

Abstract
A critical role of the amyloid precursor protein (APP) in Alzheimer's disease (AD) pathogenesis has been well established. However, the physiological function of APP remains elusive and much debated. We reported previously that the APP family of proteins is essential in mediating the developing neuromuscular synapse. In the current study, we created a conditional allele of APP and deleted APP in presynaptic motor neuron or postsynaptic muscle. Crossing these alleles onto the APP-like protein 2-null background reveals that, unexpectedly, inactivating APP in either compartment results in neuromuscular synapse defects similar to the germline deletion and that postsynaptic APP is obligatory for presynaptic targeting of the high-affinity choline transporter and synaptic transmission. Using a HEK293 and primary hippocampus mixed-culture assay, we report that expression of APP in HEK293 cells potently promotes synaptogenesis in contacting axons. This activity is dependent on neuronal APP and requires both the extracellular and intracellular domains; the latter forms a complex with Mint1 and Cask and is replaceable by the corresponding SynCAM (synaptic cell adhesion molecule) sequences. These in vitro and in vivo studies identify APP as a novel synaptic adhesion molecule. We postulate that transsynaptic APP interaction modulates its synaptic function and that perturbed APP synaptic adhesion activity may contribute to synaptic dysfunction and AD pathogenesis.
Introduction
Alzheimer's disease (AD) is the most common form of dementia in aged population. It is characterized by the deposition of β-amyloid plaques, of which the principal components are 40 and 42 aa β-amyloid peptides (Aβ) derived from proteolytic processing of the amyloid precursor protein (APP). Although β-amyloid plaques form the pathological hallmark of AD, loss of synapses is known to correlate closely with cognitive impairment. As such, it is widely accepted that synaptic dysfunction is an early and causal event in the pathogenesis of AD (for review, see Selkoe, 2002). Because of the pivotal role of APP in AD, it is essential to understand its physiological function, particularly its potential activity in synaptic regulation.
APP represents a family of conserved type I membrane proteins including APL-1 in Caenorhabditis elegans, APP-like (APPL) in Drosophila, and APP, APP-like protein 1 (APLP1), and APLP2 in mammals. Full-length APP undergoes sequential processing to generate APP extracellular and intracellular derivatives in addition to Aβ peptides (for review, see Zheng and Koo, 2006). Structural analysis revealed that the extracellular domains could form parallel or antiparallel dimers (Wang and Ha, 2004; Gralle et al., 2006). Cell culture studies also support the homodimer or heterodimer formation of the APP family members (Soba et al., 2005; Kaden et al., 2008). Significantly, the trans-dimerization of APP proteins has been reported to promote cell–cell adhesion (Soba et al., 2005; Kaden et al., 2008).
APP is highly expressed in neurons and can also be detected in numerous other tissues including the muscle. Neuronal APP undergoes rapid anterograde transport and is targeted to the presynaptic terminals (for review, see Zheng and Koo, 2006). APP can also be delivered to the somatodendritic compartments, and a recent report provides evidence that APP colocalizes with PSD95 and interacts with the NMDA receptor (Hoe et al., 2009).
Mice deficient in APP are viable exhibiting impairment in synaptic plasticity and spatial learning and memory (Zheng and Koo, 2006). The subtle phenotypes observed in APP-null mice are in part attributable to genetic redundancies. Our analysis of APP/APLP2 double knock-out (dKO) mice identified an essential role for the APP family of proteins in neuromuscular synapse patterning (Wang et al., 2005). Specifically, the dKO animals exhibit poorly formed and diffused neuromuscular synapse structure with reduced apposition of presynaptic and postsynaptic proteins and defective synaptic transmission (Wang et al., 2005). Our follow-up investigation suggests that this is likely attributable to a potent role of APP in regulating the presynaptic expression and activity of the high-affinity choline transporter (CHT), a molecule that mediates the rate-limiting step of cholinergic synaptic transmission, in both the peripheral neuromuscular junction and central cholinergic neurons (Wang et al., 2007).
By creating mice with tissue-specific deletion of APP, we reveal here that presynaptic and postsynaptic APP coordinate to mediate neuromuscular synapse assembly, and, significantly, this transsynaptic mechanism may underlie APP function in central synapse as well.
Materials and Methods
Antibodies and reagents.
Anti-neurofilament (NF), anti-MAP2, anti-CHT, anti-Mint1, anti-calcium/calmodulin-dependent serine protein kinase (Cask), and 22C11 antibodies were purchased from Millipore Bioscience Research Reagents; anti-synaptophysin and anti-SV2 antibodies were from Dako and Developmental Studies Hybridoma Bank, respectively; and anti-Flag antibody was from Sigma-Aldrich. The anti-rabbit monoclonal APP antibody used for immunostaining recognizes the C-terminal sequence of APP containing the YENPTY motif and was available from Epitomics (clone Y188). The polyclonal anti-APP C-terminal antibody APPc was described previously (Wang et al., 2007). α-Bungarotoxin (α-BTX) was from Invitrogen.
DNA constructs.
The human full-length APP (695 form), APPΔC, and APPc99 in pcDNA3 vectors were described by Wang et al. (2007). APP extracellular deletion constructs were generated from the full-length cDNA by PCR and site-directed mutagenesis. Amino acids 22-189 and 289-500 of APP were excised to generate APPΔE1 and APPΔE2 construct, respectively. To generate a glycosyl phosphatidylinositol (GPI)-anchored form of APP, the entire APP extracellular sequence was fused with exon 5 of mouse AchE (acetylcholine esterase), which encodes the 43 C-terminal amino acid residues including the signal for GPI modification. To construct the APP/synaptic cell adhesion molecule (SynCAM) chimera, the entire cytoplasmic tail of APP (amino acids 649-695) was replaced by the intracellular sequences of SynCAM1 (amino acids 428-474), which was amplified by reverse transcription (RT)-PCR of mouse brain RNA.
For constructing lentiviral expression vectors, the following primers were used to amplify the cDNAs from their pcDNA3-based vectors by PCR. The amplified fragments were subsequently cloned into the lentiviral shuttle vector FUGW(cmv)-RBN at NheI and PacI sites (Xu et al., 2008): APP FL and APPc99, 5′-ATGCTAGCGCCACCATGCTGCCCGGTTTGGCA-3′; NheI; 5′-GATTAATTAACTAGTTCTGCATCTGCTCAAAGAA-3′; PacI; APPΔC, 5′-ATGCTAGCGCCACCATGCTGCCCGGTTTGGCA-3′; 5′-GATTAATTAACTACTTCAGCATCACCAAGGTGAT-3′; APP/SynCAM, 5′-ATGCTAGCGCCACCATGCTGCCCGGTTTGGCA-3′; 5′-GATTAATTAACTAGATGAAGTACTCTTTCTTTTCTTCGGAG-3′. Mint1 and Cask constructs were kindly provided by Dr. Tom Südhof (Stanford University School of Medicine, Stanford, CA). To construct expression vectors for bimolecular fluorescence complementation (BiFC) analysis, the N-terminal 172 aa of YFP (nYFP) or the C-terminal 173-238 aa of YFP (cYFP) from the Venus-nYFP or Venus-cYFP vectors (Chen et al., 2007) were amplified by PCR and cloned into the C-terminal end of APP, and both the N terminus and the C terminus of Mint1 and Cask as in-frame fusion proteins. APP constructs with YENPTY motif deletion were generated with site-directed mutagenesis kit (Stratagene). The final constructs were verified by DNA sequencing.
Mouse production, breeding, and genotyping.
The creation of APP conditional allele is described in supplemental Methods (available at www.jneurosci.org as supplemental material). These mice, as well as all other animals used for this study, have been backcrossed onto C57BL/6J background for six generations. The neuronal double conditional knock-out (N-dCKO) and muscle double conditional knock-out (M-dCKO) mice were generated by crossing onto the APLP2-null background and from the following breeding: APPfl/+; APLP2−/− × Cre; APPfl/+; APLP2−/− → APP+/+; APLP2−/− (control), Cre; APPfl/fl; APLP2−/− (N-dCKO or M-dCKO).
The APLP2 genotyping was described previously (von Koch et al., 1997). The wild-type (WT) and APP floxed allele were identified by PCR using the following primers: p1, 5′-GAC CAT CCA GAA CTG GTG CAAG-3′, and p2, 5′-TCC CCC AGG CTT GGG ATA CAC ATT A-3′.
The two primers amplify the following: WT allele, 443 bp; floxed (fl) allele, 503 bp because of insertion of loxP and restriction sites.
The Nestin-Cre and Mck-Cre transgenic mice were identified by PCR using the following primers, which amplify a 791 bp fragment: CRE-F, 5′-GGC GTT TTC TGA GCA TAC CTG GAA-3′; and CRE-R, 5′-CAC CAT TGC CCC TGT TTC ACT ATC-3′.
For embryonic analysis, timed mating was set up, and the day when a vaginal plug was observed was considered to be embryonic day 0.5 (E0.5).
All animal experiments were performed in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee and with national regulations and policies.
Immunofluorescence staining.
Whole-mount immunostaining of the diaphragm muscle and quantification of neuromuscular phenotypes were performed as described (Wang et al., 2005). For muscle APP immunostaining, the stenomastoid muscles fixed overnight in 4% paraformaldehyde were isolated, frozen in OCT (Triangle Biomedical Sciences), and cryosectioned at 20 μm longitudinally. Sections were permeabilized with 0.1% Triton X-100 in PBS (PBST) for 30 min, blocked with 3% BSA and 4% goat serum in PBST for 1 h, and incubated with the APP rabbit monoclonal antibody Y188 (1:250) in PBST with 4% goat serum at room temperature for 30 min and then continued at 4°C for overnight. Sections were then washed six times for 5 min each in PBS and incubated with Alexa 555-conjugated goat anti-rabbit secondary (1:1000) and Alexa 488-conjugated α-BTX (1:1000) in PBST with 2% goat serum for 3 h. Sections were again washed six times for 5 min each in PBS and then mounted with Fluoromount-G mounting medium. Confocal images were obtained with a Zeiss 510 laser-scanning microscope, and quantification was done using the ImageJ program from National Institutes of Health.
Electrophysiology.
Synaptic transmission of neuromuscular junction (NMJ) was examined at 28 ± 0.5°C using intracellular recordings on acutely isolated phrenic nerve–diaphragm preparations at postnatal day 0 (P0) to P2. Muscles were dissected in ice-cold oxygenated normal Ringer's solution containing the following (in mm): 116 NaCl, 4.5 KCl, 1 MgSO4, 23 NaHCO3, 1 NaH2PO4, 11 dextrose, 2 CaCl2, pinned onto a Sylgard-coated recording chamber, and continuously superfused with oxygenated Ringer's solution at a rate of 2 ml/min. Sharp glass microelectrodes with resistance that ranged from 15 to 25 MΩ were filled with 3 m KCl. Miniature endplate potentials (mEPPs) were recorded in normal Ringer's solution containing 2.3 μm μ-Conotoxin GIIIB (Bachem) to selectively block muscle voltage-gated sodium channels and, therefore, muscle contraction. Data were collected with a MultiClamp 700B amplifier, digitized at 10 kHz, and recorded to a computer using pClamp 9 software (Molecular Devices). Off-line data analysis was performed using Clampfit 9 (Molecular Devices), MiniAnalysis (Synaptosoft), and OriginPro 7.5 (OriginLab).
Primary hippocampal/HEK293 mixed culture.
The preparation of primary hippocampal culture and coculture for synaptic puncta quantification were performed essentially as described previously (Biederer and Scheiffele, 2007). Briefly, primary hippocampal cultures were prepared from P0 C57BL/6J mice. Dissected hippocampi was suspended in trypsin-EDTA for 15 min at 37°C, washed three times with Ca2+–Mg2+-free HBSS, and triturated with a fire-polished glass pipette to dissociate the cells. Cells were resuspended in serum-free Neurobasal medium with B27 supplement (Invitrogen) and plated onto 12 mm coverslips pretreated with poly-d-lysine (5 μg/ml; Sigma-Aldrich) at a density of 30,000 cells/cm2.
HEK293 cells were transiently cotransfected with neuroligin 1 (NL1) or APP together with green fluorescent protein (GFP) at 10:1 molar ratio. After 24 h, the transfected cells were seeded at 30,000 cells/cm2 to the above hippocampal neurons cultured for 6–7 d in vitro. Forty-eight hours later, the cultures were fixed and stained with an anti-synaptophysin antibody (1:500; Dako) followed by incubation with the Alexa Fluor 594-conjugated secondary antibody. The cells were also counterstained with an anti-MAP2 antibody (1:1000; Millipore Bioscience Research Reagents), and the MAP2-negative synaptic puncta were selected for the quantification of heterologous synapse formation. Synapses were identified by the punctuate structures atop cocultured HEK293 cells and quantified by confocal microscopy followed by ImageJ analysis.
Lentiviral infection.
The production of recombinant lentivirus was done by cotransfection of the expression vector FUGW(cmv)-APP, the packaging vector CMVΔ8.9 and pVSVG into HEK293T cells by Lipofectamine 2000 (Invitrogen). HEK293 cells were split 1 d before the transfection and were ∼70–80% confluent on the day of transfection. The DMEM culture medium was replaced with neuronal culture medium (Neurobasal containing 1× B27 and 0.5 mm l -glutamine) before the transfection. For each T75 flask, 10 μg of FUGW(cmv)-APP, 7.5 μg of CMVΔ8.9 and 5 μg of pVSVG plasmid were used for transfection. Sixty hours after transfection, the supernatant was collected and filtered with 0.45 μm filter, aliquoted, and stored in −80°C. Hippocampal neurons were infected 1 d after plating using unconcentrated viral supernatant (∼50–100 μl/ml media). Five to 6 d later, one-half of infected neurons were cocultured with HEK293 cells expressing APP and GFP as described above. The APP-expressing neurons and axons were identified by GFP fluorescence. The other one-half was used for Western blot analysis of APP expression using the APPc antibody.
Western blotting and biotinylation.
Mouse brain, spinal cord, or muscle samples or cultured neuron or HEK293 cells were homogenized using RIPA lysis buffer (1% NP-40, 50 mm Tris, pH 8.0, 150 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 2 mm EDTA) containing Complete protease inhibitor mixture (Roche). After three sets of 10 pulses of sonication, the homogenates were spun at 14,000 rpm for 15 min. Protein concentrations were determined using Bio-Rad DC Protein Assay. Ten micrograms of protein were loaded on a 10% SDS-PAGE gel run at 100 V for 2 h at room temperature and transferred onto a nitrocellulose membrane (Bio-Rad) at 100 V for 1 h. Membranes were blocked 1 h using 5% nonfat dry milk in PBS containing 0.1% Tween 20 (PBS-Tween) (Sigma-Aldrich). Primary antibody incubation was done in 5% milk in PBS-Tween. After three washes with PBS-Tween, secondary antibody application was performed at room temperature for 1 h using 5% milk in PBS-Tween followed by three additional washes with PBST. Bands were visualized using Immobilon Western ECL system (Millipore).
For biotinylation of surface proteins, HEK293 cells were transiently transfected with the APP constructs. Cells were washed with ice-cold PBS containing 1.0 mm MgCl2 and 0.1 mm CaCl2 (PBS/Ca–Mg) and treated with sulfo-NHS-SS-biotin (1.5 mg/ml; Pierce) for 1 h on ice in PBS/Ca–Mg. Biotinylating reagents were removed by incubating with cold 100 mm glycine in PBS/Ca–Mg for 30 min, followed by three washes with cold PBS/Ca–Mg. Cells were then lysed in 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) lysis buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, and protease inhibitors (Roche). Biotinylated and nonbiotinylated proteins were separated by incubation with Ultralink-Neutravidin bead (Pierce) for 1 h at room temperature. Samples were subject to Western blot analysis as described above.
Results
Presynaptic and postsynaptic APP are both required for neuromuscular synapse patterning
We generated a conditional allele of APP by inserting two loxP sequences flanking exon 3 of the APP gene (supplemental Methods and Fig. S1, available at www.jneurosci.org as supplemental material). The neomycin-resistant gene, inserted in intron 2 and also flanked by the loxP sequences, was first removed by partial Cre-recombinase-mediated recombination on crossing with a weak germline deleter CMV-Cre (line B6) (Papaioannou and Behringer, 2005), creating an exon 3 floxed allele (fl) (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). As predicted, insertion of the loxP sites surrounding the exon 3 did not affect APP expression (fl/fl) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material), whereas removal of exon 3 and neo by a complete germline deleter GDF9-iCre (ΔE3) (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) disrupted the reading frame and resulted in a null allele (ΔE3/ΔE3) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material) (Lan et al., 2004). The floxed APP allele was used for subsequent tissue-specific knock-out experiments.
To address whether the function of APP in NMJ was mediated through a presynaptic or postsynaptic mechanism, we created mice with specific deletion of APP in presynaptic motor neurons [neuronal conditional knock-out (N-CKO)] or in postsynaptic muscle [muscle conditional knock-out (M-CKO)] by crossing the floxed APP allele with transgenic mice expressing the Cre-recombinase under the neuronal rat nestin promoter (Nestin-Cre) (Tronche et al., 1999) or the muscle creatine kinase promoter (Mck-Cre), which have been shown to lead to almost complete inactivation of target genes in motoneuron or muscle, respectively (Schwander et al., 2004; Tang et al., 2004). Because profound NMJ defects can be identified in APP/APLP2 double-null animals, we subsequently crossed the APP N-CKO and M-CKO mice onto APLP2-null background to create neuronal or muscle double conditional knock-out (N-dCKO or M-dCKO), respectively. Littermate APLP2-null mice were used as controls as no overt NMJ defects can be detected in these animals (Wang et al., 2005, 2007). Western blot analysis showed that, indeed, the APP protein was dramatically diminished in the spinal cord, but not in the muscle of N-dCKO mice (Fig. 1 A). Similarly, crossing with the Mck-Cre line led to a greatly reduced APP expression in the muscle, but not the spinal cord (Fig. 1 A). The residual expression in M-dCKO muscle samples is likely attributable to APP expressed in the nerve terminals.
Figure 1.

Biochemical and immunohistochemical characterization of APP expression and localization. A, Western blot analysis of APP protein levels from P0 spinal cord or muscle samples of APLP2-null control (Ctrl), neuronal or muscle conditional APP knock-out on APLP2-null background (N-dCKO and M-dCKO, respectively) using the APPc antibody. Tubulin was used as a loading control. B, Immunofluorescence staining of P0 stenomastoid muscle sections of control (Ctrl), APP/APLP2 double knock-out (dKO), N-dCKO, or M-dCKO animals using the anti-APP antibody Y188. α-BTX staining was used to mark the postsynaptic AchRs. Merge, Overlay of APP and AchR images. Scale bar, 20 μm. C, Quantification of APP signal intensity (mean ± SEM) (2 animals/genotype; 6 sections/animal) in AchR-positive endplates. **p < 0.01, ***p < 0.001 compared with control (one-way ANOVA).
APP has been shown to be enriched in the endplates of the adult muscle (Akaaboune et al., 2000). Since our studies identified a prominent role of APP proteins in the developing neuromuscular synapse, we sought to examine the APP localization in P0 NMJ. Importantly, the creation of motoneuron or muscle APP conditional knock-out mice allows us to distinguish the APP expressed from postsynaptic and presynaptic compartments, respectively. We performed immunostaining of P0 stenomastoid muscle sections using various anti-APP antibodies. Staining of APP-null and APP/APLP2 dKO tissues showed that most of the antibodies yielded background staining indicating cross-reactivity with other nonspecific proteins (data not shown). Nevertheless, we were able to identify a rabbit monoclonal antibody (Y188) that was highly specific for APP as shown by minimum background in APP-null muscle (data not shown) and no signal in dKO samples (Fig. 1 B). The specificity of the antibody was further established in primary neuronal cultures and in transfected cells (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Immunostaining using this antibody showed that APP was highly concentrated at acetylcholine receptor (AchR)-positive sites in control, N-dCKO, and M-dCKO muscles (Fig. 1 B). Significantly, APP immunoreactivity was reduced by ∼50% in both the neuronal and muscle conditional knock-out samples (Fig. 1 C). These results demonstrate that neuronal and muscle synthesized APP are targeted to the presynaptic and postsynaptic sites at 1:1 stoichiometry.
Consistent with positive expression of APP in both presynaptic and postsynaptic terminals, whole-mount staining of P0 stenomastoid muscles with an anti-synaptophysin (Syn) antibody and α-BTX, which marks presynaptic terminals and postsynaptic AchRs, respectively, demonstrated that inactivation of APP/APLP2 in either the motor neuron (N-dCKO) or the muscle (M-dCKO) resulted in diffused patterning of the presynaptic and postsynaptic terminals similar to that of germline APP/APLP2 dKO mice (Fig. 2 A). These were associated with excessive nerve terminal sprouting identified by neurofilament staining (Fig. 2 A, NF). These results establish that both presynaptic and postsynaptic APP expression is required for proper patterning of the developing NMJ. Intriguingly, quantitative comparison of neuronal and muscle conditional deletion with the dKO animals on average endplate band width (Fig. 2 B), distribution (Fig. 2 C), and number (Fig. 2 D) all showed that, whereas the N-dCKO mutant exhibited a partial phenotype (differences between N-dCKO and dKO were statistically significant), the degree of NMJ defect in M-dCKO was not statistically different from APP/APLP2 dKO in all the measurements. As such, postsynaptic APP/APLP2 deletion apparently leads to a more severe NMJ defects compared with the motor neuron inactivation. Closer examination of the synapse at higher resolution (Fig. 2 E) and quantification of the apposition of synaptophysin-positive staining with AchRs (Fig. 2 F) also showed lower coverage of the presynaptic marker with postsynaptic endplates in both neuronal and muscle conditional mutants (Fig. 2 E, arrowheads) with M-dCKO exhibiting a more severe phenotype. This is associated with extrasynaptic expression of synaptophysin in all mutant animals (Fig. 2 E, arrows).
Figure 2.
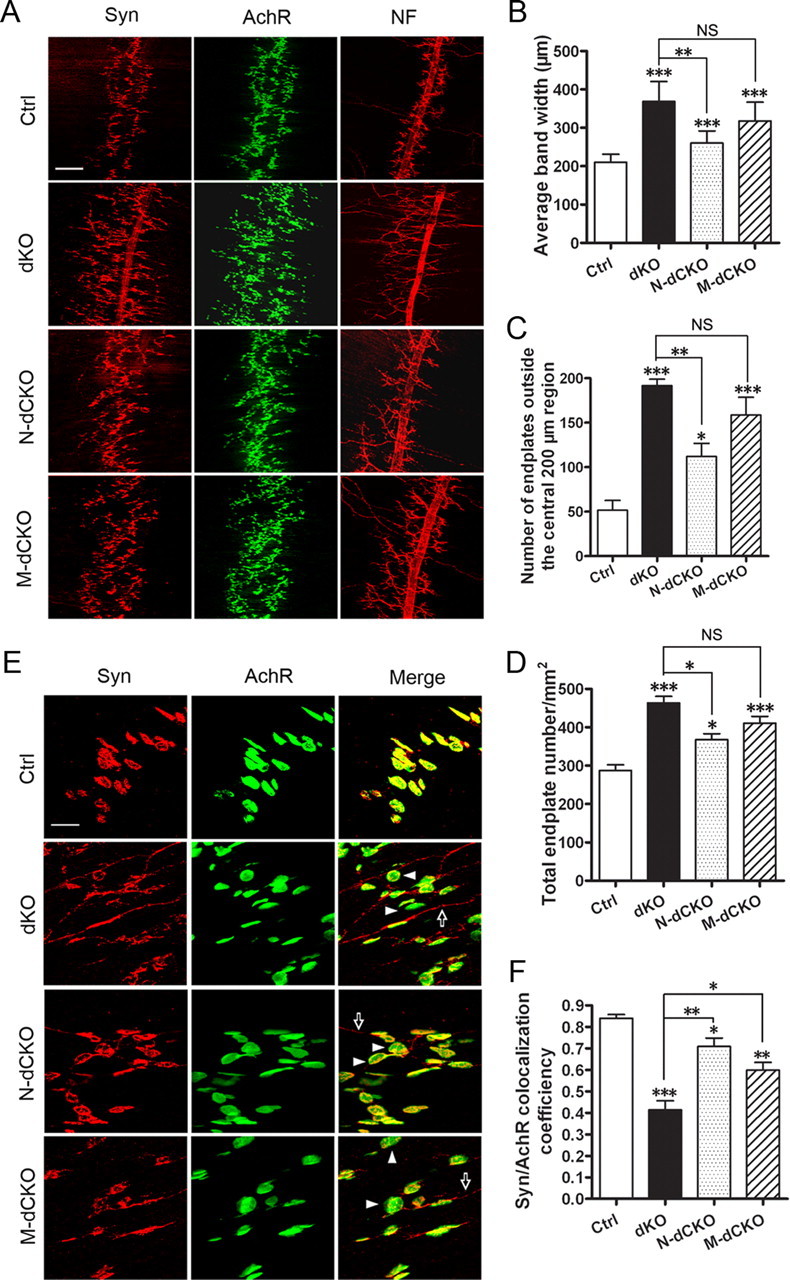
Neuromuscular synapse defects in APP/APLP2 motor neuron (N-dCKO) and muscle (M-dCKO) knock-out mice. A, Whole-mount immunostaining of control, dKO, N-dCKO, or M-dCKO P0 diaphragm muscles with antibodies against Syn or NF. α-BTX staining was used to mark the AchRs. Both the germline and conditional mutants showed diffused presynaptic and postsynaptic distribution and nerve terminal sprouting. B, Quantification of the average band width of AchR-positive endplates. C, Quantification of the number of AchR-positive endplates outside the central 200 μm band zone. D, Measurement of the number of endplate per unit area. B–D are expressed as mean ± SEM (6 animals/genotype). E, Higher magnification images of synapse structures with representative endplates poorly covered by Syn marked by arrowheads and extrasynaptic Syn staining by arrows. F, Quantification of the percentage of AchR-positive endplates covered by Syn (average ± SEM of 20 endplates/genotype). The asterisks above each bar are in comparison with the control. The asterisks on top of the brackets are in comparison with the dKO mice. *p < 0.05; **p < 0.01; ***p < 0.001; NS, nonsignificant (p > 0.05) (one-way ANOVA). Scale bars: A, 100 μm; E, 20 μm.
Nestin-Cre is known to be expressed in neural progenitors, which results in inactivation of target genes in all neuronal types as well as in glia (Tronche et al., 1999). To establish that the neuromuscular phenotypes seen in N-dCKO animals are specifically caused by the loss of APP in spinal cord motor neurons, we crossed the APP floxed allele with a strain of knock-in mice that expresses the Cre-recombinase under the choline acetyltransferase (ChAT) promoter (Chattm1(cre)Lowl) and confers cholinergic-specific neuronal expression (http://jaxmice.jax.org/strain/006410.html). On crossing onto the APLP2-null background, we show that NMJ defects similar to that of N-dCKO animals can be readily detected (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Postsynaptic APP is required for presynaptic CHT targeting and spontaneous vesicle release
The requirement for both presynaptic and postsynaptic APP expression in neuromuscular synapse assembly could result from independent APP activities in these two compartments or APP–APP interaction across the synapse. Since intercellular APP dimerization has been previously documented (Soba et al., 2005), and our immunostaining demonstrated the presence of APP in presynaptic and postsynaptic compartments at ∼1:1 stoichiometry, we favor the transsynaptic APP interaction as the functional modality. This model, contrasting to the independent mode of action mechanism, posits that presynaptic structure and function is dependent on postsynaptic APP expression and vice versa. We chose to perform immunostaining and functional recording to test this prediction. We reported previously that APP plays a potent role in regulating the localization and activity of the high-affinity CHT, a presynaptic protein that recycles choline from the synaptic cleft back to presynaptic terminals, and that defective CHT localization to presynaptic terminals in the absence of APP represents a primary effect rather than secondary to nerve sprouting or impaired synaptic transmission (Wang et al., 2007). As such, CHT serves as a unique presynaptic marker to test its dependency on postsynaptic APP expression. We performed immunohistochemical staining of CHT in the NMJ of N-dCKO and M-dCKO mutants (Fig. 3). In control NMJ, CHT was predominantly localized to presynaptic terminals and was closely apposed to the postsynaptic AchRs. In NMJs of APP/APLP2 double-null (dKO) animals, the coverage of AchR-positive endplates by CHT was greatly reduced, and this phenotype was shared in both the neuronal and muscle conditional knock-out NMJ (Fig. 3 A, quantified in B). Furthermore, consistent with the general NMJ characterization, postsynaptic deletion led a more severe CHT impairment compared with the neuronal knock-out mutant (Fig. 3 B). These data demonstrate that postsynaptic APP expression is obligatory for proper presynaptic targeting of CHT. This activity is likely mediated through neuronal APP as N-dCKO mice showed similar defects.
Figure 3.

Aberrant CHT localization in presynaptic terminals of APP mutant NMJ. A, Whole-mount diaphragm muscles from P0 control (Ctrl), dKO, N-dCKO, or M-dCKO animals were stained with anti-CHT antibody or α-BTX (AchR). The images were captured by confocal microscopy and displayed either as individual staining or merged (last row). Representative endplates with poor CHT coverage were marked by arrowheads and extrasynaptic CHT staining by arrows. Scale bar, 20 μm. B, Quantification of the percentage of AchR-positive endplates covered by CHT (average ± SEM of 20 endplates/genotype). The asterisks above each bar are in comparison with the control. The asterisks on top of the brackets are in comparison with the dKO mice. *p < 0.05; **p < 0.01; NS, nonsignificant (p > 0.05) (one-way ANOVA). C, RT-PCR analysis of APP, APLP1, and APLP2 expression in P0 brain, spinal cord, and muscle samples. RT, With reverse transcriptase; NR, the same sample and reaction in the absence of reverse transcriptase. GAPDH was used as amplification control.
Above studies using both general synaptic markers and cholinergic specific protein CHT demonstrate that postsynaptic APP deletion leads to a more severe impairment compared with the presynaptic counterpart. We reasoned that quantitative differences in presynaptic and postsynaptic knock-out mutants could be attributed by the neuronal specific expression of APLP1 since, in adult tissues, APP and APLP2 are known to be ubiquitously expressed, whereas APLP1 is considered a neuronal specific protein. Whether this applies to neonatal stage has not been established. We thus performed RT-PCR analysis of APP, APLP1, and APLP2 expression in the brain, spinal cord, and muscle samples of P0 neonates (Fig. 3 C). As expected, APP and APLP2 expression could be detected in all three tissues. In contrast, whereas APLP1 was readily detected in the brain and spinal cord, it was absent in the muscle (Fig. 3 C). As such, expression of APLP1 in N-dCKO, but not M-dCKO mutant could partially compensate for the loss of APP and APLP2 (further discussed and illustrated in Fig. 5 E).
Figure 5.
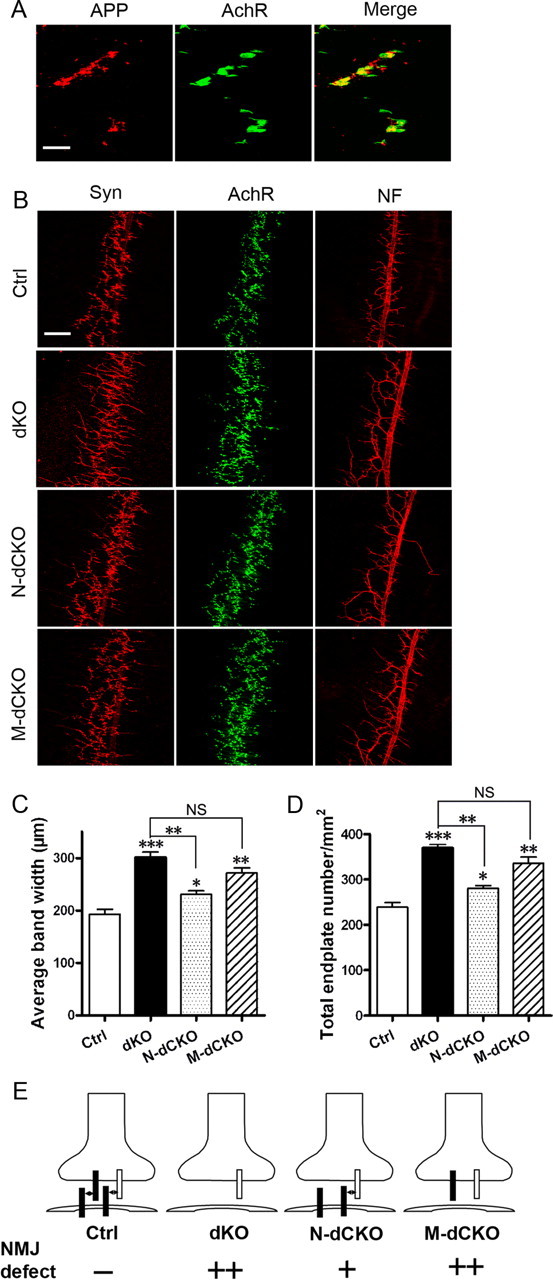
Expression analysis of APP and characterization of APP/APLP2 conditional mutant during embryonic development. A, Double labeling of E16.5 diaphragm muscles with the anti-APP antibody Y188 (APP) and α-BTX (AchR). Merge, Overlay of APP and AchR images. Scale bar, 20 μm. B, Staining of E16.5 whole-mount diaphragm muscles of control, dKO, N-dCKO, and M-dCKO embryos with anti-synaptophysin antibody (Syn), α-BTX (AchR), or anti-neurofilament antibody (NF). Scale bar, 100 μm. C, Quantification of the average endplate band width. D, Quantification of the number of endplate per unit area. All are expressed as mean ± SEM (N = 5 animals/genotype). The asterisks above each bar are in comparison with the control. The asterisks on top of the brackets are in comparison with the dKO mice. *p < 0.05; **p < 0.01; ***p < 0.001; NS, nonsignificant (p > 0.05) (one-way ANOVA). E, A transsynaptic APP interaction model and explanation for the quantitative differences between dKO, N-dCKO, and M-dCKO mutants based on the model. Filled bar, APP; open bar, APLP1.
We reported that mice lacking APP/APLP2 exhibit defective spontaneous mEPP frequency while preserving mEPP amplitude (Wang et al., 2005), indicating a functional deficit in presynaptic terminals. Having established an essential role of postsynaptic APP in mediating presynaptic CHT targeting, we next sought to determine whether postsynaptic APP deletion leads to functional deficits in presynaptic vesicle release by performing sharp-electrode intracellular recordings of P0 diaphragm muscle fibers of N-dCKO and M-dCKO mutant animals (Fig. 4). Consistent with results obtained using APP/APLP2 dKO animals (Wang et al., 2005), dramatically reduced mEPP frequency was observed in both N-dCKO and M-dCKO mice when compared with the littermate APLP2-null controls (Fig. 4 B), whereas mEPP amplitudes of sampled fibers were similar (Fig. 4 C). These data suggest a functional deficit in presynaptic terminals resulting from either motor neuron or muscle APP deficiency. Of note, we did not observe quantitative differences in N-dCKO and M-dCKO animals. This could be attributable to the limitations of experimental sensitivity associated with the specific assays.
Figure 4.

Miniature synaptic responses in NMJ of Ctrl, N-dCKO, and M-dCKO mice. A, Representative traces of mEPPs. Calibration: 1 mV, 0.5 s. B, Pooled data indicate significant reductions in the frequency of mEPPs in N-dCKO (N = 14) and M-dCKO (N = 31) NMJs compared with controls (N = 31). C, mEPP amplitude showed no significant differences among three groups tested. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01 (one-way ANOVA).
Developmental studies of APP expression and neuromuscular phenotypes
The above studies demonstrate an essential requirement for both presynaptic and postsynaptic APP expression in neuromuscular synapse assembly and an obligatory role for postsynaptic APP in mediating presynaptic protein expression and function. Although the data strongly support the transsynaptic APP interaction model, the question arises as how could transsynaptic APP interaction take place at NMJ where the presynaptic and postsynaptic terminals are separated by the basal lamina?
The mammalian NMJ undergoes dynamic changes during development. At E14.5, motor neuron axons initiate contact with muscle fibers to form rudimentary neuromuscular synapses. This is followed by active inductive interaction between the presynaptic and postsynaptic terminals, and, at P0 and onward, postsynaptic AchR clusters and presynaptic terminals are closely apposed (Sanes and Lichtman, 1999). Synaptic basal lamina is immature and subject to active remodeling during embryonic synaptogenesis, offering the potential for transsynaptic APP–APP interaction (Patton et al., 1997). To explore this possibility, we examined expression patterns of APP and performed NMJ analysis in the neuronal and muscle conditional knock-out mutants at various embryonic stages. Quantitative RT-PCR analysis of APP, APLP1, and APLP2 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) expressions in the spinal cord and muscle samples of E12.5, E14.5, E16.5, E18.5 embryos and P0 neonates showed that APP expression was low in both spinal cord and muscle lysates at E12.5 when nerve terminals are not in contact with the muscle (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). Interestingly, APP levels in both tissues were upregulated at E14.5, a stage that marks the initiation of synaptogenesis. Neuronal APP stayed relatively stable afterward while muscle APP expression declined. Similar to that of APP, quantitative RT-PCR analysis showed that APLP2 was also upregulated at E14.5 in both spinal cord and muscle (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). The similar expression patterns between APP and APLP2 and the possible compensatory activities are likely the reason for the overtly normal NMJ structures in individual knock-out animals. Although induction of APLP1 at E14.5 was also evident in spinal cord samples, consistent with the P0 result, APLP1 expression was absent in the muscle at all stages examined (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material).
Our previous analysis of neuromuscular structures of APP/APLP2 dKO mutants during embryonic development documented a clear defect in E16.5, but not E14.5, stage embryos (Wang et al., 2005). Immunostaining of APP at these developmental stages showed that, although only low and diffused APP immunoreactivity could be detected in E14.5 diaphragm muscle (data not shown), at E16.5, APP was highly concentrated at the AchR-positive endplates (Fig. 5 A), offering the potential for presynaptic and postsynaptic interaction. This synaptic localization is correlated with its function as staining of E16.5 embryos of neuronal and muscle conditional knock-out mutants along with the dKO embryos with synaptophysin, α-BTX, and neurofilament (Fig. 5 B), and quantification of endplate band width (Fig. 5 C) and number (Fig. 5 D) showed that a significant defect can be established in both N-dCKO and M-dCKO mutants compared with their littermate controls. Similar to that of P0 stage, phenotypes present in the M-dCKO and APP/APLP2 dKO embryos were statistically comparable and both displayed a more severe defect compared with the neuronal deletion mutant. Since APP is targeted to the neuromuscular synapse at E16.5, when clear NMJ phenotypes can be detected in both neuronal and muscle conditional knock-out animals at this early stage of synaptogenesis, we propose a model whereby transsynaptic APP interaction mediates its activity in neuromuscular synapse assembly and synaptic transmission (Fig. 5 E). Neuronal-specific APLP1 expression most likely contributes to the less severe phenotype in N-dCKO mutants compared with their muscle counterpart. Specifically, expression of APLP1 in the presynaptic motoneuron terminals of N-dCKO mice could interact with APP expressed in the postsynaptic muscle, yielding partial activity. This potential interaction is missing in M-dCKO animals as no APP or any of its family members are expressed in the postsynaptic compartment.
A mixed-culture system to test the synaptic adhesion properties of APP
Although the above studies provide substantial evidence to support the transsynaptic APP interaction, a direct test of this model proves to be difficult in neuromuscular synapse. We thus sought to investigate whether a similar mechanism exists in CNS where APP is also highly expressed. The complex nature of the CNS calls for simultaneous actions of multitude of transsynaptic signals (for review, see Dalva et al., 2007), making the investigation of individual proteins challenging. In this regard, the mixed-culture system offers the distinct advantage for examining and quantifying the synaptic activities of individual cell surface proteins in isolation (Scheiffele et al., 2000; Graf et al., 2004; Biederer and Scheiffele, 2007). In this assay, primary neurons are cocultured with non-neuronal cells expressing the candidate synaptic adhesion protein, and the synaptogenic effect of the candidate protein can be visualized by the recruitment of synaptic markers atop of non-neuronal cells. To examine the possible synaptogenic properties of APP, we transiently cotransfected an APP and a GFP expression construct in HEK293 cells and cocultured the transfected cells with dissociated hippocampal neurons. Cotransfection with neuroligin (NL) and GFP was performed as a positive control (Scheiffele et al., 2000), whereas transfection of the GFP vector alone was used as a negative control (Fig. 6 A). Similar to that of NL, expression of APP but not GFP in HEK293 cells potently promoted synaptic puncta as measured by both the area of HEK293 cells covered by synaptophysin (Fig. 6 B) and the number of synaptic puncta per transfected HEK293 cell (Fig. 6 C). Overall, the synaptic promoting activity of APP is not as strong as that of NL. This quantitative difference could be attributed by their intrinsic adhesion properties or the expression levels.
Figure 6.
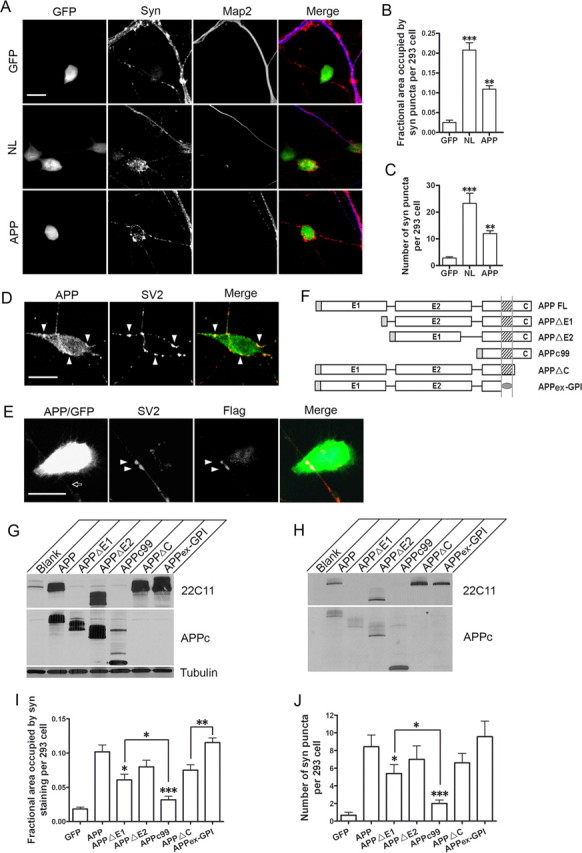
Mixed-culture analysis of APP synaptogenic activity. A, Representative images of HEK293 cells transfected with GFP (GFP), neuroligin/GFP (NL), or APP/GFP (APP) (each at 10:1 construct/GFP molar ratio), cocultured with wild-type hippocampal neurons, and stained with anti-synaptophysin (Syn) and anti-MAP2 (Map2) antibodies. Syn-positive, but Map2-negative puncta could be seen atop of NL and APP transfected HEK293 cells (marked in green), but not GFP vector control. The images are displayed either individually or merged (last column, GFP, green; Syn, red; Map2, blue). Scale bar, 20 μm. B, Quantification of the average area of HEK293 cells covered by synaptophysin immunoreactivity. Error bars indicate SEM. C, Quantification of average number of Syn-positive puncta per transfected HEK293 cell. **p < 0.01; ***p < 0.001 (t test). D, Double immunostaining of APP transfected HEK293/hippocampal cocultures with anti-APP and anti-SV2 antibodies. The images are displayed either individually or merged (SV2, red; APP, green). Selected SV2 and APP double-positive punctas are marked by arrowheads. Scale bar, 20 μm. E, Introduction of a Flag-tagged APP in neurons and coculture with APP-expressing HEK293 cells (APP/GFP). APP-expressing axon is marked by arrow. SV2, Immunostaining with the anti-SV2 antibody to identify the synaptic puncta atop HEK293 cells; Flag, immunostaining with the anti-Flag antibody to recognize neuronal expressed APP. SV2- and Flag-positive punctas are highlighted by arrowheads. Merge, Overlap of the three panels (293 cells, green; SV2, red; Flag, blue with overlap of the three in white). F, Schematic diagram of the APP constructs. Shaded rectangle at the N terminus of each construct represents the APP signal peptide; E1 and E2, the E1 (amino acids 22-189) and E2 (amino acids 289–500) domains of the APP extracellular sequences; shaded square, APP transmembrane domain; shaded oval, GPI anchor. G, Western blot analysis of APP expression in transfected HEK293 cells using the 22C11 and APPc antibodies, which recognize residues within the E1 domain and the C-terminal end of APP respectively. Tubulin was used as a loading control. H, Cell surface APP expression assayed by biotinylation experiment. I, J, Quantification of the average area of HEK293 cells covered by synaptophysin immunoreactivity (I) and the average number of Syn-positive puncta per transfected HEK293 cell (J) induced by APP and its derivatives. The asterisks above each bar are in comparison with the full-length APP. The asterisks on top of the brackets are comparison between the specific pair. *p < 0.05; **p < 0.01; ***p < 0.001 (t test).
To establish that APP is indeed localized to the synaptic puncta as expected for a synaptic adhesion protein, we performed double labeling of the cocultures with anti-SV2 and anti-APP antibodies, which showed that, although the transfected HEK293 cells were overall positive for APP (Fig. 6 D, APP), the APP immunoreactivity at the synaptic sites marked by SV2 (arrowheads) was significantly stronger. We next asked whether neuronal APP was recruited to the synaptic puncta by introducing a Flag-tagged APP to primary neurons and, on coculture with APP-expressing HEK293 cells, performed costaining with anti-SV2 and anti-Flag antibodies (Fig. 6 E). Indeed, there was significant colocalization of neuronal APP, marked by the anti-Flag immunoreactivity, with SV2-positive puncta (Fig. 6 E, arrowheads). These data, combined with our neuromuscular synapse staining result (Fig. 1 B), provide strong support for the synaptic localization of APP and the transsynaptic APP interaction model in both peripheral and central synapses.
Having established a synaptogenic property of full-length APP, we sought to identify the functional domains by creating the following mutants (Fig. 6 F): APP with deletions of the E1 or E2 domains of the extracellular sequences (APPΔE1 and APPΔE2, respectively); APPc99, which links the β-secretase cleaved C-terminal fragment with the APP signal peptide and excludes both the E1 and E2 domains; APP deleting the intracellular sequences (APPΔC); and the entire APP extracellular sequences attached to the membrane with a GPI anchor (APPex-GPI). Transfection of these constructs to HEK293 cells followed by Western blotting detected appropriate expression of the APP proteins (Fig. 6 G). Their membrane localization was confirmed by biotinylation of surface proteins (Fig. 6 H). Coculture of HEK293 cells expressing the full-length APP or the deletion constructs with wild-type hippocampal neurons revealed that deleting the E1 (APPΔE1) or both the E1 and E2 domains (APPc99) resulted in a significant reduction in synaptic puncta formation (Fig. 6 I,J), suggesting that the extracellular sequences are essential and that the synaptogenic activity is mainly conferred by the E1 domain of APP. This assessment is supported by Soba et al. (2005), who reported that the E1 domain engages the APP–APP intercellular interaction. Nevertheless, significantly higher degree of synaptic puncta induced by APPΔE1 compared with APPc99 indicates that the E2 domain likely also participates in this activity (Fig. 6 I,J, APPΔE1 vs APPc99, p < 0.05). Deletion of the APP intracellular domain (APPΔC and APPex-GPI) both resulted in prominent synaptogenic activity not statistically different from that of the full-length APP. Generally speaking, APPex-GPI resulted in higher puncta compared with APPΔC, presumably because of better membrane anchoring of the APPex-GPI protein. Overall, these deletion studies establish that the APP extracellular domains, in particular the E1 domain, but not the intracellular sequences, are essential for synaptogenesis when expressed in HEK293 cells.
Neuronal full-length APP is necessary in mediating synaptogenesis
Having established that neuronal APP was recruited to the synaptic sites, we next asked whether it plays a functional role in synaptogenesis in the coculture assay. Since the APP family of proteins has been shown to form heterodimers (Soba et al., 2005), to bypass the compensatory activity of APLP2, we prepared primary hippocampal neurons from APP/APLP2 dKO neonates and used neurons from APLP2-null littermates as controls. Comparison of wild-type and APLP2-null neurons cocultured with APP transfected HEK293 cells revealed similar synaptic puncta profiles (data not shown), suggesting that loss of APLP2 alone does not lead to adverse effect, an observation that is consistent with the neuromuscular system. We cocultured the APLP2-null control or APP/APLP2 dKO neurons with GFP-, NL-, or APP-expressing HEK293 cells, respectively (Fig. 7 A,B). Comparison of synaptic puncta between the control and APP/APLP2 dKO neurons showed no difference in NL-induced puncta (Fig. 7 A,B, NL). However, both the area of HEK293 cells covered by synaptophysin immunoreactivity and the number of puncta formed per transfected HEK293 cell were greatly reduced in dKO neurons when cocultured with APP-expressing HEK293 cells (Fig. 7 A,B, APP, Ctrl vs dKO, p < 0.001). Therefore, loss of neuronal APP/APLP2 specifically impairs APP- but not NL-induced synaptogenic property. Expression of APP in dKO neurons by lentiviral infection of human APP (dKO + APP) rescued the reduced synaptic puncta measured by both the area of coverage and the number of puncta per transfected HEK293 cell (Fig. 7 A,B, Ctrl vs dKO + APP, nonsignificant). This rescue assay provides direct support that the reduced synaptogenic property in dKO neurons is caused by APP deficiency.
Figure 7.
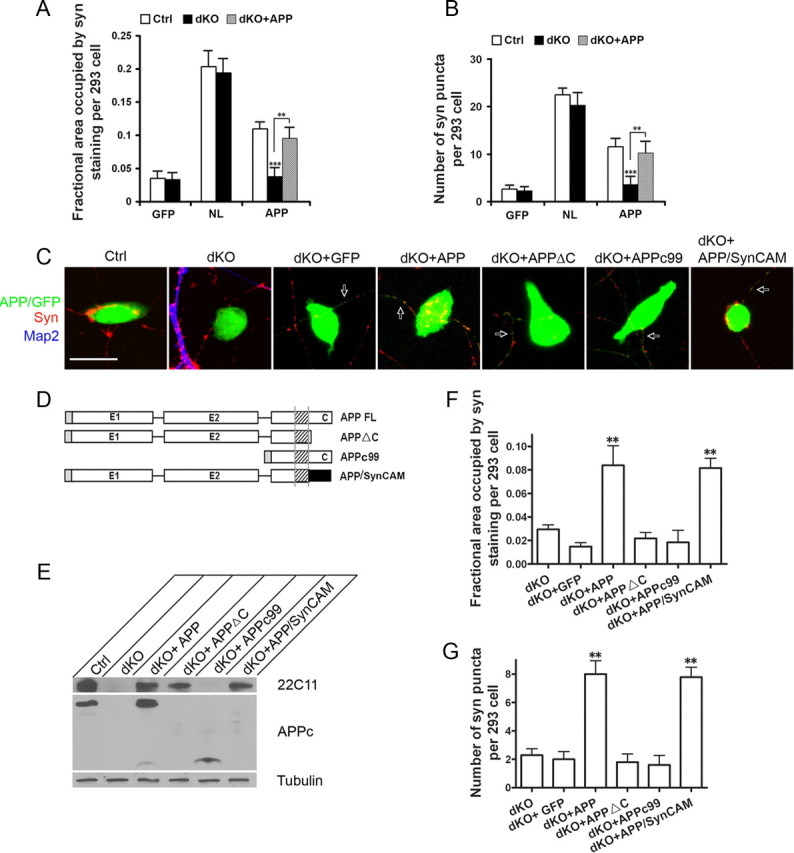
Effect of neuronal APP on APP-induced synaptic puncta. The APP/APLP2-null neurons (dKO) and littermate APLP2-null controls (Ctrl) were cocultured with GFP, NL, or APP transfected HEK293 cells. dKO + APP, dKO neurons infected with APP-expressing lentivirus. The average area of transfect HEK293 cells covered by synaptophysin (A) and the average number of synaptic puncta per HEK293 cell (B) were quantified. APP-induced puncta is significantly reduced when cocultured with dKO neurons (Ctrl vs dKO, p < 0.001) (t test). This impairment is completely rescued by neuronal expression of APP (Ctrl vs dKO + APP, nonsignificant, p > 0.05). Error bars indicate SEM. C, Representative images of control (Ctrl), APP/APLP2 double knock-out (dKO), or dKO with lentiviral expression of GFP vector (dKO + GFP), human full-length APP (dKO + APP), intracellular domain deleted APP (dKO + APPΔC), extracellular sequence deleted APP (dKO + APPc99), or the APP/SynCAM chimera (dKO + SynCAM) cocultured with APP-transfected HEK293 cells. The cultures were stained with anti-synaptophysin (Syn; red) and anti-MAP2 (Map2; blue) antibodies. Both the transfected HEK293 cells and APP infected axons (arrows) are GFP-positive. Scale bar, 20 μm. D, Schematic diagram of the APP constructs. Black rectangle, SynCAM C-terminal sequences (amino acids 428-474). E, Western blot analysis of neuronal lysates from APLP2−/− control, dKO, and dKO neurons infected with APP and derivatives using the N-terminal and C-terminal antibodies 22C11 and APPc, respectively. Tubulin was used as a loading control. F, Quantification of the average area of HEK293 cells covered by synaptophysin immunoreactivity. G, Quantification of average number of Syn-positive puncta per transfected HEK293 cell. **p < 0.01 (t test) in comparison with the dKO + GFP control.
Using this neuronal rescue system, we investigated the requirement of APP functional domains by introducing the APPΔC or APPc99 mutant constructs to dKO neurons (Fig. 7 C, dKO + APPΔC and dKO + APPc99), and determining their rescue activities using the full-length APP and GFP vector infected neurons as positive (dKO + APP) and negative (dKO + GFP) controls, respectively. Physiological expression of the APP constructs in APP/APLP2 dKO neurons were confirmed by Western blot analysis (Fig. 7 E). As expected, no rescue activity was detected by expressing APPc99 in dKO neurons (Fig. 7 F,G, dKO + APPc99). However and in contrast to the dispensable role of the APP intracellular domain in HEK293 cells, expression of APPΔC also failed to show appreciable rescue (dKO + APPΔC), suggesting that both the extracellular and intracellular sequences are necessary for neuronal APP-induced synaptogenesis. To provide additional support for this notion, we constructed an APP/SynCAM chimeric molecule by fusing the APPΔC with the SynCAM intracellular sequences (Fig. 7 D,E). When introduced into the dKO neurons, the APP/SynCAM fusion protein effectively rescued the synaptogenic activity (Fig. 7 F,G, dKO + APP/SynCAM).
Formation of APP/Mint1/Cask complex visualized by BiFC assay
It has been proposed that presynaptic differentiation induced by neurorexin/neuroligin (NX/NL) and SynCAM class of synaptic adhesion proteins involve intracellular association with Cask and Munc18 interacting protein Mint1 (also termed X11a, herein referred as Mint1) (Hata et al., 1996; Biederer and Südhof, 2000; Biederer et al., 2002). Since the SynCAM intracellular domain can replace the APP intracellular sequences in the coculture assay, and since the highly conserved YENPTY sequence of the APP intracellular domain is known to interact with the PTB (phosphotyrosine-binding) motif of Mint1 (Zhang et al., 1997), we wondered whether APP/Mint1/Cask could also form a presynaptic complex similar to that of SynCAM. We thus explored the possible interactions of these proteins in live cells using the BiFC assay (Kerppola, 2006) by introducing various combinations of APP-, Mint1-, and Cask-split YFP (nYFP or cYFP) fusion constructs (Fig. 8). Proper expression of the fusion proteins were established by Western blot analysis on transfection to HEK293 cells (Fig. 8 A). As expected, coexpression of nYFP or cYFP vectors (nYFP/cYFP) or APP-nYFP/cYFP did not yield appreciable fluorescence signal (Fig. 8 B). Expression APP-nYFP with Fe65-cYFP, a known APP binding protein, resulted in significant fluorescence. Consistent with the notion that APP forms homodimers and that Mint1 can interact with Cask and APP, respectively, coexpression of APP-nYFP/APP-cYFP, Mint1-cYFP/Cask-nYFP, or APP-nYFP/Mint1-cYFP all resulted in strong fluorescent signals. Significantly, cotransfection of APP-nYFP and Cask-cYFP with nontagged Mint1 also resulted in prominent fluorescence, suggesting that Mint1 functions as a scaffold to bring APP and Cask into one complex (Fig. 8 B, APP-nYFP/Mint1/Cask-cYFP). Indeed, consistent with the notion that APP–Mint1 interaction is mediated through the YENPTY sequence of APP, deletion of the YENPTY sequence abolished both APP/Mint1 and APP/Mint1/Cask interactions (APPΔYENPTY-cYFP/Mint1-nYFP and APPΔYENPTY-cYFP/Mint1/Cask-cYFP) (Fig. 8 B). These results support the view that APP may mediate presynaptic function through the recruitment of Mint1 and Cask similar to other synaptic adhesion proteins (Hata et al., 1996; Biederer and Südhof, 2000; Biederer et al., 2002).
Figure 8.
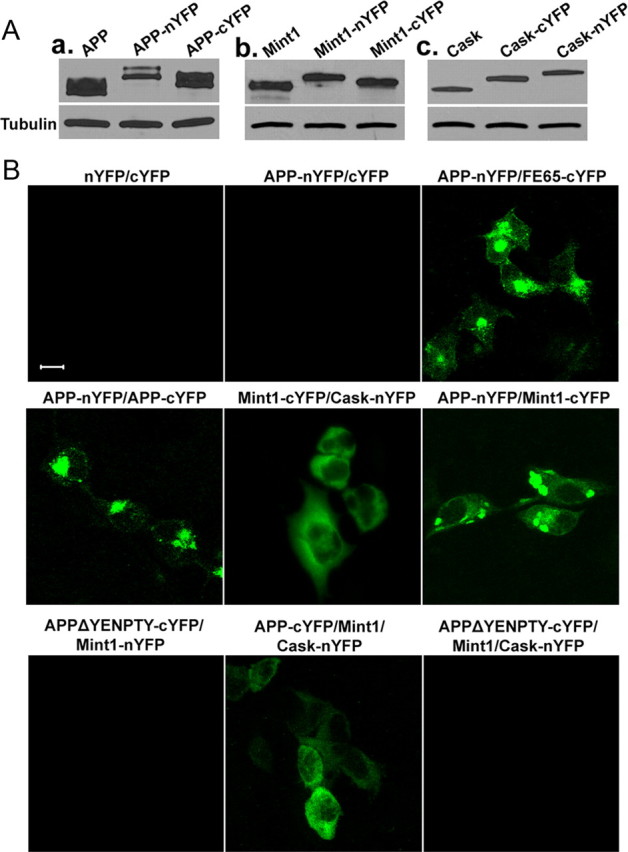
Visualization of APP-associated protein complexes by BiFC analysis. A, Western blotting of HEK293 cells transfected with DNA constructs encoding APP, Mint1, or Cask proteins fused to complementary fluorescent protein fragments (nYFP or cYFP). Total protein lysates were blotted with the APPc antibody (a), anti-Mint1 (b), and anti-Cask (c) antibodies. B, HEK293 cells were cotransfected with the BiFC constructs indicated above the images. BiFC complexes were imaged 24 h after transfection. Scale bars, 10 μm.
Discussion
Several novel findings are presented in the current study: (1) APP is targeted to the synaptic sites of both peripheral and central synapses; (2) presynaptic and postsynaptic APP cooperate to regulate the NMJ structure and function; (3) APP induces synaptogenesis in a mixed-culture assay; (4) this synaptic promoting property requires neuronal full-length APP and may involve the formation of the APP/Mint1/Cask complex. These studies identify APP as a novel synaptic adhesion molecule, and we postulate that transsynaptic APP interaction modulates central and peripheral synapse formation and function.
Presynaptic and postsynaptic APP interaction in neuromuscular synapse development
We reported previously that the APP family of proteins plays essential roles in the developing NMJ (Wang et al., 2005, 2007). By creating the motor neuron or muscle conditional knock-out mutants, we reveal here that APP is targeted to presynaptic and postsynaptic sites at 1:1 stoichiometry and that APP is required in both compartments for NMJ structure and function. Furthermore, presynaptic mediated CHT targeting and neurotransmission is dependent on postsynaptic APP expression. Since the NMJ defects can be detected in both neuronal and muscle conditional knock-out animals at an early stage of synaptogenesis (E16.5) before the formation of mature basal lamina, we propose that a direct interaction of neuronal and muscle APP at synaptic sites is the underlying mechanism. Among the long list of proteins that has been identified to contribute to mammalian neuromuscular synapse formation and differentiation (for review, see Fox and Umemori, 2006; Kummer et al., 2006), this is, to our knowledge, the first to demonstrate the interdependency for presynaptic and postsynaptic expression of a single protein.
It is important to note, however, that a direct interaction of APP across developing neuromuscular synapse has not been established. It remains possible that this interaction is mediated through an intermediate molecule in the synaptic cleft or through other adhesion proteins. In this regard, APP has been reported to interact with other cell adhesion molecules including neural cell adhesion molecule (NCAM) (Ashley et al., 2005), NgCAM (neuron–glia cell adhesion molecule) (Osterfield et al., 2008), and TAG 1 (Ma et al., 2008). Of particular interest, studies of Drosophila NMJ revealed that transsynaptic interaction of Fasciclin II, a fly homolog of NCAM, promotes synaptic bouton formation, and this activity requires APPL and its binding partner dX11/Mint (Ashley et al., 2005). Therefore, it is conceivable that APP may mediate synaptic adhesion through interaction with NCAM. Although this mechanism cannot be formally excluded, our in vitro and in vivo results argue against this NCAM dependency: (1) unlike APP/APLP2-null animals, mice deficient in all forms of NCAM are viable and do not exhibit overt morphological abnormalities at the developing NMJ (Moscoso et al., 1998); (2) whereas NCAM is negative in the coculture assay (Sara et al., 2005), APP shows a prominent synapse promoting activity (discussed below).
A recent report by Nikolaev et al. (2009) revealed that soluble APPβ and derivatives bind to DR6 receptor and mediate axon pruning. The authors provided physiological relevance of this pathway by showing that DR6-deficient mice exhibit NMJ phenotypes similar to that of APP/APLP2-null animals. Therefore, an alternative explanation, independent of adhesion, is that soluble, secreted APP is the functional moiety for NMJ development through DR6 interaction. Although attractive, the cell-autonomous model tested and put forward by Tessier-Lavigne and colleagues predicts that neuronal APP would be sufficient to produce the secreted forms of APP independent of muscle, and such a prediction would be incompatible with the interdependency of presynaptic and postsynaptic APP revealed by our conditional knock-out study. However, it is worth noting that the APP–DR6 interaction is mediated specifically through sAPPβ, whereas the entire APP, in particular both sAPPα and sAPPβ, are simultaneously affected in our conditional knock-out system. The possible distinct or even contrasting activities of sAPPα and sAPPβ may complicate the interpretation of the results. A direct test of the role of DR6–APP in vivo awaits the creation of a sAPPβ-specific allele.
Transsynaptic APP interaction in central synaptogenesis
Supporting the model whereby transsynaptic APP interaction mediates neuromuscular synapse development, using a primary neuron/HEK293 coculture assay, we demonstrate that expressing APP in HEK293 cells promotes synaptogenesis in contacting axons. Among the numerous synaptic adhesion proteins identified, including NX/NL, EphB/ephrin, SynCAM, NCAM, and N-cadherin, which mediate various aspects of synaptic processes through transsynaptic heterophilic or homophilic interactions (for review, see Akins and Biederer, 2006; Dalva et al., 2007), NX/NL and SynCAM have been well documented to promote synapse formation in the coculture assay (Scheiffele et al., 2000; Biederer et al., 2002; Graf et al., 2004; Sara et al., 2005; Fogel et al., 2007). APP, like NX/NL and SynCAM, are type I membrane proteins, and our coculture studies revealed remarkable similarities between APP and NX/NL and SynCAM class of synaptic adhesion proteins. For example, like NL (Scheiffele et al., 2000), the APP intracellular domain is not required to induce presynaptic structure in contacting axons. Similar to that of SynCAM (Biederer et al., 2002), neuronal expression of full-length APP is necessary for presynaptic puncta formation. That the SynCAM intracellular domain can functionally replace the corresponding APP sequence and that the latter can also form a complex with Mint1 and Cask, presynaptic complexes associated with neurorexin and SynCAM intracellular domains (Hata et al., 1996; Biederer and Südhof, 2000; Biederer et al., 2002), further provide strong support that the APP synaptic function is mediated through similar mechanisms. In addition to formation of the common Mint1/Cask complex, specific regulation by the adhesion proteins may be conferred through recruitment of additional molecules. For instance, we previously reported that APP potently regulates CHT in both NMJ and in central cholinergic neurons (Wang et al., 2007). Our current finding supports the idea that this is mediated through a concerted action of presynaptic and postsynaptic APP expressed in cholinergic terminals and their target neurons, respectively. Since APP and its family members are abundantly expressed in CNS, we propose that this transsynaptic APP interaction is not limited to cholinergic synaptic transmission, but represents a functional moiety in regulating multiple neuronal circuits in central synapses. The specific proteins involved in these processes and the precise nature of the synaptic regulation remain to be elucidated.
The transsynaptic model calls for the necessity of expressing membrane anchored APP. This assessment appears to contradict studies by Hornsten et al. (2007) and Ring et al. (2007), who reported that the lethal phenotype in apl-1-null worm and spatial learning and long-term potentiation impairment in APP deficient mice can be rescued by expressing only their soluble extracellular derivatives, respectively. The lethality of the apl-1-deficient worm is likely caused by a molting instead of a synaptic defect. The lack of apparent involvement of the APP intracellular domain in APP-mediated synaptic plasticity reported by Ring et al. (2007) could be attributable to the fact that the synaptogenic activity of APP studied here and those mediating synaptic plasticity are executed through independent mechanisms. Indeed, neuronal APP and its cleavage products have been shown to be differentially sorted and independently transported (Muresan et al., 2009). Of interest, Muresan and Muresan (2005) provided data to show that APP phosphorylated at the conserved Thr668 site is preferentially transported to the nerve terminals, and this process is kinesin dependent and can be distinguished from the nonphosphorylated APP. Therefore, it is conceivable that a distinct pool of full-length APP, possibly marked by its phosphorylation status, is targeted to the synaptic plasma membrane and engages transsynaptic interaction, whereas the soluble, secreted APP derivatives are produced from other compartments.
Synaptic adhesion and CNS diseases
APP, NL/NX, and SynCAM each contains multiple family members and isoforms. This creates a multitude of possible combinations of transsynaptic interactions, which could confer the complex and precise nature of the central synapses; and their dysregulation may lead to neurodevelopmental and neurodegenerative disorders. The critical importance of synaptic adhesion proteins in brain function is highlighted by the finding that mutations in neurexins and neuroligins are associated with autism spectrum disorders (Jamain et al., 2003; Autism Genome Project Consortium et al., 2007). Of direct relevance to APP and AD, impairment of cholinergic neurons is a cardinal feature of AD pathogenesis. Our finding that transsynaptic APP–APP interaction regulates CHT function indicates that dysregulation of APP synaptic adhesion activity may contribute to cholinergic neuronal vulnerability. Furthermore, APP dimerization has been shown to affect APP processing (Kaden et al., 2008), and activity-dependent APP processing and Aβ production has been documented to suppress excitatory synaptic transmission by postsynaptic removal of AMPA receptors (Kamenetz et al., 2003; Chang et al., 2006; Hsieh et al., 2006). Whether transsynaptic APP interaction is involved in this process is an interesting question that awaits additional investigation. Nevertheless, it is worth noting that Aβ has been reported to be produced and released at nerve terminals in CNS and that synaptically generated Aβ has been shown to contribute to amyloid pathology (Lazarov et al., 2002). On a boarder perspective, synaptic dysfunction is widely viewed as causative to AD pathogenesis. Our finding that APP plays a direct role in regulating synaptogenesis through a transsynaptic mechanism makes it a legitimate possibility that misregulation of APP synaptic adhesion activity, in an Aβ-dependent or -independent manner, may contribute to synaptic dysfunction and AD pathogenesis.
Footnotes
This work was supported by National Institutes of Health Grants AG032051 and AG033467, Alzheimer's Association Grant IIRG-06-25779, and American Health and Assistance Foundation Grant A2008-052. We thank P. Scheiffele (Columbia University, New York, NY) for providing the neuroligin construct, Hongmei Li and Tom Südhof (Stanford University School of Medicine) for Mint1 and CASK cDNA vectors, C. Rosenmund [Baylor College of Medicine (BCM)] for the gift of lentiviral vectors and S. Hamilton (BCM), A. Cooney (BCM), and R. Behringer (M. D. Anderson Cancer Center, Houston, TX) for the Mck-Cre, GDF9-iCre, and CMV-Cre animals, respectively. We are indebted to E. Lorenzo of the Darwin Transgenic Mouse Core, R. Atkinson of the Confocal Core, and Eunice Kennedy Shriver Intellectual and Developmental Disabilities Research Center (Grant HD024064) at BCM for support in ES cell targeting and injection and confocal imaging, respectively. We are grateful to X. Chen for expert technical support, Tamara Milakovic for initial construction of the BiFC vectors, and members of the Zheng laboratory for stimulating discussions.
References
- Akaaboune M, Allinquant B, Farza H, Roy K, Magoul R, Fiszman M, Festoff BW, Hantaï D. Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol Cell Neurosci. 2000;15:355–367. doi: 10.1006/mcne.2000.0834. [DOI] [PubMed] [Google Scholar]
- Akins MR, Biederer T. Cell-cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Ashley J, Packard M, Ataman B, Budnik V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci. 2005;25:5943–5955. doi: 10.1523/JNEUROSCI.1144-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism Genome Project Consortium. Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- Biederer T, Südhof TC. Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Südhof TC. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Liu D, Songyang Z. Telomere maintenance through spatial control of telomeric proteins. Mol Cell Biol. 2007;27:5898–5909. doi: 10.1128/MCB.00603-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel AI, Akins MR, Krupp AJ, Stagi M, Stein V, Biederer T. SynCAMs organize synapses through heterophilic adhesion. J Neurosci. 2007;27:12516–12530. doi: 10.1523/JNEUROSCI.2739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Umemori H. Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J Neurochem. 2006;97:1215–1231. doi: 10.1111/j.1471-4159.2006.03834.x. [DOI] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralle M, Oliveira CL, Guerreiro LH, McKinstry WJ, Galatis D, Masters CL, Cappai R, Parker MW, Ramos CH, Torriani I, Ferreira ST. Solution conformation and heparin-induced dimerization of the full-length extracellular domain of the human amyloid precursor protein. J Mol Biol. 2006;357:493–508. doi: 10.1016/j.jmb.2005.12.053. [DOI] [PubMed] [Google Scholar]
- Hata Y, Butz S, Südhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on post-synaptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, Daigle I, Markowitz M, O'Connor G, Plasterk R, Li C. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci U S A. 2007;104:1971–1976. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T Paris Autism Research International Sibpair Study. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden D, Munter LM, Joshi M, Treiber C, Weise C, Bethge T, Voigt P, Schaefer M, Beyermann M, Reif B, Multhaup G. Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect beta-secretase cleavage of APP. J Biol Chem. 2008;283:7271–7279. doi: 10.1074/jbc.M708046200. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Lan ZJ, Xu X, Cooney AJ. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod. 2004;71:1469–1474. doi: 10.1095/biolreprod.104.031757. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, Xu RX, Bagnard D, Schachner M, Furley AJ, Karagogeos D, Watanabe K, Dawe GS, Xiao ZC. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol. 2008;10:283–294. doi: 10.1038/ncb1690. [DOI] [PubMed] [Google Scholar]
- Moscoso LM, Cremer H, Sanes JR. Organization and reorganization of neuromuscular junctions in mice lacking neural cell adhesion molecule, tenascin-C, or fibroblast growth factor-5. J Neurosci. 1998;18:1465–1477. doi: 10.1523/JNEUROSCI.18-04-01465.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-β precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009;29:3565–3578. doi: 10.1523/JNEUROSCI.2558-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-β precursor protein. J Neurosci. 2005;25:3741–3751. doi: 10.1523/JNEUROSCI.0152-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–990. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Osterfield M, Egelund R, Young LM, Flanagan JG. Interaction of amyloid precursor protein with contactins and NgCAM in the retinotectal system. Development. 2008;135:1189–1199. doi: 10.1242/dev.007401. [DOI] [PubMed] [Google Scholar]
- Papaioannou VE, Behringer R. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2005. Mouse phenotypes: a handbook of mutation analysis. [Google Scholar]
- Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Müller UC. The secreted β-amyloid precursor protein ectodomain APPs α is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sara Y, Biederer T, Atasoy D, Chubykin A, Mozhayeva MG, Südhof TC, Kavalali ET. Selective capability of SynCAM and neuroligin for functional synapse assembly. J Neurosci. 2005;25:260–270. doi: 10.1523/JNEUROSCI.3165-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schwander M, Shirasaki R, Pfaff SL, Müller U. β1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J Neurosci. 2004;24:8181–8191. doi: 10.1523/JNEUROSCI.1345-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Ingalls CP, Durham WJ, Snider J, Reid MB, Wu G, Matzuk MM, Hamilton SL. Altered excitation-contraction coupling with skeletal muscle specific FKBP12 deficiency. FASEB J. 2004;18:1597–1599. doi: 10.1096/fj.04-1587fje. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Wang B, Yang L, Wang Z, Zheng H. Amyolid precursor protein mediates presynaptic localization and activity of the high-affinity choline transporter. Proc Natl Acad Sci U S A. 2007;104:14140–14145. doi: 10.1073/pnas.0704070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ha Y. The x-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee CH, Mandiyan V, Borg JP, Margolis B, Schlessinger J, Kuriyan J. Sequence-specific recognition of the internalization motif of the Alzheimer's amyloid precursor protein by the X11 PTB domain. EMBO J. 1997;16:6141–6150. doi: 10.1093/emboj/16.20.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]