

Abstract
Background
Despite the existence of over 50 subtypes and circulating recombinant forms of human immunodeficiency virus type-1 (HIV-1), subtype-C dominates the heterosexual pandemic causing 56% of all infections.
Objective
To evaluate whether viral genetic factors may contribute to the observed subtype-C predominance.
Methods
Chimeric viruses were generated using V1-V3 envelope fragments from a subtype-A/C dually-infected woman with preferential genital replication of subtype-C. Viral adaptation, spread and cell fusion ability were evaluated in vitro using peripheral blood mononuclear cells (PBMC) and HeLa-CD4-CCR5 cell lines, sequencing and cloning. Structural modeling was performed using a crystal structure of gp120-CD4-X5. Phylogenetic analysis was done using subtype-A, -B and -C sequences from blood and cervix of 37 infected women and database sequences.
Results
We identified two envelope motifs, compact V1-V2 loops and V3-316T, which are found at high frequency throughout subtype-C evolution, and affect gp120 interactions with CD4 and CCR5, respectively. When a V1-Δ5 deletion or V3-A316T was incorporated into subtype-A, each increased viral fusion and spread in PBMC and cell-lines with low CCR5-expression several fold. Structural modeling suggested the formation of an additional hydrogen bond between V3 and CCR5. Moreover, we found preferential selection of HIV with 316T and/or extremely short V1-V2 loops in cervices of three women infected with subtype-A/C, -B or −C.
Conclusions
As CD4+-CCR5+-T-cells are key targets for genital HIV infection, and cervical selection can favor compact V1-V2 loops and 316T, which increase viral infectivity, we propose that these conserved subtype-C motifs may contribute to transmission and spread of this subtype.
Keywords: HIV-1, subtype-C, subtype-A, evolution, CCR5, cervix, transmission, phylogeny
Introduction
The current HIV pandemic is dominated by subtype-C, which accounts for approximately 56% of the estimated 33 million infections worldwide, despite the concurrent existence of over fifty other subtypes and CRFs[1]. First identified in Ethiopia, subtype-C has spread through Southern and Eastern Africa during the last two decades, invading areas formerly dominated by subtypes-A and -D, and is currently causing devastating epidemics in parts of Sub-Saharan Africa, Brazil, India and China[2-5].
These transmissions are largely through heterosexual contact, or from mother-to-child. While founder effects probably can explain the more recent subtype-C epidemics in India and China, subtyping of early HIV sequences from Southern and Eastern Africa strongly suggest that subtype-C was introduced into these areas subsequent to subtype-A and -D (www.hiv.lanl.gov). As no epidemiological studies have shown any subtype-related differences between heterosexual transmission networks, it has been suggested that subtype-C virus may have a transmission advantage over subtypes-A and -D[6-8]. Moreover, epidemiological studies have demonstrated differences between these subtypes with respect to vaginal shedding and in utero transmission, subtype-C associating with increased risks of both[9, 10]. However, no functional mechanism explaining these differences has so far been identified.
While many studies have successfully focused on identifying human genetic factors influencing HIV-1 disease progression, these seem to account for just around 10% of the variability in disease progression rates and only some have been demonstrated to affect transmission[11]. Viral genetic variation is also likely to influence pathogenesis and transmission, and short V1-V4 loops, and more neutralisation sensitive viruses, have been demonstrated to be selected during, or soon after, many sexual transmissions caused by subtype-A and -C, but not -B[12-16].
Here we asked whether conserved, subtype-specific sequence motifs in subtype-C could affect viral replication and transmission. Our results highlight the functional importance of two common subtype-C motifs, short V1-V2 loops and V3-316T, and demonstrate that these are at high frequency throughout subtype-C evolution and may be specifically selected in the genital tract of infected women.
Methods
Study subjects
HIV-1 infected women were recruited at Rigshospitalet, Denmark, 1995-96. Most were untreated, however some had received mono-therapy (AZT (8/37), ddI (1/37)) or dual therapy (AZT and ddI (2/37)) for over 6 months[17, 18]. The study was approved by the Danish Board of Medical Ethics and patients gave informed consent[17].
Limiting dilution, nested PCR and sequencing
DNA was extracted from PBMC and cervical cell pellets using the QIAamp®Blood kit(Qiagen). V1-V3, C2-C4 and gp160 envelope fragments were amplified by limiting-dilution nested PCR using Advantage 2 Polymerase mix(Clontech) and sequenced (GenBank accession numbers XXXX-YYYY)[19].
Construction of recombinant HIV-1 clones
Infectious HIV-A and -C chimeric clones with V1-V3 env fragments obtained from blood or cervix of a dually-infected patient were generated as described previously for HIV clones 81A-4 and 49-5[20-23].
PCR amplification, sequencing and cloning of PBMC-adapted virus
Virus-containing supernatants were samples throughout the PBMC infection experiment. Viral RNA was purified using the QIAamp Viral RNA Mini Kit(Qiagen), reverse-transcribed and amplified using the SuperScript One-Step RT-PCR Kit for Long Templates(Invitrogen), sequenced and cloned. The reconstructed PBMC-adapted chimeric subtype-A clones were named A20-V1Δ5, A2-T146I, A1-A316T, and A1-N302K, according to envelope region and type of mutation.
Virus stocks and infection
Proviral HIV constructs were transfected into RC49 HeLa cells and viral stocks were generated for PBMC, HeLa cell and fusion experiments; titers were expressed as focus-forming units (FFU) per ml[24, 25]. Different virus dilutions (Multiplicity of infection (MOI) 0.004-0.1) were used to infect PBMC, and cultures continued for 16 days; p24 levels were measured using a antigen capture assay as recommended(AIDS Vaccine Program, NCI). Infected HeLa cell monolayers were stained for p24 and foci counted as described previously[24, 25].
Fusion experiments and immunohistochemical detection
293T cells, which do not express CD4 or CCR5, were transfected with subtype-A, -C or adapted-A chimeric HIV constructs and relative envelope expression levels compared using pooled antisera from HIV-infected patients and fluorescently-labeled anti-human IgG (Invitrogen). Data were acquired on a FACSCalibur flow cytometer (Becton Dickinson). Transfected 293T cells were subsequently overlaid onto JC37, or RC49, monolayers for 24 hours and stained for p24 antigen.
Sequence analysis
Phylogenetic trees were constructed by maximum likelihood using PAUP* 4.0b10 and phyML software[26, 27]. Non-parametric bootstrap support estimates were generated using 1000 replicates under neighbour-joining using the maximum likelihood substitution model. Tree figures were produced using FigTree1.1(tree.bio.ed.ac.uk/software/figtree/). Distance analysis was done using MEGA and Kimura-2 parameters; standard errors were estimated using 100 bootstrap runs[28]. The variation analyses were computed using a counting algorithm (http://bioafrica.mrc.ac.za/JoeTool/counting.html). Frequencies of 316A and T were calculated using 1671 subtype-A and 4165 subtype-C sequences. To decrease the risk of sampling bias in these database derived sequences, we only used one sequence from each patient or transmission cluster from as many countries as possible (i.e. multiple sequences from known transmission clusters such as wife/husband or mother/baby were excluded). Sequences with translational problems (i.e. frameshift or stop codons) were furthermore excluded as these may either represent non-functional virus or could indicate poor sequence quality.
Molecular modelling and disorder analysis
The A316T and N302K single amino acid point mutations were modelled on the crystal structure of gp120-CD4-X5 [29](PDB ID 2B4C) by manually substituting the individual side chains using COOT [30], ensuring by visual inspection that the resulting model maintained good local stereochemistry. The increase in local positive charge at the region of the N302K mutation was confirmed by mapping the molecular electrostatic potential onto the solvent-accessible surface of gp120 (PDB ID 2B4C) and the gp120 N302K model (not shown) using the APBS [31] plug-in for PyMOL (DeLano Scientific) with default parameters. The propensity of gp120 residues to be disordered was predicted from the sequences of primary isolates and adapted strains using the neural-network pattern recognition program RONN[32] with default parameters. Molecular graphics were prepared using PyMOL (DeLano Scientific).
Results
Generation of infectious HIV molecular clones
We previously reported preferential genital replication of HIV subtype-C in an asymptomatic, untreated woman dually-infected by subtype-A and -C in blood; her blood- and cervix-derived C sequences were indistinguishable and subtype-A appeared to have infected her prior to -C[33].
We hypothesized that preferential cervical subtype-C infection was associated with viral genetic traits favoring genital infection. To compare the biological characteristics of HIV sequences found in blood and cervix, we amplified and sequenced single-copy V1-V3 envelope fragments from these tissues, as this region contains key determinants of cell tropism, infectivity and chemokine sensitivity[34-36](Fig. 1A). Sequences were inserted into the pNL4-3ΔV1-V3 vector to generate infectious molecular clones (subtype-C: C57, C64, C66, C68, C69, and C85, subtype-A: A1, A2, and A20). Infection studies using HeLa cells expressing CD4 and CXCR4 plus or minus CCR5, showed that all virus clones required the CCR5 co-receptor for infection. Furthermore, all viruses were able to infect and spread slowly in monocyte-derived macrophage cultures (data not shown).
Figure 1.
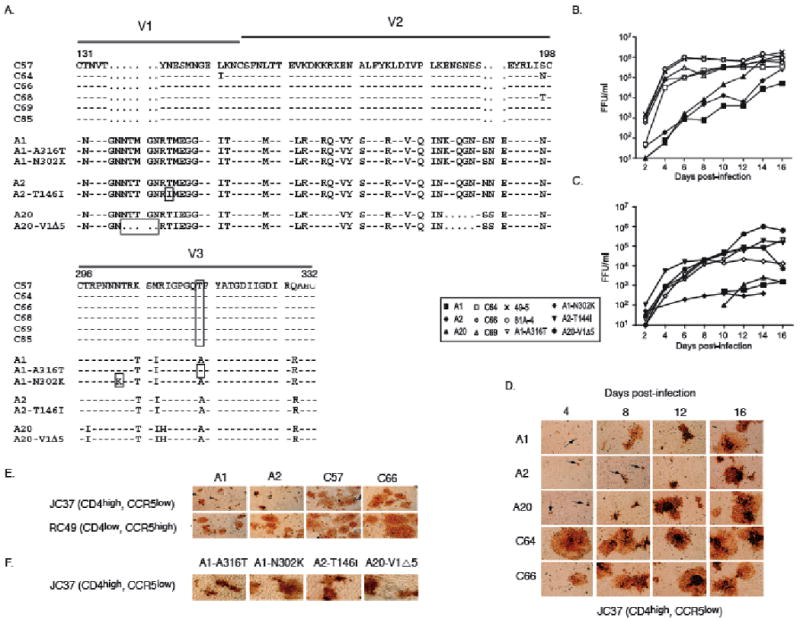
A. Amino acid alignment of the V1-V2 and V3 region of subtypes-A, -C and the PBMC-adapted A1-A316T, A1-N302K, A2-T144I and A20-V1Δ5 viruses numbered according to HIV(HXB2). Patient-derived viral sequences had constant V1-V2 loop lengths (A=68, C=58) and the V3-loop charge within and between subtypes was identical (+4). The Δ5 deletion in A20, 144I in A2, 302K in A1 and the 316T residue in A1 and subtype-C are boxed. As HXB2 has a unique 2-amino acid insertion at position 309, the following amino acid is number 312.
B. The influence of patient subtype-A and -C V1-V3 envelope sequences on PBMC infection and spread. Live viral output was measured as focus forming units per ml (FFU/ml) over time using chimeric subtype-A, -C and subtype-B control viruses (49-5 and 81A-4) at an MOI of 0.02. Results are representative of six independent infections.
C. Live viral output, measured as FFU/ml, over time after PBMC infection at an MOI of 0.02 using chimeric A, PBMC-adapted A and subtype C66 virus. The PBMC donor differs from the one used in B. Results are representative of six independent infections.
D. Virus titration from supernatants of infected PBMC on JC37 cells followed by p24 antibody staining. Arrows signify single foci consisting of 1-2 nuclei.
E. Fusion experiment. 293T cells (CD4-, CCR5-) were transfected with either subtype-A or -C chimeric proviral DNA, overlaid with either JC37 or RC49 cells and stained using p24 antibodies. The HIV foci sizes are representative of general differences between the subtypes.
F. As in d), but using A1-A316T, A1-N302K, A2-T146I and A20-V1Δ5 and JC37 cells.
Bioinformatic analysis demonstrated that these sequences were common subtype representatives as they clustered within their respective subtypes on phylogenetic trees, and the distance and amino acid variation between them and other subtype-A and -C sequences was comparable to the medium intra-subtype distance and variation (supporting documentation, data not shown). Thus, the viral sequences from our patient may provide information on how subtype-A and -C compare functionally in vivo.
In vitro PBMC replication and cell fusion
In PBMC cultures, we noted marked subtype-specific differences in live virus production. Whereas subtype-C titers peaked at 4-6 days post-infection, subtype-A titers rose more slowly over the course of 16 days (Fig. 1B). Throughout the PBMC infection experiment, viral supernatants were removed and used to infect JC37 cells, which expressed low levels of CCR5. Regardless of sampling time, supernatants from the subtype-C infections produced large foci (10-20 nuclei/focus) (Fig. 1D). In contrast, supernatants from the subtype-A infections at day 4-8 generated mostly small foci (1-2 nuclei/focus). However, subtype-A foci increased markedly in size at days 12-16, coincident with increased replication in PBMC, suggesting that adaptation occurred during in vitro culture of these viruses.
Fusogenicity studies
We examined the ability of the viral envelope protein to fuse with cell lines expressing CD4 and CCR5 by transfecting 293T (CD4-, CCR5-) cells with plasmid DNA of subtype-A and -C infectious clones. The transfected cells expressed similar cell surface viral envelope levels by flow cytometry analysis at 48h post-transfection. When these cells were overlaid for 24 hours on RC49 cells (CD4low, CCR5high), both subtypes-A and -C induced a high level of fusion (Fig. 1E). By contrast, when they were overlaid onto JC37 cells (CD4high, CCR5low), subtype-A viruses generated small HIV-positive fusion foci ranging in size from 1 to 9 nuclei (mean ∼2 nuclei per focus), whereas subtype-C foci contained from 1 to >100 nuclei per focus (mean ∼20 nuclei). Thus, HeLa cells with low CCR5 levels were fused more efficiently by subtype-C viruses than by subtype-A viruses, but both subtypes resulted in extensive fusion on cells expressing high CCR5 levels.
Selection of adapted subtype-A mutants in PBMC
During replication in PBMC (Fig. 1B), subtype-A viruses only slowly attained the high titers seen early on in subtype-C infection, and the early and late supernatants from subtype-A infections differed in their ability to fuse JC37 cells, suggesting that mutant subtype-A viruses had been selectively amplified during in vitro culture. To search for evidence of adaptation, we reverse-transcribed, amplified, and sequenced viral subtype-A RNA from day 16 and found four distinct envelope sequence changes targeting the V1 and V3 loops. Using these sequences, we generated new chimeric viruses, A1-A316T, A1-N302K, A2-T146I and A20-V1Δ5, which, apart from the indicated changes, were otherwise identical to their parental virus(Fig. 1A). These adapted viruses displayed a distinct functional phenotype, which differed from their parental subtype-A clones but was similar to the chimeric subtype-C clones, i.e. they spread rapidly in PBMC and produced large foci in JC37 cells(Fig. 1C,F), cell types that both express low levels of CCR5.
The influence of CCR5 expression levels on virus titers
To determine whether CCR5 expression levels influenced viral replication in CD4+, CCR5+ HeLa cells, subtype-A, -C and PBMC-adapted subtype-A clones were used to infect JC37 and RC49 cells. In RC49CCR5high cells, all viruses gave high titers (2.5×105-3.7×106 FFU/ml) at 5 days post-infection(Table 1). In contrast, in JC37CCR5low cells, all three subtype-A clones produced low virus titers (1.9×103-1.7×105 FFU/ml), whereas subtype-C and all four PBMC-adapted subtype-A clones produced high titers similar to those seen on RC49 cells.
Table 1.
Comparison of chimeric virus infectivity titers in supernatant fluid from infected JC37 and RC49 cells.
| Titer | (FFU/ml) | ||
|---|---|---|---|
| Clone | JC37 CCR5low | RC49 CCR5high | Ratio (RC49/JC37) |
| A1 | 9.7×104 | 1.0 ×106 | 10.31 |
| A2 | 1.7×105 | 1.6 ×106 | 9.41 |
| A20 | 1.9×103 | 2.5 ×105 | 131.58 |
| C66 | 1.6 ×106 | 3.7 ×106 | 2.31 |
| A1-V3-A316T | 6.9 ×106 | 6.7 ×106 | 0.97 |
| A1-V3-N302K | 4.2 ×106 | 5.4 ×106 | 1.29 |
| A2-V1-T146I | 7.9 ×106 | 6.2 ×106 | 0.78 |
| A20-V1Δ5 | 9.3 ×106 | 9.9 ×106 | 1.06 |
Structural modeling of envelope variation
Structural modeling was performed to help understand the molecular consequences of the four subtype-A in vitro adaptations(supplementary documentation). The V1-V2 envelope region lies close to the CD4-binding site, while the V3 region interacts with two parts of CCR5. The V3-A316T substitution may strengthen HIV-CCR5 interactions as the additional polar group is in an appropriate position to form a new hydrogen bond to CCR5, most likely independently of the surrounding genomic context. Similarly, the N302K substitution adds another positive charge to the V3-stem, presumably strengthening CCR5-binding, as a crystal structure of V3 and the N-terminal negatively-charged CCR5-tail has shown it to directly interact with position 302[37].
The interactions between the CD4 receptor and the V1-V2 envelope region are less precisely understood as no high-resolution structures of V1-V2-containing gp120 envelopes exist. The loss of a V1-glycosylation site in A2-T146I and the A20-V1Δ5 deletion might enhance CD4 receptor contact, perhaps by decreasing steric hindrance. We used a neural network technique to analyze the propensity for disorder of the envelope proteins, as disordered regions can combine high binding specificity with modest affinity[32, 38](supplementary documentation). We found significant differences in disorder probability between subtype-A, A20-V1Δ5 and -C in the V1-V2 region, suggesting variation in the way these sequences might bind to CD4 and/or conformationally mask the CD4 binding site; however, both wild-type-A and -C chimeric viruses efficiently utilized the CD4 receptor in our functional assay.
Analysis of in vivo occurrence of mutations selected in vitro
We examined the frequency of the four subtype-A in vitro adaptations in natural HIV infections. Two substitutions were found occasionally in natural infections in some subtypes, however rarely (146I, 2%) or never (302K) in subtype-A(Table 2). By contrast, V3-316T was seen in 68% of all subtype-C sequences, but at much lower frequencies, if at all, in the other subtypes (25-0%)(Table 2, Fig. 2). Furthermore, the V1-Δ5 subtype-A deletion, which shortens the loop, resembled the short V1-V2 loops typically found in subtype-C but not in subtypes-A, -B and −D(Table 2). The high frequency of V3-316T and short V1-V2 loops in subtype-C, which both increase HIV spread in vitro, suggest these motifs may affect HIV replication in infected patients.
Table 2. In vivo frequencies of mutations selected in vitro.
| HIV Subtype | |||||
|---|---|---|---|---|---|
| Residue | Region | A | B | C | D |
| 316Ta | V3 | 0.25 (13/53)c | 0.05 (27/528) | 0.68 (318/469) | 0.11 (7/61) |
| 316Ab | V3 | 0.68 (36/53) | 0.84 (443/528) | 0.25 (119/469) | 0.77 (47/61) |
| 302Ka | V3 | 0 (0/53) | 0.02 (9/528) | 0.004 (2/469) | 0.08 (5/61) |
| 302Nb | V3 | 0.98 (52/53) | 0.98 (521/528) | 0.98 (461/469) | 0.80 (49/61) |
| 146Ia,d | V1 | 0.02 (1/53) | 0.05 (28/528) | 0.01 (4/469) | 0.07 (4/61) |
| 146T/Sb,d | V1 | 0.30 (16/53) | 0.18 (94/528) | 0.14 (66/469) | 0.28 (17/61) |
| V1-Δ5a | V1-V2 | -e | - | + | - |
| V1-no del | V1-V2 | +e | + | - | + |
Mutant (a) and wild type (b) residues. (c) Frequency of mutant and wild type amino acid residues in HIV subtype-A (A1), -B, -C and −D sequences from the Los Alamos database; number of sequences over number examined in brackets. (d) As V1 is very difficult to align, numbers are approximations. (e) V1-V2 length comparison: regions with long loops were common (+) in subtypes −A, B, D, and rare (-) in subtype-C. In contrast, regions with shorter loops were common (+) in subtype-C and rare (-) in the other subtypes (the consensus subtype-C V1-V2 loop was 13 amino acid shorter than that of any other subtype)(www.hiv.lanl.gov).
Figure 2.
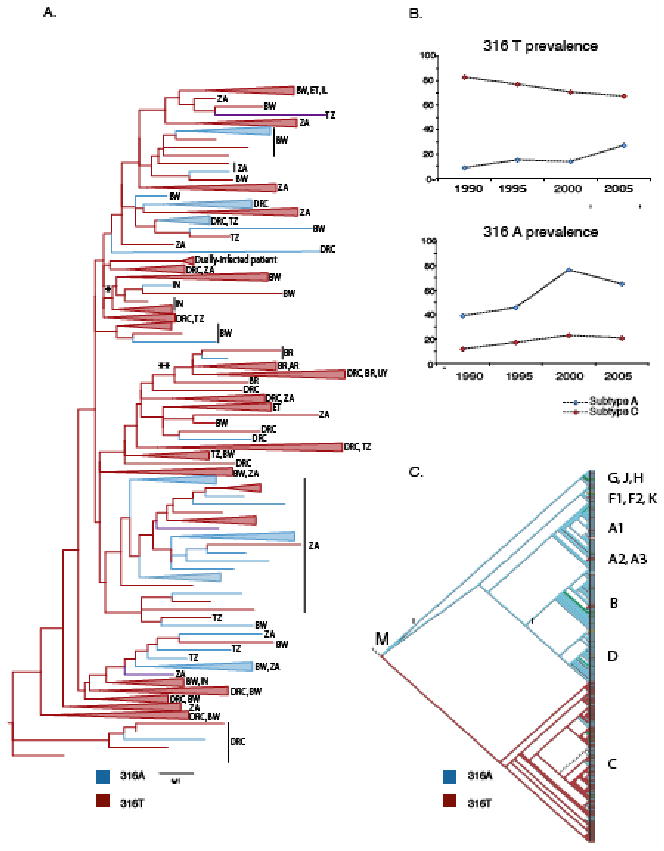
A. Phylogenetic tree of a 219 base pair env region (HXB2 7050-7271) from 163 subtype-C sequences (1985-2005), including our patient sequences. The scale bar shows the branch length equal to 5 nucleotide changes per 100 bases (0.05). Sequences carrying 316 A and T are blue and red, respectively, branches where all sequences have the same amino acid at position 316 are collapsed and shown as colored triangles (uncollapsed tree with GenBank accession numbers, supplementary documentation, Figure 2). Ancestral branches for India and South America are labeled with * and **, respectively. AR = Argentina, BR= Brazil, BW= Botswana, DRC= Democratic Republic of Congo, ET= Ethiopia, IL = Israel, IN= India, KE = Kenya, SE=Sweden, TZ = Tanzania, UG=Uganda, UY = Uruguay, ZA= South Africa.
B. Graphs show the prevalence of 316A and T in subtype A (blue) and C (red), respectively, from 1990 to 2005 (number of sequences, subtype A/C: 1985-90, n= 82/261; 1991-95, n=147/356; 1996-2000, n= 614/1519; 2001-2005, n=166/993).
C. Cladogram of 263 V3 env sequences showing the distribution of 316A (blue) and T (red) in all subgroup M subtypes (M = subgroup M root).
Phylogenetic analysis of 316T selection
While deletions cannot be incorporated in phylogenetic analysis at present, we could examine V3-316T selection. We generated a phylogenetic tree based on subtype-C sequences from 1985-2005 and superimposed the nature of the amino acid at position 316(Fig. 2A). The earliest sequences are from the Democratic Republic of Congo (DRC) where HIV is thought to have evolved and diversified. The phylogenetic tree demonstrates that both 316A and 316T could be found in the DRC sequences at the base of the tree. However, the deep lineages and branches of the tree mostly carry 316T suggesting that this residue has been generally advantageous to subtype-C spread. While 316V can be found sporadically, alanine seems to spread locally, especially in more recent sequences from South Africa and Botswana. We studied the distribution of 316A and 316T in larger alignments of shorter envelope sequences dating from 1990 to 2005 to see if this observation could be substantiated(Fig. 2B). We found that the frequency of 316T indeed had decreased from around 80% in 1990 to approximately 68% in 2005. This suggests that there may be alternate selective advantages for 316A in some situations in vivo and/or that other changes sometimes are sufficient to favor transmission of subtype-C.
Analysis of HIV-316T distribution in blood and cervix
To analyze the prevalence of 316T in blood and cervix, we obtained samples from 36 additional women infected by HIV subtype-A(n=3), -B(n=28) or -C(n=5). While single-copy envelope sequences could be amplified from PBMC from all women, no HIV was found in the cervices of four[19].
Most subtype-B infected women carried virus with V3-316A, but four had either 316T, 316T/M, 316S or 316V virus in both samples(Fig. 3A). Moreover, we found V3-316 compartmentalization between blood and cervix in one subtype-B infected woman (1/28, 4%); all genital virus carried 316T (5/5) while only 316A could be found in PBMC (7/7)(Fishers exact test, p<0.001)(Fig. 3B).
Figure 3.
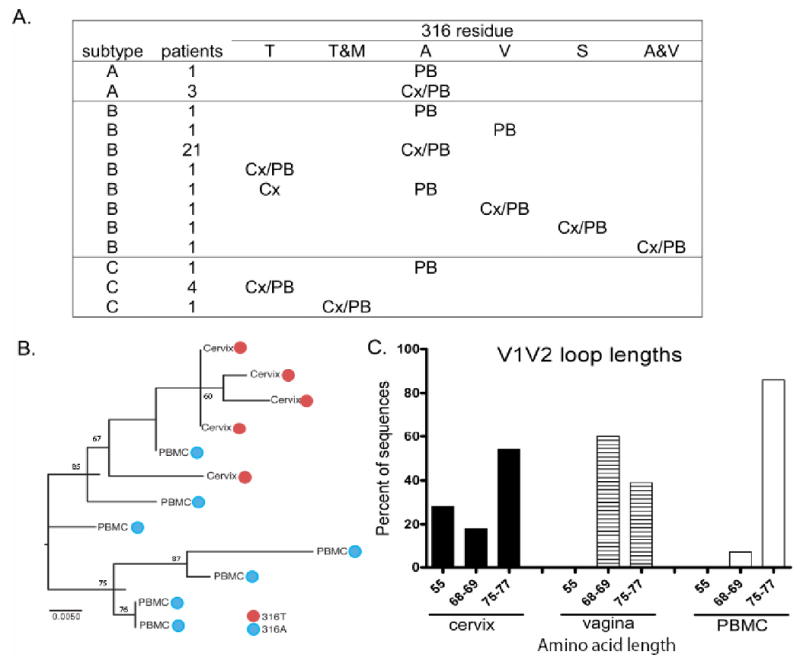
A. The patient's HIV subtype and the nature of the amino acid in position 316 in PBMC (PB) and cervix (Cx) is listed; A= Alanine, M= Methionine, S-= Serine, T= Threonine, and V= Valine (n=38, as the dually-infected woman is shown as both subtype-A and -C).
B. Phylogenetic tree demonstrating genital selection of 316T (red circle) and blood selection of 316A (blue circle) in a subtype-B infected woman. Bootstrap estimates above 60 are shown. Statistically significant values are generally considered to be ≥70, although no established significance threshold exist.
C. Percentage of different V1-V2 loop lengths in gp160 sequences from vagina, cervix and blood from a subtype-C infected woman with V3-316T.
Of the five subtype-C infected women, only one carried HIV-316A, and no virus could be amplified from her genital sample; the rest had either 316T or 316T/M in both PBMC and cervix. In one of these patients with HIV-316T, we were able to analyze gp160 from vagina as well as cervix and PBMC(Fig. 3C). While 28% of the cervical sequences had very short V1-V2 loops (55 amino acids), this was the case for only 5% of those from blood; most vaginal sequences had intermediate V1-V2 loops. Overall, V1-V2 loop lengths differed significantly between genitalia and blood when grouped into either short (≤69 amino acids) or long (>69 amino acids) loops (Fisher's exact test, p=0.02).
While our results demonstrate that subtype-A and -B virus with 316A can replicate in the cervix, they also highlight that selective forces in the genital tract may favor replication of virus with 316T even in subtype-B infected women. This is reminiscent of the preferential cervical replication of subtype-C virus in our dually-infected woman. Furthermore, we demonstrate that in a woman carrying only HIV-316T, selective forces in the genital tract may favor shorter V1-V2 loops than selection in blood.
Discussion
Subtype-C is responsible for an ever-increasing fraction of new HIV infections worldwide, especially in areas where heterosexual transmission predominates, and it now prevails even in regions where subtype-A and/or D used to be common[1, 2, 39, 40]. This suggests that there are biological factors favouring transmission of subtype-C, but no plausible mechanism has been suggested.
Our study of infectious, chimeric HIV clones carrying V1-V3 sequences from a woman dually-infected with subtype-A and -C, but with preferential cervical subtype-C replication, combined with the analysis of HIV sequences from blood and cervix from 36 additional women, lead to four findings suggesting that subtype-C generally has a relative replication advantage compared to most subtype-A and -B viruses in the female genital tract.
Firstly, we identify a V3-envelope signature sequence (316T) at high frequency in subtype-C, which might strengthen the interaction between HIV and CCR5, the main co-receptor in the female genital tract[41-45]. We find that the effect of 316T may be independent of the surrounding genomic context and show the effect of an A316T substitution in subtype-A. Secondly, we find that selection for 316T can occur even in the cervix of a minority of untreated subtype-B infected women. Thirdly, we demonstrate that a subtype-A V1-deletion augment virus production in cell lines expressing low levels of CD4 or CCR5 and propose that the shortening of the V1-loop increases viral interactions with the CD4-receptor. Fourthly, we find that genital selection in a subtype-C infected woman favors outgrowth of very short V1-V2 loops, despite subtype-C already having the most compact V1-V2 loops of all HIV subtypes.
We found that subtype-C had a greater ability to replicate in PBMC cultures than subtype-A, and mediated more extensive virus-induced fusion of cells expressing low CCR5 levels. However, we observed marked changes in subtype-A replication patterns over time and identified adaptations in either the V1 (T114I, V1-Δ5) or V3 (N302K, A316T) region, which are known to interact with the CD4 and CCR5 receptors, respectively. These adaptations enhanced virus replication and fusion comparable to what we found using the wild-type subtype-C clones. While two were found rarely, if at all, in HIV sequences obtained from infected persons, two others (A316T and V1-Δ5) resembled sequence motifs in the subtype-C sequences from our patient and in most subtype-C sequences from natural infections. In other subtypes, and in most CRFs, alanine is preferred at position 316, with the notable exception of CRF_02_AG, which carries threonine. Analogous to subtype-C spread in Southern and Eastern Africa, CRF_02_AG has spread throughout Western Africa, primarily via heterosexual contact[1, 46].
Both subtype-A and -C chimeric viruses interacted efficiently with the CD4-receptor; however, the V1-Δ5 adaptation augmented this interaction and increased infectivity several-fold in both cell lines and PBMC. The CD4-binding region is predicted to be shielded by the V1-V2 loop and shorter loops have been associated with increased susceptibility to antibody neutralization (13-15). As short V1-V4 loops and more neutralisation-sensitive HIV variants have been found in early subtype-A and -C infections, it was suggested that these were either preferentially transmitted, or rapidly selected in the recipient (12-15). We demonstrate that genital selection pressures in subtype-C infected women may sometimes favor virus with more compact V1-V2 loops than what is commonly found in blood. Consequently, whether transmission of such variants is preferential, or at times simply occurs by chance, remains to be elucidated.
Structural modeling suggested that the A316T-effect might be independent of the surrounding genomic background, as a new hydrogen-bond may form between the polar threonine and CCR5. The importance of position 316 in gp120-CCR5 interactions is indicated by the induction of drug-resistance mutations at this site by two different CCR5-inhibitors (A316T, Maraviroc; A316V, AD101)[47, 48]. CCR5-binding studies have demonstrated that these polar and non-polar substitutions crucially affect HIV-CCR5 interactions through different mechanisms[47, 48].
Our phylogenetic analysis demonstrated that 316T must have been favorable for subtype-C transmission. However, it also revealed that 316A seems to spread locally, especially in recent sequences from South Africa and Botswana. This implies that viruses with 316T incur a viral fitness cost even in subtype-C, as has been directly demonstrated for Maraviroc-resistant subtype-B virus with 316T in in vitro replication assays[47]. Additionally, in some situations in vivo, there could be selective advantages associated with 316A and/or other changes resulting in effective transmission. The A316T substitution could also affect virus susceptibility to host antibodies or CTL responses, as the V3-crown is a prime target for neutralizing antibodies and encompasses CTL-epitopes restricted by at least five different HLA alleles.
While HIV carrying 316A is fully replication competent and can be found in cervix, virus with 316T may sometimes have a relative replication advantage as CCR5 expression has been found on cells sampled from vagina, ecto- and endocervix [41-45, 49], the proportion of cervicovaginal T-cells expressing CCR5 is markedly expanded as compared with peripheral blood [41], HIV primarily propagates in vagina by T-cell infection[50], and cell-to-cell transfer is the predominant mode of HIV spread[51]. This relative replication advantage may be greatest in women with low genital CCR5-expression levels such as pre-menopausal women with normal genital mucosa, and less in women with high expression levels due to e.g. sexually-transmitted diseases (STD), cellular infiltrates, menopause or oral contraceptive use[42, 52]. Indeed, our patients with preferential cervical replication of HIV subtype-C or -B with 316T had normal genital epithelium and no STD[17].
We propose that the efficient HIV-316T-CCR5 interaction may increase viral transmissibility. That a polymorphism at a single amino acid site can have such profound consequences agrees with the first report of species-specific selection in primate lentiviruses, which demonstrate that efficient spread of SIVcpz in human cells depend on a single amino acid change in Gag[53]. Our proposal is indirectly supported by transmission studies in discordant couples with known CCR5 genotype; if the uninfected partner were heterozygous for the Δ32-CCR5 allele, and thus had lower levels of CCR5 on T cells and monocytes[43], HIV transmission decreased during both heterosexual[45] and homosexual[43, 44] contact, although one study found no effect[54].
Our results raise the possibility of subtype-specific differences in the efficacy of topical genital CCR5-inhibitors, and highlight the need for clinical trials in patients infected with different subtypes. Furthermore, they suggest that the Maraviroc-induced drug mutation A316T could result in viruses that are more easily transmitted through sexual contact.
In conclusion, our study suggests that subtype-C has evolved in ways compatible with increased sexual transmissibility. While other envelope regions, additional viral genes and LTR promotor differences [55] in combination with epidemiological dynamics, such as founder effects, also must be important factors, these subtype-C motifs may contribute to its current dominance in Southern and Eastern Africa, which facilitated subsequent worldwide spread.
Supplementary Material
Supplemental Figure 1
Structural modeling.
A. Proposed gp120/CD4/CCR5 complex. The gp120 V3 loop makes two independent contacts with CCR5: the V3 crown interacts with the extra-cellular surface loops of CCR5, while the stem and base interact with the CCR5 tail. Only a monomer of the presumed trimeric interaction is shown.
B. Structure of HIV gp120 bound to the C-terminal 2 domains of CD4 and the antigen-binding fragment of the neutralizing antibody X5, which targets the CCR5 binding site[27](PDB-ID 2B4C). Green spheres denote N302 and A316 in the V3 loop stem and crown, respectively.
C. RONN disorder predictions of the V1-V3 region of subtype-C (black), subtype-A (grey) and PBMC-adapted viruses; A2-T144I and A20-V1Δ5 sequences are shown in green while A1-A316T, A1-N302K were indistinct from their parent virus. Values above 0.5 are predictive of disorder29.
Figure 2
Uncollapsed phylogenetic tree with GenBank accession numbers.
Acknowledgments
We thank the patients, Andrew Rambaut, Kim Hasenkrug, Rachel Lacasse, Angela Vincent and Nick Willcox for helpful comments, Anita Mora for graphics, Kevin Braughton and Ron Messer for technical assistance.
Funding was provided by the Danish AIDS Foundation, the Nuffield Dominions Trust, Cancer Research UK, the EU (LSHG-CT-2006-031220) and MRC, UK.
Footnotes
The authors have no competing financial interests.
Supplementary documentation can be obtained on request
Author contributions: AKNI initiated and designed the overall study, AKNI and BC planned and supervised different parts of the project, AKNI, BC and BLW analyzed data and wrote the manuscript, BLW, AEA and AKNI performed experiments, SCG, EYJ and DIS performed the structural modeling and RONN analyses, TdO and AKNI performed the phylogenetic analysis, AKNI and PS provided patients, clinical data and reagents, AJM contributed reagents. All authors discussed the results and approved the final version of the manuscript.
References
- 1.Osmanov S, Pattou C, Walker N, Schwardlander B, Esparza J. Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000. J Acquir Immune Defic Syndr. 2002;29:184–190. doi: 10.1097/00042560-200202010-00013. [DOI] [PubMed] [Google Scholar]
- 2.Morison L, Weiss HA, Buve A, et al. Commercial sex and the spread of HIV in four cities in sub-Saharan Africa. Aids. 2001;15 4:S61–69. doi: 10.1097/00002030-200108004-00007. [DOI] [PubMed] [Google Scholar]
- 3.Shankarappa R, Chatterjee R, Learn GH, et al. Human immunodeficiency virus type 1 env sequences from Calcutta in eastern India: identification of features that distinguish subtype C sequences in India from other subtype C sequences. J Virol. 2001;75:10479–10487. doi: 10.1128/JVI.75.21.10479-10487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu XF, Wang X, Mao P, et al. Characterization of HIV type 1 heterosexual transmission in Yunnan, China. AIDS Res Hum Retroviruses. 2003;19:1051–1055. doi: 10.1089/088922203322588422. [DOI] [PubMed] [Google Scholar]
- 5.Soares EA, Martinez AM, Souza TM, et al. HIV-1 subtype C dissemination in southern Brazil. Aids. 2005;19 4:S81–86. doi: 10.1097/01.aids.0000191497.00928.e4. [DOI] [PubMed] [Google Scholar]
- 6.Carswell W. HIV in South Africa. Lancet. 1993;342:132. doi: 10.1016/0140-6736(93)91342-j. [DOI] [PubMed] [Google Scholar]
- 7.Hunter DJ. AIDS in sub-Saharan Africa: the epidemiology of heterosexual transmission and the prospects for prevention. Epidemiology. 1993;4:63–72. [PubMed] [Google Scholar]
- 8.Morison L, Buve A, Zekeng L, et al. HIV-1 subtypes and the HIV epidemics in four cities in sub-Saharan Africa. Aids. 2001;15 4:S109–116. doi: 10.1097/00002030-200108004-00012. [DOI] [PubMed] [Google Scholar]
- 9.Renjifo B, Gilbert P, Chaplin B, et al. Preferential in-utero transmission of HIV-1 subtype C as compared to HIV-1 subtype A or D. Aids. 2004;18:1629–1636. doi: 10.1097/01.aids.0000131392.68597.34. [DOI] [PubMed] [Google Scholar]
- 10.John-Stewart GC, Nduati RW, Rousseau CM, et al. Subtype C Is associated with increased vaginal shedding of HIV-1. J Infect Dis. 2005;192:492–496. doi: 10.1086/431514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- 12.Chohan B, Lang D, Sagar M, et al. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol. 2005;79:6528–6531. doi: 10.1128/JVI.79.10.6528-6531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Decker JM, Johnson RW, et al. Evidence for potent autologous neutralizing antibody titers and compact envelopes in early infection with subtype C human immunodeficiency virus type 1. J Virol. 2006;80:5211–5218. doi: 10.1128/JVI.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sagar M, Wu X, Lee S, Overbaugh J. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J Virol. 2006;80:9586–9598. doi: 10.1128/JVI.00141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derdeyn CA, Decker JM, Bibollet-Ruche F, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 16.Frost SD, Liu Y, Pond SL, et al. Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J Virol. 2005;79:6523–6527. doi: 10.1128/JVI.79.10.6523-6527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iversen AK, Larsen AR, Jensen T, et al. Distinct determinants of human immunodeficiency virus type 1 RNA and DNA loads in vaginal and cervical secretions. J Infect Dis. 1998;177:1214–1220. doi: 10.1086/515266. [DOI] [PubMed] [Google Scholar]
- 18.Iversen AK, Fugger L, Eugen-Olsen J, et al. Cervical human immunodeficiency virus type 1 shedding is associated with genital beta-chemokine secretion. J Infect Dis. 1998;178:1334–1342. doi: 10.1086/314433. [DOI] [PubMed] [Google Scholar]
- 19.Iversen AK, Learn GH, Fugger L, Gerstoft J, Mullins JI, Skinhoj P. Presence of multiple HIV subtypes and a high frequency of subtype chimeric viruses in heterosexually infected women. J Acquir Immune Defic Syndr. 1999;22:325–332. doi: 10.1097/00126334-199912010-00002. [DOI] [PubMed] [Google Scholar]
- 20.Adachi A, Gendelman HE, Koenig S, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–6554. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesebro B, Nishio J, Perryman S, et al. Identification of human immunodeficiency virus envelope gene sequences influencing viral entry into CD4-positive HeLa cells, T-leukemia cells, and macrophages. J Virol. 1991;65:5782–5789. doi: 10.1128/jvi.65.11.5782-5789.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology. 1995;213:70–79. doi: 10.1006/viro.1995.1547. [DOI] [PubMed] [Google Scholar]
- 24.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter BL, Wehrly K, Swanstrom R, Platt E, Kabat D, Chesebro B. Role of low CD4 levels in the influence of human immunodeficiency virus type 1 envelope V1 and V2 regions on entry and spread in macrophages. J Virol. 2005;79:4828–4837. doi: 10.1128/JVI.79.8.4828-4837.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swofford DL. PAUP* 4.0: Phylogenetic Analysis Using Parsimony (*and Other Methods) Sunderland, MA: 1999. [Google Scholar]
- 27.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 28.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 29.Huang CC, Tang M, Zhang MY, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 33.Iversen AK, Learn GH, Skinhoj P, Mullins JI, McMichael AJ, Rambaut A. Preferential detection of HIV subtype C′ over subtype A in cervical cells from a dually infected woman. Aids. 2005;19:990–993. doi: 10.1097/01.aids.0000171418.91786.ad. [DOI] [PubMed] [Google Scholar]
- 34.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 35.Cormier EG, Dragic T. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J Virol. 2002;76:8953–8957. doi: 10.1128/JVI.76.17.8953-8957.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabatov AA, Pollakis G, Linnemann T, Kliphius A, Chalaby MI, Paxton WA. Intrapatient alterations in the human immunodeficiency virus type 1 gp120 V1V2 and V3 regions differentially modulate coreceptor usage, virus inhibition by CC/CXC chemokines, soluble CD4, and the b12 and 2G12 monoclonal antibodies. J Virol. 2004;78:524–530. doi: 10.1128/JVI.78.1.524-530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang CC, Lam SN, Acharya P, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulz GE. Molecular mechanism of biological recognition. New York: Elsevier/Noth-Holland Biomedical Press; 1979. [Google Scholar]
- 39.Janssens W, Buve A, Nkengasong JN. The puzzle of HIV-1 subtypes in Africa. Aids. 1997;11:705–712. doi: 10.1097/00002030-199706000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Walker PR, Worobey M, Rambaut A, Holmes EC, Pybus OG. Epidemiology: Sexual transmission of HIV in Africa. Nature. 2003;422:679. doi: 10.1038/422679a. [DOI] [PubMed] [Google Scholar]
- 41.Hladik F, Lentz G, Delpit E, McElroy A, McElrath MJ. Coexpression of CCR5 and IL-2 in human genital but not blood T cells: implications for the ontogeny of the CCR5+ Th1 phenotype. J Immunol. 1999;163:2306–2313. [PubMed] [Google Scholar]
- 42.Patterson BK, Landay A, Andersson J, et al. Repertoire of chemokine receptor expression in the female genital tract: implications for human immunodeficiency virus transmission. Am J Pathol. 1998;153:481–490. doi: 10.1016/S0002-9440(10)65591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hladik F, Liu H, Speelmon E, et al. Combined effect of CCR5-Delta32 heterozygosity and the CCR5 promoter polymorphism -2459 A/G on CCR5 expression and resistance to human immunodeficiency virus type 1 transmission. J Virol. 2005;79:11677–11684. doi: 10.1128/JVI.79.18.11677-11684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas SM, Tse DB, Ketner DS, et al. CCR5 expression and duration of high risk sexual activity among HIV-seronegative men who have sex with men. Aids. 2006;20:1879–1883. doi: 10.1097/01.aids.0000244207.49123.ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffman TL, MacGregor RR, Burger H, Mick R, Doms RW, Collman RG. CCR5 genotypes in sexually active couples discordant for human immunodeficiency virus type 1 infection status. J Infect Dis. 1997;176:1093–1096. doi: 10.1086/516519. [DOI] [PubMed] [Google Scholar]
- 46.Laurent C, Bourgeois A, Faye MA, et al. No difference in clinical progression between patients infected with the predominant human immunodeficiency virus type 1 circulating recombinant form (CRF) 02_AG strain and patients not infected with CRF02_AG, in Western and West-Central Africa: a four-year prospective multicenter study. J Infect Dis. 2002;186:486–492. doi: 10.1086/341833. [DOI] [PubMed] [Google Scholar]
- 47.Westby M, Smith-Burchnell C, Mori J, et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81:2359–2371. doi: 10.1128/JVI.02006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuhmann SE, Pugach P, Kunstman KJ, et al. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol. 2004;78:2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McClure CP, Tighe PJ, Robins RA, et al. HIV coreceptor and chemokine ligand gene expression in the male urethra and female cervix. Aids. 2005;19:1257–1265. doi: 10.1097/01.aids.0000180096.50393.96. [DOI] [PubMed] [Google Scholar]
- 50.Hladik F, Sakchalathorn P, Ballweber L, et al. Initial Events in Establishing Vaginal Entry and Infection by Human Immunodeficiency Virus Type-1. Immunity. 2007;26:257–270. doi: 10.1016/j.immuni.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol. 2007;81:1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prakash M, Patterson S, Gotch F, Kapembwa MS. Ex vivo analysis of HIV-1 co-receptors at the endocervical mucosa of women using oral contraceptives. Bjog. 2004;111:1468–1470. doi: 10.1111/j.1471-0528.2004.00283.x. [DOI] [PubMed] [Google Scholar]
- 53.Wain LV, Bailes E, Bibollet-Ruche F, et al. Adaptation of HIV-1 to its human host. Mol Biol Evol. 2007;24:1853–1860. doi: 10.1093/molbev/msm110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lockett SF, Alonso A, Wyld R, et al. Effect of chemokine receptor mutations on heterosexual human immunodeficiency virus transmission. J Infect Dis. 1999;180:614–621. doi: 10.1086/314918. [DOI] [PubMed] [Google Scholar]
- 55.Centlivre M, Sala M, Wain-Hobson S, Berkhout B. In HIV-1 pathogenesis the die is cast during primary infection. Aids. 2007;21:1–11. doi: 10.1097/QAD.0b013e3280117f7f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Structural modeling.
A. Proposed gp120/CD4/CCR5 complex. The gp120 V3 loop makes two independent contacts with CCR5: the V3 crown interacts with the extra-cellular surface loops of CCR5, while the stem and base interact with the CCR5 tail. Only a monomer of the presumed trimeric interaction is shown.
B. Structure of HIV gp120 bound to the C-terminal 2 domains of CD4 and the antigen-binding fragment of the neutralizing antibody X5, which targets the CCR5 binding site[27](PDB-ID 2B4C). Green spheres denote N302 and A316 in the V3 loop stem and crown, respectively.
C. RONN disorder predictions of the V1-V3 region of subtype-C (black), subtype-A (grey) and PBMC-adapted viruses; A2-T144I and A20-V1Δ5 sequences are shown in green while A1-A316T, A1-N302K were indistinct from their parent virus. Values above 0.5 are predictive of disorder29.
Figure 2
Uncollapsed phylogenetic tree with GenBank accession numbers.