pentikäinen O. T., Osborn T. M., Kasza K. E., Weitz D. A., Kupiainen O., Permi P., Kilpeläinen I., Ylänne J., Hartwig J. H., Stossel T. P. (2009) PLOS One 4, e4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lad Y., Kiema T., Jiang P., Pentikäinen O. T., Coles C. H., Campbell I. D., Calderwood D. A., Ylänne J. (2007) EMBO J. 26, 3993–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azam M., Andrabi S. S., Sahr K. E., Kamath L., Kuliopulos A., Chishti A. H. (2001) Mol. Cell. Biol. 21, 2213–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raynaud F., Jond-Necand C., Marcilhac A., Fürst D., Benyamin Y. (2006) Int. J. Biochem. Cell Biol. 38, 404–413 [DOI] [PubMed] [Google Scholar]
- 18.Heuzé M. L., Lamsoul I., Baldassarre M., Lad Y., Lévêque S., Razinia Z., Moog-Lutz C., Calderwood D. A., Lutz P. G. (2008) Blood 112, 5130–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson C. P., Tang H. Y., Carag C., Speicher D. W., Discher D. E. (2007) Science 317, 663–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorlin J. B., Yamin R., Egan S., Stewart M., Stossel T. P., Kwiatkowski D. J., Hartwig J. H. (1990) J. Cell Biol. 111, 1089–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) J. Appl. Crystallogr. 40, S190–S194 [Google Scholar]
- 22.Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guinier A. (1939) Ann. Physics 12, 161–237 [Google Scholar]
- 24.Svergun D. (1992) J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 25.Porod G. (1982) in Small Angle X-ray Scattering ( Glatter O., Kratky O. eds) pp. 17–51, Academic Press, New York [Google Scholar]
- 26.Svergun D. I. (1999) Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franke D., Svergun D. I. (2009) J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volkov V. V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 860–864 [Google Scholar]
- 30.Svergun D., Barberato C., Koch M. H. J. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 31.Heikkinen O. K., Permi P., Koskela H., Ylänne J., Kilpeläinen I. (2009) Biomol. NMR Assignments 3, 53–56 [DOI] [PubMed] [Google Scholar]
- 32.Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 33.Herrmann T., Güntert P., Wüthrich K. (2002) J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 34.Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 36.Reynolds C., Damerell D., Jones S. (2009) Bioinformatics 25, 413–414 [DOI] [PubMed] [Google Scholar]
- 37.Koradi R., Billeter M., Wüthrich K. (1996) J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 38.Case D. A., Darden T. A., Cheatham T. E., III, Simmerling C. L., Wang J., Duke R. E., Luo R., Merz K. M., Wang B., Pearlman D. A., Crowley M., Brozell S., Tsui V., Gohlke H., Mongan J., Hornak V., Cui G., Beroza P., Schafmeister C., Caldwell J. W., Ross W. S., Kollman P. A. (2004) AMBER 8, University of California, San Francisco, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.