

Abstract
Brain-derived neurotrophic factor (BDNF) acts through TrkB, a receptor with kinase activity, and mitigates light-induced apoptosis in adult mouse rod photoreceptors. To determine whether TrkB signaling is necessary for rod development and function, we examined the retinas of mice lacking all isoforms of the TrkB receptor. Rod migration and differentiation occur in the mutant retina, but proceed at slower rates than in wild-type mice. In postnatal day 16 (P16) mutants, rod outer segment dimensions and rhodopsin content are comparable with those of photoreceptors in P12 wild type (WT). Quantitative analyses of the photoreceptor component in the electroretinogram (ERG) indicate that the gain and kinetics of the rod phototransduction signal in dark-adapted P16 mutant and P12 WT retinas are similar. In contrast to P12 WT, however, the ERG in mutant mice entirely lacks a b-wave, indicating a failure of signal transmission in the retinal rod pathway. In the inner retina of mutant mice, although cells appear anatomically and immunohistochemically normal, they fail to respond to prolonged stroboscopic illumination with the normal expression of c-fos. Absence of the b-wave and failure of c-fos expression, in view of anatomically normal inner retinal cells, suggest that lack of TrkB signaling causes a defect in synaptic signaling between rods and inner retinal cells. Retinal pigment epithelial cells and cells in the inner retina, including Müller, amacrine, and retinal ganglion cells, express the TrkB receptor, but rod photoreceptors do not. Moreover, inner retinal cells respond to exogenous BDNF with c-fos expression and extracellular signal-regulated kinase phosphorylation. Thus, interactions of rods with TrkB-expressing cells must be required for normal rod development.
Keywords: retina, rod photoreceptors, development, c-fos, ERK kinase, neurotrophins, BDNF, electroretinograms, A-wave
Neurotrophins play important roles in both early and late stages of neural development. Recently it has been shown that brain-derived neurotrophic factor (BDNF), basic fibroblast growth factor (FGF-2), and ciliary neurotrophic factor (CNTF) mitigate light-induced cell death in adult rodent photoreceptors (LaVail et al., 1998). During normal development, trophic support for the photoreceptors is thought to come from two retinal support cells, the retinal pigment epithelium (RPE) and Müller glia cells, with which the photoreceptors are in close apposition. To explore the role of TrkB-dependent signaling in rod photoreceptor differentiation and maturation, we investigated rod development in mice genetically manipulated to knock out the expression of both the full-length and the truncated forms of the TrkB receptor.
TrkB is one of three receptors with tyrosine kinase activity activated by neurotrophins. Nerve growth factor activates TrkA; neurotrophin 3 activates TrkC; and BDNF and neurotrophin 4/5 (NT-4/5) activate TrkB (for review, see Reichardt and Fariñas, 1997). Each of the neurotrophins also interacts with an unrelated receptor, the neurotrophin receptor p75. TrkB isoforms include proteins with the intracellular tyrosine kinase domain and truncated proteins that lack this domain. Activation of the TrkB kinase leads to stimulation of ras and other second messenger systems, which in turn activates expression of an immediate early gene, such as c-fos, and also other signaling events (Segal and Greenberg, 1996). Although it has been assumed that the truncated proteins may not signal directly, experiments in cell lines demonstrate that the truncated isoforms signal effectively (Baxter et al., 1997).
In the rat retina, at postnatal day 10 (P10), TrkB is expressed in retinal ganglion cells and the inner plexiform layer, with additional expression in the adult outer plexiform layer and Müller cells. The receptor is not expressed in rod or cone photoreceptors (Rickman and Brecha, 1995; Ugolini et al., 1995), and the absence of either TrkB or BDNF does not appear to affect photoreceptor survival (Rickman and Rickman, 1996; Cellerino et al., 1997). Yet, short-term exposure to BDNF prevents light-induced apoptosis in adult photoreceptors (LaVail et al., 1998). Also, photoreceptor development in Xenopus laevis is affected by the expression of a dominant negative construct of TrkB, probably because of an arrest in retinal pigment epithelium development (Liu et al., 1997).
Neurotrophin-dependent signaling has also been shown to regulate development of the electrical properties and synaptic activity of other neurons. Retinal ganglion cells in BDNF knock-out (KO) mice are developmentally delayed, and, at a given stage, they display reduced spontaneous and elicited activity characteristic of less mature cells (Rothe et al., 1996). In the same mice, in hippocampal neurons, long-term potentiation (Korte et al., 1995) is impaired. In TrkB KO mice, the number of synaptic contacts and the production of synapse-associated proteins in the hippocampal region are reduced (Martínez et al., 1998).
We generated a TrkB KO mouse in which both the full-length and the truncated forms of TrkB are missing. The animals live as long as 3 weeks, during which they actively move about but never appear able to orient themselves visually. Even though rods do not express detectable levels of TrkB receptors, their function and development are impaired in the mutant mice. Migration and differentiation of rods and the development of their transduction machinery, although slowed down, appears generally normal. On the other hand, light-elicited signal transmission to an otherwise normally appearing inner retina fails, suggesting a deficit in rod synapse function. Because rods do not normally express TrkB receptors, the developmental failures we observe likely arise from a defect in a required signaling path between TrkB expressing retinal cells and the photoreceptors.
MATERIALS AND METHODS
Materials. All chemicals were obtained from Sigma (St. Louis, MO) and were at least cell culture grade, unless otherwise noted.
TrkB knock-out mouse. The TrkB KO mouse used in this study was generated in our laboratory by targeting the first coding exon in the TrkB gene (B. Xu and L. F. Reichardt, unpublished results). Both full-length and the truncated forms of TrkB proteins are removed in this mouse. This mouse therefore differs from the original TrkB KO mouse reported by Klein et al. (1993), in which only the kinase domain was eliminated, generating an allele from which there was expression of the truncated form of TrkB. TrkB heterozygous mice were crossed to obtain homozygous mutants, identified by PCR of tail DNA. This mutation was maintained on an ICR (Institute for Cancer Research) strain background because litter size is larger, and pup survival improved on this background than on C57/Bl6 or 129 mice.
Mice were maintained under a 12 hr light/dark cycle in the animal facilities of the University of California San Francisco, with food and water ad libitum. All procedures were approved by the University of California San Francisco Animal Care Committee and conformed to Association for Research in Vision and Ophthalmology guidelines for animal research.
BDNF injections. To determine which cells express TrkB receptors on the cell surface and can therefore respond to BDNF stimulation, BDNF (1 μl; 1 mg/ml in sterile PBS; a gift from Amgen Inc., Thousand Oaks, CA) was injected into the right eye and PBS vehicle was injected into the left eye of P12 wild-type (WT) animals. Because there are no reports on NT-4/5 in the mouse retina, and because NT-4/5 activates the same receptors as BDNF, we did not repeat the experiments with NT-4/5. Animals were killed 1 hr after the injection, and eyes were enucleated and fixed in 4% paraformaldehyde (in PBS, pH 7.4) containing 4% sucrose and 100 μm sodium orthovanadate to block endogenous phosphatases. Tissue was then processed as summarized below in Paraffin sections and immunocytochemistry.
Semithin sections. Animals were deeply anesthetized with CO2 and perfused transcardially with Karnovsky fixative (2% paraformaldehyde and 4% glutaraldehyde in PBS, pH 7.4). Eyes were isolated and hemisected through three landmarks (superior oblique muscle, optic nerve, and inferior oblique muscle) to guarantee the same orientation in all eyes. After tissue osmication eyecups were embedded in Epon and Araldite. Semithin (1 μm) sections were cut with a glass knife and stained with toluidine blue solution (1% toluidine blue and 1% borax in distilled water). Photographs were taken on a Nikon (Tokyo, Japan) microscope using Ektachrome 64T film (Eastman Kodak, Rochester, NY).
Paraffin sections and immunocytochemistry. Animals were decapitated, and eyes were enucleated and immersion-fixed in Carnoys fixative for 2 hr, after which they were dehydrated over several hours and embedded in paraffin in transverse orientation. Seven micrometer sections of the eye were cut in dorsoventral orientation and dried onto poly-l-lysine-coated glass slides. Sections were dewaxed and rehydrated through xylene and a graded series of ethanol.
For visualization of the antigens, we used the peroxidase method. Endogenous peroxidase was first quenched for 10 min in 3% hydrogen peroxide in TBS (100 mm Tris, pH 7.5, and 150 mm NaCl) plus 10% methanol. Nonspecific binding was blocked by incubating sections for 1 hr in blocking solution (3% bovine serum albumin, 10% normal goat serum, and 0.4% Triton-X in TBS). Primary antibodies (see below) were applied overnight in blocking solution, followed by biotinylated secondary antibodies and the avidin and biotinylated horseradish peroxidase complex (ABC; Vector Laboratories, Burlingame, CA) for 1 hr each. Slides were developed in DAB (0.05% diaminobenzidine in 0.1 m Tris, pH 7.5, and 0.003% hydrogen peroxide as substrate) for 1–5 min, dehydrated, and mounted in DPX medium. Nickel enhancement (0.2% nickel chromium in DAB, pH 8.0) was used for some experiments. Cell counts of immunolabeled AII amacrine cells were performed in sections from the center of the retina. Sections were photographed using a Nikon microscope and 100 ASA or 64T Ektachrome film.
For fluorescence immunocytochemistry of RPE flat mounts, relaxation cuts were made on the sclera, choroid, and RPE; the tissue was then mounted flat, RPE side up, on small pieces of nitrocellulose filter (Millipore, Bedford, MA; 8 μm pore size). The blocking step was performed as above, whereas primary and secondary antibodies were applied for 48 and 24 hr at 4°C, respectively. For viewing, the complex was placed onto a slide inside of two stacked Avery rings (Avery Dennison Corp., Diamond Bar, CA), covered in Fluoromount (Southern Biotechnology Associates, Inc., Birmingham, AL), coverslipped, and examined by confocal microscopy (600 MRC series; Bio-Rad, Hercules, CA). Images were false-colored and superimposed using Photoshop (Adobe Systems, Mountain View, CA).
Antibodies. Nine primary antibodies were used in this study. Two antibodies recognizing TrkB, one of which was raised against the extracellular domain of TrkB, and therefore does not distinguish between the kinase and the truncated forms of TrkB (1:400; obtained from G. Wilkinson, University of California San Francisco; Meyer-Franke et al., 1998), whereas the second one was raised against the kinase domain of TrkB and is therefore specific for the full-length TrkB (1:1000; generously given by D. Kaplan, University of Montreal, Montreal, Quebec Canada; Allendoerfer et al., 1994). A monoclonal antibody against protein kinase C (α and β subspecies) was used to label rod bipolar cells (1:100; Amersham, Arlington Heights, IL;Grünert et al., 1994), whereas an anti-recoverin antibody was used to label cone OFF bipolar cells (1:5000; obtained from A. Dizhoor; University of Washington, Seattle, WA; Milam et al., 1993). A polyclonal antibody raised against the metabotropic glutamate receptor 6 (mGluR6) was obtained to label the glutamate receptors on bipolar cells (1:1000; given by R. Shigemoto, University of Kyoto, Kyoto, Japan; Nomura et al., 1994). AII amacrine cells were identified with an antibody against a calcium binding protein parvalbumin (1:2000; Swant, Bellinzona, Switzerland), and the inwardly rectifying potassium channel Kir4.1 on the Müller cells was labeled with an anti-Kir4.1 polyclonal antibody (1:1000; generously given by Y. Kurachi, Osaka University, Osaka, Japan; Ito et al., 1996). Immediate early gene expression was analyzed with a polyclonal antibody against c-fos (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), and short-term TrkB signaling was analyzed with a polyclonal antibody raised against the phosphorylated from of extracellular signal-regulated kinase (ERK) kinase ppERK (1:2000; Promega, Madison, WI; Liu et al., 1998). FITC-coupled anti-rabbit antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Biotinylated anti-mouse and anti-rabbit antibodies were purchased from Vector Laboratories.
Spectrophotometry. All experiments were conducted in darkness under infrared illumination with the aid of a television camera and monitor. Animals were dark-adapted overnight and killed by cervical dislocation. Eyes were enucleated, and retinas were dissected free of RPE in Ringer’s solution (in mm: 137 NaCl, 2.7 KCl, 1.36 CaCl2, 0.5 MgCl2, 1 NaH2PO4, 10 glucose, and 10 Tris-Cl, pH 7.2; 283 mOsm). The central piece of the retina (photoreceptor side up) was mounted flat, using relaxation cuts, in a specially designed chamber with a transparent glass floor (number 1 coverslip); the retina was then immersed in Ringer’s solution and covered with a second coverslip. The chamber was placed in the measuring light path within the modified measuring compartment of a Cary 118 spectrophotometer. The light path was masked (1.5 mm diameter) to ensure that absorption spectra were collected from a well defined, flat area of central retina. The retina was bleached without removing it from the sampling compartment using high-intensity light-emitting diodes of 510 nm and white light (Nichia America Corp., Mountville PA). Difference spectra (dark bleach) were fit with a nomogram with λmax = 507 nm (Lamb, 1995).
| Equation 1 |
Best fits were obtained with A = 0.88,B = 0.924, C = 1.104, D= 0.655, a = 70, b = 28.5, andc = 14.1.
To determine the absorbance per rod outer segment (OS), we measured the number of rod photoreceptors per square millimeter and the length and diameter of the OS at the various ages studied. To determine the number of rods, we counted photoreceptor nuclei in the central retina in semithin plastic sections. To determine the OS dimensions, we measured freshly dissociated OS at high magnification using a calibrated reticle. Cells were dissociated by mechanical trituration in Ringer’s solution (see above) in which 10 mm glucose was replaced by 5 mm pyruvic acid. OS were attached to concanavalin A-coated slides (Cherr and Cross, 1987; Babashak and Phillips, 1988). To compute the concentration of rhodopsin per retina, we used two additional numbers: the total number of rod photoreceptors per retina (6.4 × 106; Jeon et al., 1998), and the mouse rhodopsin molar extinction coefficient (ε = 42,000m/cm; Eder and Williams, 1973).
Stroboscopic illumination. Stroboscopic illumination was used to study light-induced protein modification and gene expression in the inner retina. For exposure to stroboscopic light we used a Grass stimulator (PS22; Grass Instrument Co., Quincy, MA) set at its maximum intensity (1.5 × 106 cd) that presented 10 μsec flashes of white light at 2 Hz. Animals, dark-adapted for >4 hr, were placed at 25 cm from the face plate of the strobe light and exposed to the flashing light for 2 hr to allow for induction of protein expression. Eyes were collected and processed as described above for BDNF injections.
Electroretinogram recordings. Animals were dark-adapted overnight and anesthetized under dim red light with a mixture of ketamine (10 mg/kg body weight) and xylazine (25 mg/kg) or avertine alone (15 mg/kg), after which their pupils were dilated with 1% atropine (Bausch and Lomb Pharmaceuticals Inc., Tampa, FL) and 2.5% phenylepinephrin (Alcon Inc., Humacao, Puerto Rico). Mice were secured on a heated block held at 37°C within a light-tight Faraday cage, where they were allowed to dark adapt for an additional 10 min before commencing recordings.
The eye was illuminated with a device of our construction, based on the design of Lyubarsky and Pugh (1996), which provides uniform, full-field retinal illumination. The device consisted of a clear Plexiglas rod (1.8 × 3.5 cm), which ends in a tip of 4 mm diameter and 1.0 cm length. Light was delivered to the other end of the rod through a 4-mm-diameter liquid light. The tip of the plastic rod was shaped to approximate the front of the mouse eye and was then treated with chloroform to produce a light-diffusing surface. A platinum wire (0.1 mm diameter) was secured on the rim of the Plexiglas tip and served as the recording electrode. The reference and the ground electrodes were sharpened tungsten wires (0.5 mm diameter) placed under the animals skin at the neck and tail, respectively. The diffusing tip of the Plexiglas rod was gently placed against the cornea with a micromanipulator, using care not to exert pressure onto the eye. Electrical contact with the cornea was ensured with a drop of methylcellulose solution (Gonisol; CibaVision Ophthalmics, Atlanta, GA) placed between rod tip and the cornea. Lids of P12 animals were forced open and kept from closing by the Plexiglas rod itself, making surgery unnecessary. A photostimulator that included a 250 W tungsten source, narrow-band interference filters, neutral density filters, and an electromechanical shutter (Vincent Associates, Rochester, NY) was used to deliver light to the end of the liquid light guide. Stimuli consisted of 10 msec flashes of varying intensity. Light intensity was measured using a calibrated photodiode placed in the position of the eye at the tip of the plastic rod (United Detector Technology, Hawthorne, CA). Analog electrical signals were differentially amplified 5000-fold with a high common mode rejection amplifier (Princeton Applied Instruments, Princeton, NJ), bandpassed between 0.1 and 1000 Hz (two-pole Bessel filter), and digitized on-line at 2 KHz with 12 bit accuracy (Indec, Capitola, CA). Digital signals were signal-averaged (up to four waveforms, depending on the noise level. The interval between repeat flashes was set to allow complete recovery of the b-wave between the flashes.
Data analysis. The leading edge of the a-wave in the electroretinogram (ERG) can be quantitatively analyzed to gain an understanding of the gain and kinetics of the phototransduction cascade. We analyzed the light intensity dependence of the peak amplitude and the normalized initial slope of the a-wave. The initial slope was measured as the slope of the tangent that best fit the initial rise of the normalized waveform. In addition, we fit the time course of the rising phase of the a-wave with the kinetic model developed by Lamb and Pugh. In this process we fit with computer-assisted nonlinear least square minimization (origin, Microcal Software, Northampton, MA) an analytical function to the experimental data. This function is a Gaussian function with two adjustable parameters, teff and A, that estimate a combined time delay generated by the biochemical reactions that lead to the photoresponse and an amplification parameter (A), which represents the combined gains of four biochemical activation stages (Lamb, 1994). A depends on light intensity and like all biochemical reactions has a rate-saturating profile. Thus to obtain both A andteff, the rising phase of the a-wave,f(t), was fit with the function:
| Equation 2 |
where φ is light intensity (photons per square millimeter). Responses to flashes below φ = 1.4 × 1010photons/mm2 could be fit simultaneously, whereas responses to flashes of higher intensities were individually fit. The amplification parameter A, when plotted against light intensity, exhibits a Weber-like saturation function, described by the following function:
| Equation 3 |
where F and Q are adjustable parameters. Q is the maximum possible value of A, and F is the intensity at which A is at half its maximum value. Values for A andteff were fit to data from each animal tested. These values were averaged, and comparison was made among the averaged values. Errors are presented as ±SD. To determine statistical significance, Student’s two tailed t test was used, accepting differences to be significant when p < 0.05.
RESULTS
Generation of TrkB knock-out mutant
The elimination of the first coding exon, which encodes the signal peptide and the start codon for TrkB and is common to both the full-length and the truncated forms of TrkB, led to a complete absence of TrkB protein expression in the homozygous transgenic mice. Kinase and nonkinase isoforms of TrkB receptors were undetectable by either Northern or Western blotting (Xu and Reichardt, unpublished results). Progeny of heterozygous parents have a normal mendelian distribution of genotypes, arguing that TrkB is not required for survival to term. Although most of the mutant homozygotes die within 48 hr of birth, some survived up to 3 weeks, which is sufficiently long to allow us to analyze some aspects of the postnatal development of the visual system.
Wild-type mice are born with their eyes closed, the lids separate between P13 and P14, after which time the pups orient themselves visually and score positive in the visual cliff avoidance test (Zhong et al., 1996). In the homozygous mutant animals, the lids separate, but they do not open entirely. The animals display a pupillary light reflex but with a very slow response time compared with WT littermates. Moreover, they show no visually guided orientation behavior and do not turn away from a visual cliff (B. Rohrer, personal observations).
As expected, the phenotype of the TrkB KO animals appears generally similar to that of BDNF KO (Jones et al., 1994), although we did not compare animals in great detail. TrkB KO are smaller than their littermates, reaching only one-third of the body weight of their littermates [P16 WT/Het, 12.0 ± 1.8 gm (n = 10) vs P16 KO, 4.9 ± 1.2 gm (n = 6)]. In addition, they have problems righting themselves, which in the BDNF KO has been attributed to a vestibular defect (Jones et al., 1994). TrkB KO also exhibit irregularly patterned breathing, which in the BDNF KO has been attributed to an absence of sensory innervation of the carotid body (Brady et al., 1999).
Developmental formation of the photoreceptor and other layers in the retina
We investigated whether the development of the retina, and in particular the development of rods, was perturbed in the TrkB KO. Figure 1 illustrates representative steps in the development of rods and the maturation of the different layers of the retina. The top panel depicts several developmental stages in the WT retina. At P2, the outer half of the retina is occupied by a mantle of poorly differentiated cells (Fig.1A), some of which are destined to become rods. The outer plexiform layer (OPL) starts to form at ∼P4–P5 and is visible as a gap between the photoreceptor nuclei (Fig. 1B). The rod precursors migrate through the OPL into the outer photoreceptor layer (arrows), a process that is complete in the central retina by ∼P10 (Fig. 1C) and in the periphery by P12 (data not shown). Thus by P10, in the central retina all the cells are organized in a layered structure, resembling the adult pattern. This developmental sequence occurs in the TrkB KO but it is slowed down and is not completed until P12 in the central retina (Fig. 1, middle panel) and P14 in the periphery (data not shown). Thus, the absence of TrkB signaling does not prevent the differentiation and migration of retinal cells and their assembly into defined layers but slows down these processes.
Fig. 1.
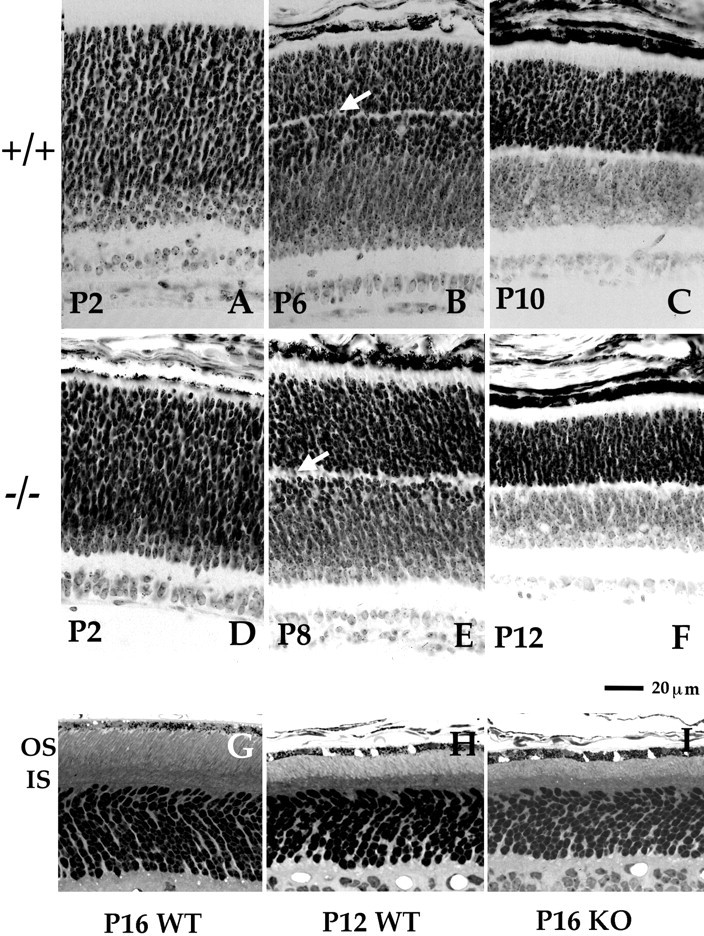
Early (A–F) and late (G–I) steps of photoreceptor development in WT and TrkB KO retina. A–F, Photographs of 7 μm sections stained with toluidine blue were taken in the central retina for comparison. Photoreceptor nucleus migration occurs over 10 d in WT animals (A–C) or 12 d in KO animals (D–F), including the period over which the nuclei have to migrate through the forming OPL (B,E, arrows) until they get to their final destination in the ONL (C,F).G–I, Photoreceptor outer segment development was analyzed in a 1 μm semithin section stained with toluidine blue. In WT animals, photoreceptors start to grow outer segments after P10. OS development is slowed in the TrkB mutant, such that by P16 (H) they are as short as at P12 in the WT and approximately one-third of the length of P16 WT (G).
In the WT retina, after the rods reach their final position at ∼P10, OS begin to elongate (Fig. 1, bottom panel) and reach their mature length after P21 (LaVail, 1973). Rods from TrkB KO mice do not form OS of normal length in the comparable time scale. Rod outer segments remain short over the life span of the mutant animals. At P16, the OS of the TrkB KO retina are approximately the same length as those of P12 WT mice (Fig. 1H,I). In freshly dissociated cells, the length of OS of P16 WT were 14.04 ± 1.69 μm in comparison with 6.42 ± 1.37 and 6.34 ± 1.49 μm in P12 WT and P16 KO, respectively (Table1). Thus by P16, anatomically, the mutant photoreceptors resemble those from a P12 WT animals.
Table 1.
Dimensions and visual pigment content of the photoreceptors
| Animals | Rod density (no. per mm2) | ROS diameter (μm) | ROS length (μm) | ROS volume (μm3) | Packing density | Visual pigment per eye (nmol/μm3) | Concentration [μm3(nmol)] | Rod axial absorbance1-a |
|---|---|---|---|---|---|---|---|---|
| P16 WT | 5.06 × 105 | 1.26 ± 0.32 | 14.04 ± 1.69 | 17.51 ± 1.71 | 0.63 | 0.1720 ± 0.030 | 0.00982 ± 0.0017 | 0.869 |
| P12 WT | 7.16 × 105 | 1.06 ± 0.26 | 6.42 ± 1.37 | 5.67 ± 1.50 | 0.62 | 0.0493 ± 0.011 | 0.00869 ± 0.0019 | 0.351 |
| P16 KO | 6.90 × 105 | 1.11 ± 0.30 | 6.34 ± 1.49 | 6.14 ± 1.51 | 0.66 | 0.0563 ± 0.010 | 0.00917 ± 0.0016 | 0.366 |
Rod density was derived by counting rows of rod nuclei per millimeter from 1 μm plastic sections. Rod outer segment width and length were measured from freshly dissociated photoreceptors, using a calibrated reticule. Those numbers were used to derive the total volume of the photoreceptors and their packing density (area of the mask minus the area of all the photoreceptors in the mask). Visual pigment concentration of the entire retina was derived from the color measured per mask area, a value that could then be used to determine the concentration of visual pigment per cubic micrometer and the resulting rod axial absorbance. Note that the values derived for the P12 and the P16 KO are virtually indistinguishable and significantly different from the P16 WT animals.
Absorbance = ɛ × c × length × 3/2; ɛ, extinction coefficient for mouse rhodopsin, 4.2 × 104; c, concentration of rhodopsin per cubic micrometer (nanomoles) length, ROS length (centimeters), 3/2, orientation factor (Witkovsky et al., 1997).
Cellular expression of TrkB in the retina
Figure 2 shows the distribution of TrkB receptors in the developing WT retina. TrkB staining was obtained with two different antibodies, one of which was raised against the extracellular domain of TrkB and therefore recognizes all isoforms of TrkB (pan-TrkB), whereas the other was raised against the kinase domain of TrkB and is therefore specific for the kinase isoform (TrkB-kinase). Almost identical staining patterns were seen with both antibodies, suggesting that all cells expressing TrkB express either the kinase or both the kinase and the truncated isoforms of TrkB. TrkB staining changes with maturation of the retina. Initial expression is in retinal ganglion cells and the developing inner plexiform layer (IPL) at P0 (Fig. 2A,E), followed by expression in the inner nuclear layer (INL), OPL, and Müller cells by P6 and later (Fig.2B–D,F–H). The staining with the pan-TrkB antibody revealed multiple strata in the IPL (Fig.2D, small arrowheads), which may correspond to some of the sublamina of the IPL described by Veruki and Wässle (1996). The differentiating horizontal cells (P6) are strongly labeled by the TrkB antibodies (Fig. 2B,F,arrows). Müller cells, spanning the retina radially, are strongly stained, with their microvillous processes forming a TrkB-positive layer at the level of the inner segments (Fig.2C,G, large arrowheads). Notably, there is no staining in the photoreceptor layer with either of the two antibodies. Localization of TrkB receptor was analyzed in RPE whole mounts using fluorescently labeled secondary antibodies. With a confocal microscope, it was possible to resolve that the pan-TrkB antibody stained the lateral surfaces and the apical surface of RPE cells (Fig.2I,K). In summary, the photoreceptors do not express detectable levels of TrkB receptors, but two of the cells types known to be important for photoreceptor differentiation and maintenance, Müller glia cells and RPE cells, do express TrkB receptors.
Fig. 2.

Developmental profile of TrkB staining. Two different antibodies, a pan-TrkB (A–D,I, J; raised against the extracellular domain of TrkB) and a TrkB kinase antibody (E–H; raised against the kinase domain of TrkB), were compared in their staining pattern. The two antibodies gave basically identical staining, arguing that all cells either express the full-length form or both the full-length and the truncated forms of TrkB. TrkB expression follows the maturation of the retina from inner (A,E) to outer retina (D,H). Note the staining of horizontal cells by P6 (B, F, arrows), the radial Müller glia cells (with their micrivillous processes forming a layer at the level of the inner segments of the photoreceptors) at all stages (e.g., C, G, large arrowheads), and the strata in the inner plexiform layer (D,small arrowheads). Whole-mounts of RPE demonstrate labeling of TrkB receptors on the lateral surfaces of the RPE cells (I, J) and in clusters on the apical surface of the cells (I,J,arrows). HC, Horizontal cells; IS, inner segments; PR,photoreceptors; RGC, retinal ganglion cells; other abbreviations are defined in Results.
Rhodopsin content in normal and mutant retinas
To determine whether the mutants have a defect in visual pigment production, we measured the concentration of functional rhodopsin in KO and WT animals by spectrophotometry, using whole mounts of retinas isolated from dark-adapted mice. Difference spectra were obtained by analyzing the absorbance before and after complete photobleaching and were fit by an absorbance nomogram (Lamb, 1995; see Materials and Methods). Measurements of OS dimensions were used to calculate the amounts of rhodopsin in the whole retina and individual photoreceptors and the resulting rod axial absorbance (Table 1).
Results summarized in Table 1 show that the P16 WT retina contains 0.172 ± 0.03 nmol/retina in comparison with 0.0493 ± 0.011 nmol/retina in the P12 WT. These values match well those previously estimated from digitonin extracts of whole retina (P16, 0.160 nmol; P12, 0.065 nmol; Carter-Dawson et al., 1986). The KO retinas at P16 contain rhodopsin at a concentration similar to that in P12 WT retina (0.0563 ± 0.01 nmol; Table 1, sixth column).
The efficiency with which rhodopsin captures photons depends on the rod axial absorbance, a function of both rhodopsin content and OS dimensions. Rod outer segment volumes were threefold higher in P16 WT retinas than in P12 WT and P16 KO retinas (17.51 ± 1.71, 5.67 ± 1.50, and 6.14 ± 1.51 μm3, respectively; Table 1, third column). Computations using the absorbance and dimension data indicate that rhodopsin concentration (nanomoles per cubic micrometer) does not change significantly between P12 and P16 (P12 WT, 0.00869 ± 0.0019 nmol/μm3; P16 WT, 0.00982 ± 0.0017 nmol/μm3; and P16 KO, 0.00917 ± 0.0016 nmol/μm3; Table1, seventh column). Thus as OS elongate, the axial absorbance increases proportionately with length (P12 WT, 0.351; P16 WT, 0.869; and P16 KO, 0.366; Table 1, seventh column). Taken together, the data show that photoreceptors in P16 KO animals contain functional rhodopsin in normalized amounts similar to those in P12 WT animals.
ERG recordings
The ERG is a light-evoked mass potential composed of several waves generated by the sum of the light response of different cell types within the eye. The cornea-negative a-wave is generated by the cessation of a current that flows in the dark along the rods from the inner to the outer segment. In the mouse retina, the rising phase of the a-wave is reflects the activation of the rod photoresponses without any contribution from the cone photoresponse (Hetling and Pepperberg, 1999). The cornea-positive b-wave reflects the activity of the rod-driven depolarizing bipolar (RDB) cells and is generated by the Müller glia cells (for review, see Pugh et al., 1997).
ERGs generated by 10 msec flashes of light were recorded from fully dark-adapted WT, TrkB heterozygous (Het), and KO mice. Because there was no difference in the responses of WT and Het animals, the data for these two groups are combined and referred to as WT. In Figure3, we illustrate ERGs from three individual mice recorded in a single session. P16 WT (Fig. 3,left panel) and P12 WT (Fig. 3, middle panel) are compared with P16 KO mice (Fig. 3, right panel). Most strikingly, at all light intensities, the ERG from the KO retina (Fig. 3, right panel) consisted of a small a-wave but no b-wave (six of six animals). This was in sharp contrast to the ERG from immature P12 WT animals, in which a b-wave could be reliably recorded (Fig. 3, middle panel). By P16, the ERG in the WT animals was dominated by the b-wave and its oscillatory potentials (Fig. 3, left panel). Note that in Figure 3, responses to equal light intensities are marked by anasterisk (∼1.5–1.8 × 1011photons/mm2 at the level of the cornea). Thus, in the mutant mouse the transduction signal of the rod photoreceptors develops, albeit at a slower rate than in the normal retina, but signal transmission along the retinal rod pathway is absent.
Fig. 3.

Family of representative ERG responses, elicited by short light flashes of increasing light intensities (∼0.6 log unit steps) for P16 WT (left panel), P12 WT (middle panel), and P16 KO (right panel) animals. Each trace is an average of two to four responses. Left panel, In the P16 WT the ERG consists of the negative a-wave and the fast, positive b-wave with the overlying oscillatory potentials (light intensities, 2.6 × 109; 9.8 × 109; 4.2 × 1010; and 1.5 × 1011photons/mm2). Middle panel, The P12 WT ERG is characterized by a small a-wave and a small and slowly rising b-wave (light intensities, 7.5 × 1010; 1.8 × 1011; 1.1 × 1012; and 5.5 × 1012photons/mm2). Right panel, By contrast, the P16 KO ERG consists only of a recordable a-wave but no b-wave (compare light intensities of middle panel). The negative potential only slowly reverts to baseline. Note that the asterisk indicates responses to equal light intensities (1.5–1.8 × 1011photons/mm2).
We analyzed the light dependence of the amplitude and the initial rate of rise of the a-wave in the three groups of animals. To compare among groups, the rate of rise was normalized by dividing, in each animal, the rate measured at a given intensity by the maximum rate measured in the same animal. As illustrated in Figure4, left panel, stimulation of both P12 WT and P16 KO retinas resulted in small potentials, even at their maximum value (78 ± 15 and 111 ± 53 μV; compare Table 2, second column), making it difficult to determine the threshold or the light intensity to produce half-Vmax. P16 WT a-waves reached a limiting value at 437 ± 143 μV, requiring ∼2.22 × 109 ± 1.51 × 109photons/mm2 for half-Vmax (Table 2, first and second columns). Results in Figure 4, right panel, show that the variability of responses between animals is reduced as animals get older. The results also demonstrate that the P12 WT and the P16 KO photoresponses require approximately sevenfold more photons to reach a constant initial slope in comparison with the P16 WT photoresponses (1.49 × 1010 ± 4.40 × 109 vs 1.87 × 1011 ± 1.65 × 1011 and 2.31 × 1011 ± 2.02 × 1011photons/mm2; Table 2, fourth column); their slopes can be as steep as in the P16 WT (Table 2, third column). These values are comparable with the differences in light sensitivity reported for isolated juvenile rods. Ratto et al. (1991) reported that photocurrents elicited from isolated P13 rat photoreceptors are ∼6.5-fold less sensitive than those measured in P17 rods. The similarity of data measured in the intact animals and the isolated rod photoreceptors suggest that the major differences in a-wave photosensitivity reflect changes in the maturation of the photoreceptors and not changes in the optics and/or light transparency of the ocular media.
Fig. 4.

A-wave amplitudes (left panel) and normalized initial slopes of the a-wave (right panel) for P16 WT, P12 WT, and P16 KO animals in response to increasing light intensities. Light intensity is expressed as photons per square millimeter on the surface of the cornea. Left panel, a-wave amplitudes saturate very quickly in the young (P12) as well as in the KO animals, whereas in the P16 WT amplitudes increase over a range of ∼3 log units. Right panel, The initial slope of the a-wave describes how the a-wave peaks faster with increasing number of photons captured. All three groups studied reach the same value (>20%/msec), but the younger (P12) and defective (KO) photoreceptors require approximately sevenfold more light to reach a constant initial slope.
Table 2.
Summary of the a-wave analysis
| Animals | A-waves | Normalized slopes | |||
|---|---|---|---|---|---|
| Vmax (μV) | φ at 1/2Vmax2-a | Limiting values (%/msec) | φ at constant slope2-a | Gain values | |
| P16 WT | 437 ± 143 | 2.22 × 109 ± 1.51 × 109 | 22.1 ± 3.1 | 1.49 × 1010 ± 4.40 × 109 | 3.88 × 10−6 ± 2.31 × 10−7 |
| P12 WT | 78 ± 152-b* | ND | 21.5 ± 6.4 (NS)2-b | 1.87 × 1011 ± 1.65 × 1011 b** | 1.63 × 10−6 ± 1.05 × 10−7 b** |
| P16 KO | 111 ± 53 (NS)c,d* | ND | 22.5 ± 3.4 (NS)c,d | 2.31 × 1011 ± 2.02 × 1011 c** (NS)2-d | 8.14 × 10−7 ± 4.25 × 10−7 c* (NS)2-d |
P12 WT and P16 KO photoreceptors produce a-waves that are comparable in amplitude (first column) and kinetics (third column). They are approximately seven-fold less sensitive then the P16 WT photoreceptors as demonstrated by the number of photons required for a constant normalized initial slope (set at 11%/msec; fourth column). In addition, the gain, as determined by the Lamb and Pugh (1992) analysis, is significantly greater in the P16 photoreceptors.
φ, Light intensity in photons per square millimeters.
P16 WT versus P12 WT.
P16 WT versus P16 KO.
P12 WT versus P16 KO.
*P < 0.005.
**P < 0.05.
We fit the rising phase of the a-wave with an analytical function that can be derived from a molecular model of the activation reactions underlying rod phototransduction (Lyubarsky and Pugh, 1996). This function has two adjustable parameters,teff and A, that can be argued to measure the kinetics and gain of the activation of the phototransduction biochemical cascade. Figure5, top panel, illustrates the application of this kinetic analysis to families of a-waves for one individual animal each. The amplification parameters A for all animals derived from this analysis were plotted against light intensity and fitted by a Weber-like saturation function (Fig. 5,bottom panel), giving a measure of the gain of the photocurrent of the rod photoreceptors. The gain of the rod photocurrent Q has a value of 3.88 × 10−6 ± 2.16 × 10−7 in the P16 WT animals, in comparison with the 2.5-fold smaller gain of 1.63 × 10−6 ± 8.07 × 10−7 in P12 WT (p< 0.05) and an ∼4.5-fold smaller gain of 8.14 × 10−7 ± 8.24 × 10−7 in the P16 KO (p < 0.005; Table 2, fifth column). On the other hand, the values of teff, which provides a measure for the delay in the activation reactions, are not significantly different among the three groups, (2.5–5.4 msec), which suggests that the kinetics of the enzymatic reactions are similar in all three sets of animals. Taken together, these results demonstrate that the sensitivity, kinetics, and gain of the a-wave in the P16 KO are similar to those in the P12 WT retina, suggesting that maturation of the transduction function in the outer segments is delayed by ∼4 d in the mutant animals.
Fig. 5.

Lamb and Pugh (1992) analysis of the rising phase of the photocurrent. Top panel, The leading edge of the a-wave was fitted by Equation 2 with all parameters free to vary. The solid lines in A–C represent the first 10–30 msec of the original recording of the light response of three individual animals, whereas the dotted linesshow the fit of the equation, demonstrating that the Lamb and Pugh model is appropriate to describe even the most immature photoresponses.Bottom panel, In D–F the amplification parameter A derived from the fitting of all animals was plotted against light intensity, and the curves for the individual animals were fitted by the Weber-like saturation function (Eq. 3).Q, which represents the gain of the responses, is significantly lower in the P12 and P16 KO in comparison with the P16 WT animals.
The slow development of rod phototransduction could reflect the general slowdown in development in the TrkB mouse; however, this is unlikely because P16 WT animals with body weight comparable with that of the KO animals [WT runts, 4.25 ± 0.37 gm (n = 3) vs P16 KO, 4.86 ± 1.22 gm (n = 6)] displayed both a- and b-waves (data not shown).
Light-driven c-fos expression
The absence of the b-wave suggests a functional defect either in the signal transmission from rod photoreceptors to the inner retinal cells or in the responsiveness of RDB or Müller cells. We reasoned that in the KO mice rod photoreceptor synaptic transmission is defective because the ERG in these mice is similar to that recorded in mammals in which rod photoreceptor synaptic transmission is blocked, either pharmacologically (Knapp and Schiller, 1984; Wakabayashi et al., 1988) or by knocking out the mGluR6 receptors on the rod ON-bipolar cells (Nomura et al., 1994).
Signal transmission in the retinal rod pathway has also been studied through the discovery that long-term exposure to light (minutes to hours) activates the expression of immediate early gene expression, such as c-fos, in cells of the inner retina (Rohrer et al., 1995;Yoshida et al., 1996), although the mechanism of this effect is unknown. To obtain additional data on whether the rod photoreceptors of the KO retina can transmit information to the inner retina, we examined induction of c-fos expression in response to prolonged, stroboscopic illumination. In P16 WT mice, this illumination induced c-fos expression in ganglion cells and cells in the proximal INL (likely amacrine cells; Fig.6A). Stroboscopic illumination also induced c-fos expression in the central retina of the P12 WT (Fig. 6B), indicating that rod to inner retina communication is established by this age. In contrast, no induction of c-fos expression could be detected in the P16 mutant retina under the same experimental conditions (Fig. 6C). These results further indicate that a defect in signal transmission from rods prevents other retinal cells from responding to light.
Fig. 6.

After complete dark adaptation, animals were exposed to 2 hr of 2 Hz stroboscopic illumination to analyze whether the inner retina can respond to a sustained signal from the photoreceptors. Two hertz strobe light induced c-fos expression in the retinal ganglion cells and cells in the inner half of the INL in WT animals (A, B), whereas no c-fos expression could be demonstrated in the KO animals (C) using this paradigm. RGC, Retinal ganglion cells; other abbreviations are defined in Results.
Immunohistochemical analysis of the rod pathway
Results obtained from ERG recordings and stroboscopic illumination suggest that the photoreceptors from KO mice fail to communicate with cells in the inner retina. This could be because the necessary postsynaptic elements in the rod pathway may not be present. In mouse, the b-wave reflects principally the light-driven activity of RDB and Müller cells, yet other cells are also involved in the processing of rod responses, including AII amacrine and cone OFF bipolar cells (for review, see Wässle and Boycott, 1991). Figure7 illustrates all these cell types identified by specific immunoreagents in retinal sections. The immunohistochemical sections compare the pattern of specific staining in P16 WT and P16 KO. RDB cells were visualized by staining with either protein kinase C (Fig. 7A,B; Zhang and Yeh, 1991) or the metabotropic glutamate receptor mGluR6 (Fig. 7C,D; Nakajima et al., 1993; Nomura et al., 1994). AII amacrine cells are immunopositive for the calcium-binding protein parvalbumin (Fig.7E,F; Wässle et al., 1993). The calcium-binding protein recoverin labels a class of OFF cone bipolar cells (flat midget bipolar cells; Milam et al., 1993; Euler and Wässle, 1995), which are considered to be one of the five classes of cone OFF-bipolar cells that receive the rod OFF signal (Fig. 7G,H). At the light microscope level, the staining pattern, including shape of cell bodies and localization of axon terminals of the RDB cells as well as localization of the mGluR6 receptors, appears comparable in the WT and KO retinas. In addition, the inwardly rectifying K+ channel Kir4.1 on the Müller glia cells, which is involved in the generation of the b-wave, is present in the KO retina (Fig. 7I,K). The number of AII amacrine cells seems to be slightly but significantly reduced in the KO retina (P16 WT, 140 ± 14 cells in the equatorial plane from dorsal to ventral poles; P16 KO, 115 ± 17 cells; n = 5;p < 0.001). This trend is similar to the results reported of Rickman and Rickman (1996), who observed a 50% reduction in the number of parvalbumin-labeled cells in rat retinas in which TrkB expression was reduced by antisense expression. One class of cone OFF-bipolar cells, however, either fail to express recoverin or are missing altogether. Thus, rod RDB and Müller cells, the cells that generate the b-wave, appear normal, but AII amacrine and recoverin-positive OFF-cone bipolar cells are altered in the KO mouse.
Fig. 7.
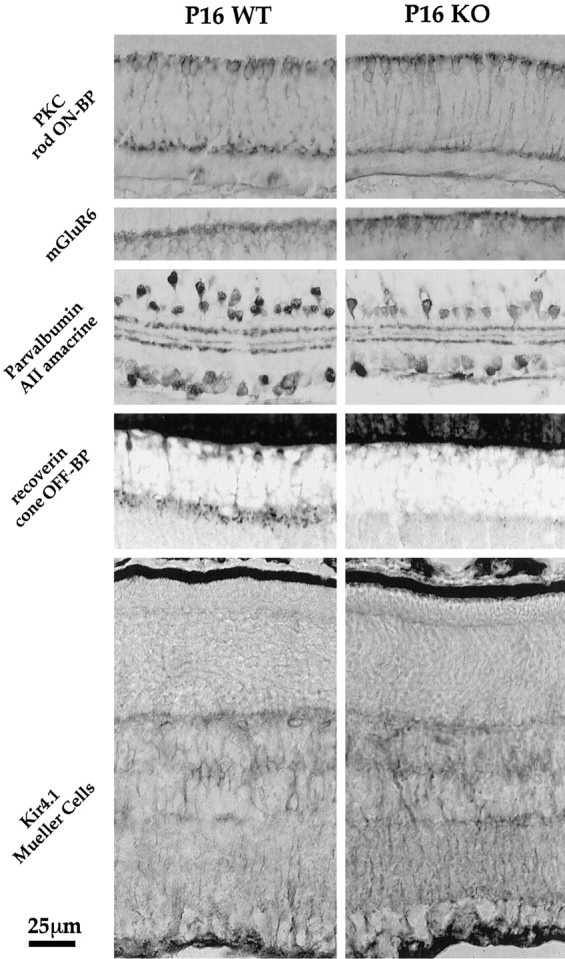
Immunocytochemical analysis of the rod pathway in WT (left column) and KO retina (right column). First row, Rod ON-bipolar cells can be stained with an antibody against protein kinase C (PKC). Their cell bodies are localized to the outermost part of the INL, and their axons terminate in the inner third of the IPL. Second row, The glutamate receptor on bipolar cells is the metabotropic glutamate receptor mGluR6. Third row, The rod signal gets transmitted to AII amacrine cells, which are immunopositive for parvalbumin. Their cell bodies are localized in the inner INL and stratify in both the OFF-lamina (outer IPL) and the ON-lamina (inner IPL) of the IPL. Fourth row, The OFF-component is signaled to cone OFF-bipolar cells. One class in rodent retina is immunopositive for recoverin, which terminates in the outer half of the IPL. Fifth row, The inwardly rectifying potassium channel Kir4.1 expressed in Müller glia cells is involved in K+ homeostasis and generates the b-wave. Note that the main components of the rod pathway, with exception of the recoverin-positive cone OFF-bipolar cells, seem to be present in the KO mouse retina.
BDNF-activated cellular responses
Rod photoreceptors do not express TrkB receptors, yet the development of their OS as well as signal transduction to the RDB cells is affected by the lack of TrkB signaling, indicating that photoreceptor development and function depend on interactions with TrkB-expressing cells. The expression of TrkB (Fig. 2), however, is widespread, making it impossible to identify a single candidate to mediate this cell–cell interaction. To identify retinal cells that do respond to BDNF and might be candidates to mediate the influence of TrkB on photoreceptors, we injected BDNF into the eyes of P12 WT mice and analyzed the retinal expression of c-fos (Fig.8, top panel) as well as phosphorylation of ERK kinase (Fig. 8, bottom panel). BDNF induced c-fos expression in nuclei of retinal ganglion cells and cells of the INL (Fig. 8A), whereas PBS alone had no effect (Fig. 8B). The majority of c-fos-immunoreactive cells in the INL are amacrine cells, which are located in the inner aspect of the INL, as well as some Müller cells, which were identified by their characteristic trapezoid shape of their cell bodies (arrows). In addition, BDNF injections resulted in a large increase in ERK kinase phosphorylation in the cytoplasm of the same cell types as well as in the horizontal cells (Fig. 8C,D). We were unable to determine the effect of exogenous BDNF on RPE cells because of their endogenous pigmentation. Taken together, these data indicate that BDNF activates known TrkB-regulated pathways in Müller glia, retinal ganglion cells, and amacrine cells in the juvenile mouse retina.
Fig. 8.
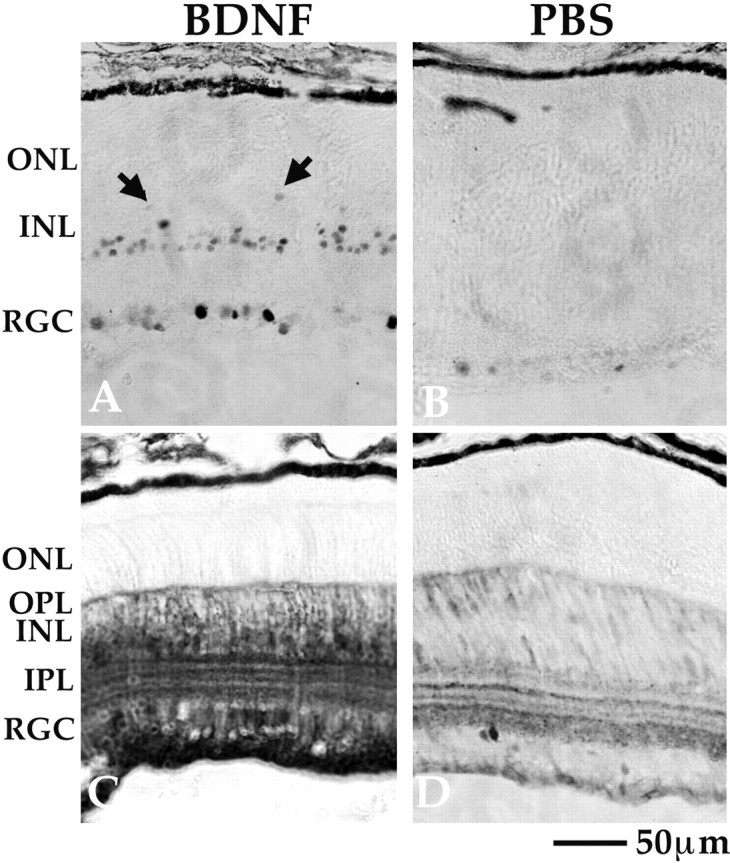
Juvenile retina responds to BDNF with ERK phosphorylation and immediate early gene expression. One microgram of BDNF was injected in one eye, whereas the contralateral eye was injected with vehicle (PBS) only. The level for c-fos expression (B) and ERK phosphorylation (D) was low in the vehicle-injected control eyes. BDNF injection led to an increase in nuclear c-fos induction predominantly in the inner retina (A) and to ERK kinase phosphorylation distributed throughout the cytoplasm of cells also in the inner retina (C). This includes amacrine, bipolar, Müller, and horizontal cell bodies. Note that the arrows in A indicate Müller cell bodies, identified by their characteristic shape.RGC, Retinal ganglion cells; other abbreviations are defined in Results.
DISCUSSION
By deletion of the first coding exon, we generated a TrkB allele from which none of the TrkB isoforms is expressed. Studies using the retinas of homozygous mice demonstrate that absence of TrkB slows postnatal rod development. In mutant P16 retinas, OS are similar in length and rhodopsin content than those in P12 wild-type eyes. Quantitative analyses of a-wave properties, which reflect rod photocurrents, also indicate that P16 KO and P12 WT photoreceptors are similar. Transmission of light-induced signals from photoreceptors to the rod pathway in the inner retina, however, appears to be absent in the KO retinas. No b-waves can be recorded in the ERG, and no light-induced c-fos expression can be detected in cells in the inner retina.
Although many cells in the retina were observed to express TrkB receptor isoforms and to respond to BDNF application with c-fos expression and ERK kinase phosphorylation, neither TrkB nor a direct response to BDNF could be detected in photoreceptors. It thus appears that interactions with TrkB-expressing cells are required for the normal developmental maturation of rods. In addition, because TrkB is expressed widely outside the retina, systemic effects may contribute to the delay in photoreceptor maturation and the defect in signal transmission in the rod pathway.
Effects of TrkB deficiency on light responses in photoreceptors
Because TrkB mutant homozygotes do not survive beyond 3 weeks postnatally, it has not been possible to examine retinal function in mutant adults. Instead, it has been necessary to examine light responses at ages before complete maturation of the retina. In previous work of others (Fulton et al., 1995; Dodge et al., 1996), the juvenile retina has been shown to be significantly less light sensitive, partially because of a reduced quantum catch of photons by smaller photoreceptors. If the reduced sensitivity of the juvenile or mutant rod response is attributable solely to reduced quantum catch, both the sensitivity and the rod axial absorbance should change by the same factor. In our experiments, rod axial absorbance increased approximately threefold comparing P12 WT or P16 KO with P16 WT, similar to a threefold increase in functional rhodopsin (Table 2). However, approximately sevenfold more light was required to produce a constant initial slope of the a-wave in the P12 WT or P16 KO than in the P16 WT retina (Table 1). Having excluded the possibility that the reduced sensitivity is attributable to changes in optics (see Results), these observations suggest that an additional 2.3-fold increase in gain in transduction is necessary to account for the sevenfold overall gain in efficiency of P16 photoreceptors (see below).
The results from the data analyses using the model of Lamb and Pugh (1992) also suggest that a reduced rhodopsin concentration as well as a reduced gain in signal transduction after photon capture contribute to the reduced light sensitivity of the a-wave generated in mutant compared with P16 WT animals. Although time delays were variable in all animals, as has been reported previously (Lyubarsky and Pugh, 1996), they did not differ significantly among P16 WT, P16 KO, and P12 WT animals. This argues that the kinetics of the reactions in the signal transduction cascade after photon capture are not affected by the absence of TrkB. In contrast, the calculated gain is ∼2.5- to 4.8-fold larger in the P16 WT than in either the P12 WT or P16 KO.
Effects of TrkB deficiency on signal transmission to the inner retina
Activity in rod photoreceptors of TrkB KO retinas did not lead to a recordable b-wave in ERG recordings. This could be attributable to a deficit in transmission from photoreceptors to RDB cells or the inability of the rod bipolar and/or Müller cells to signal. Absence of c-fos induction also indicated that there is a defect in signaling from the photoreceptors to the inner retina. However, because stroboscopic light does not elicit a c-fos response in bipolar cells but only in amacrine and ganglion cells, the data do not localize the deficit to the RDB cell synapse.
At least four mechanisms could explain our observations: (1) signal transmission occurs but is too weak to be detected; (2) the onset of signal transmission from rods to the inner retina is developmentally delayed beyond the survival time of the animal; (3) TrkB function is essential for expression of one of the proteins essential for synaptic transmission, and (4) TrkB function is required for signaling in RDB and/or Müller cells. Although the lack of a recordable b-wave suggests that transmission between photoreceptors and RDB cells is defective in the TrkB mutant, we have not obtained anatomical evidence of synaptic dysfunction. Moreover, RDB cells, which are necessary to produce a recordable b-wave, are present and express the metabotrophic glutamate receptor mGluR6, which mediates the synaptic response (Fig.7). Interestingly, despite the findings of Rickman and co-workers (1998) who observed no significant changes in synaptic vesicle distribution at photoreceptor ribbon synapses in P15 BDNF KO animals, we found that photoreceptors from BDNF KO mice at ages up to P17 also do not transmit signals to the inner retina (results not shown).
Is it possible that the development of rod signal transmission is slowed but not prevented in the TrkB KO? In the WT mouse, the development of the ERG can be divided into three phases: a-wave only, immature a- and b-waves, and finally a mature ERG. Thus, in very young animals, there is a period during which the ERG consists exclusively of the photoreceptor current-driven a-wave. The a-wave can be recorded as early as P10 (Dowling and Sidman, 1962), and its onset is controlled by the expression of rhodopsin, which trails the expression of the other components of the photoreceptor signal transduction cascade (Fulton et al., 1995). By ∼P11, the b-wave, which reflects the activity of ON-bipolar cells, can be detected (el Azazi and Wachtmeister, 1993; our unpublished observations). Thus, before P12 in wild-type animals, a functional rod pathway is established. Because an ERG lacking a b-wave is characteristic of a P10 retina, it is possible that development of the a-wave and the establishment of synaptic connections with the inner retina are partially uncoupled and that synaptic transmission would eventually appear in the mutants if they did not have such limited life spans.
Possible mechanisms of neurotrophin action
Mouse rods do not express TrkB receptors, yet their function is severely compromised in their absence. This suggests that the lack of TrkB affects development of cells that, in turn, support the development of rods. Candidates include retinal pigment epithelial, Müller, horizontal, and amacrine cells, all of which express TrkB receptors (Fig. 2).
As one possibility, TrkB signaling may regulate basic FGF-2 synthesis or secretion by RPE cells. Rods express FGF-2 receptors and bind FGF-2 (Mascarelli et al., 1989), and their death after light damage can be mitigated by FGF-2 (Faktorovich et al., 1992). BDNF has been shown to promote FGF-2 expression and secretion from cultured rat RPE cells (Hackett et al., 1997). In vivo, photoreceptor elongation has been shown to require close contact with the RPE (Kaplan et al., 1990). Consistent with a role for RPE cells, expression within the eye of a dominant negative mutant of TrkB has been shown to disrupt development of the RPE and to impair photoreceptor development inXenopus laevis (Liu et al., 1997).
Alternatively, Müller cells also express TrkB receptor (Fig. 2) and have been shown to secrete factors important for photoreceptor survival in culture (Wen et al., 1995). In this paper, we have shown that BDNF induces c-fos expression and phosphorylation of ERK kinase in these cells.
Two neuronal populations in the retina are known to provide local or long-range feedback to the photoreceptors, horizontal cells and dopaminergic amacrine cells, respectively (Linberg and Fisher, 1988;Daw et al., 1990; Udovichenko et al., 1998). Although both cell types express TrkB receptors (Fig. 2), only the dopaminergic amacrine cells were found to be affected by the absence of TrkB. In both the BDNF and the TrkB KO mouse retina, the expression of tyrosine hydroxylase is significantly reduced, and the dendritic arborizations are diminished (Cellerino et al., 1998; our unpublished observations), whereas the number and immunogenicity of horizontal cells for calbindin, parvalbumin, and GABA are not affected (B. Rohrer, unpublished observations). Lowering the dopamine concentration enhances rod responses (Dowling, 1991), making dopamine not a likely candidate to mediate the suppression of the b-wave. In contrast, GABAergic feedback has been found to reduce the amplitude of the b-wave (Arnarsson and Eysteinsson, 1997); however, GABA does not seem to be increased in the inner retina in the TrkB KO mice (Rohrer, unpublished observations).
Additional candidate molecules include taurine (Altshuler et al., 1993), S-laminin (Libby et al., 1996), sonic hedgehog (Levine et al., 1997), and members of the CNTF family (Fuhrmann et al., 1995;Kirsch et al., 1996; Ezzeddine et al., 1997; Neophytou et al., 1997), which have been demonstrated in in vitro experiments to be involved in photoreceptor differentiation.
The absence of TrkB may also inhibit the development of RDB cells and the Müller cells, because the lack of signal transduction to the inner retina in the KO could also be attributable to postsynaptic defects in these cells. TrkB labeling, however could not be detected in RDB cells by immunocytochemistry (data not shown). Any deficit in bipolar cells could also reflect regulation of FGF-2 secretion from other cells by TrkB signaling, because BDNF has been shown to promote survival of RDB cells by increasing release of FGF-2 from Müller cells in vitro (Wexler et al., 1998). In the future, it will be interesting to determine consequences on photoreceptor maturation and synaptic transmission of elimination of TrkB within specific retinal cell populations.
Footnotes
This study was supported in part by National Institutes of Health Research Grants EY11349, EY01919, EY02162, and MH48200 and funds from the Howard Hughes Medical Institute, the Foundation Fighting Blindness, and That Man May See, Inc. L.F.R. is an investigator and B.R. and B.X. are research associates of the Howard Hughes Medical Institute, and M.M.L. is a Research to Prevent Blindness senior scientist investigator. We thank Roger Pedersen, Juanito Menesses, Douglas Yasumura, Cathy Lau-Villacorta, Ward Peterson, and Laszlo Bocskai for advice and technical assistance.
Correspondence should be addressed to Dr. Baerbel Rohrer, Howard Hughes Medical Institute, University of California San Francisco, 533 Parnassus Avenue, Room U-332, San Francisco, CA 94143-0723.
REFERENCES
- 1.Allendoerfer KL, Cabelli R, Escandón E, Kaplan DR, Nikolics K, Shatz CJ. Regulation of neurotrophin receptors during the maturation of the mammalian visual system. J Neurosci. 1994;14:1795–1811. doi: 10.1523/JNEUROSCI.14-03-01795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshuler D, Lo Turco JJ, Rush J, Cepko C. Taurine promotes the differentiation of a vertebrate retinal cell type in vitro. Development. 1993;119:1317–1328. doi: 10.1242/dev.119.4.1317. [DOI] [PubMed] [Google Scholar]
- 3.Arnarsson A, Eysteinsson T. The role of GABA in modulating the Xenopus electroretinogram. Vis Neurosci. 1997;14:1143–1152. doi: 10.1017/s0952523800011834. [DOI] [PubMed] [Google Scholar]
- 4.Babashak JV, Phillips TM. Use of avidin-coated glass beads as a support for high-performance immunoaffinity chromatography. J Chromatogr. 1988;444:21–28. doi: 10.1016/s0021-9673(01)94005-1. [DOI] [PubMed] [Google Scholar]
- 5.Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB isoforms, trkB.T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breton ME, Schueller AW, Lamb TD, Pugh EN., Jr Analysis of ERG a-wave amplification and kinetics in terms of the G-protein cascade of phototransduction. Invest Ophthalmol Vis Sci. 1994;35:295–309. [PubMed] [Google Scholar]
- 8.Carter-Dawson L, Alvarez RA, Fong SL, Liou GI, Sperling HG, Bridges CD. Rhodopsin, 11-cis vitamin A, and interstitial retinol-binding protein (IRBP) during retinal development in normal and rd mutant mice. Dev Biol. 1986;116:431–438. doi: 10.1016/0012-1606(86)90144-2. [DOI] [PubMed] [Google Scholar]
- 9.Cellerino A, Carroll P, Thoenen H, Barde YA. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci. 1997;9:397–408. doi: 10.1006/mcne.1997.0641. [DOI] [PubMed] [Google Scholar]
- 10.Cellerino A, Pinzaon-Duarte G, Carroll P, Kohler K. Brain-derived neurotrophic factor modulates the development of the dopaminergic network in the rodent retina. J Neurosci. 1998;18:3351–3362. doi: 10.1523/JNEUROSCI.18-09-03351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cherr GN, Cross NL. Immobilization of mammalian eggs on solid substrates by lectins for electron microscopy. J Microsc 145 1987. 341 345[erratum (1987) 147:357]. [PubMed] [Google Scholar]
- 12.Daw NW, Jensen RJ, Brunken WJ. Rod pathways in mammalian retinae. Trends Neurosci. 1990;13:110–115. doi: 10.1016/0166-2236(90)90187-f. [DOI] [PubMed] [Google Scholar]
- 13.Dodge J, Fulton AB, Parker C, Hansen RM, Williams TP. Rhodopsin in immature rod outer segments. Invest Ophthalmol Vis Sci. 1996;37:1951–1956. [PubMed] [Google Scholar]
- 14.Dowling JE. Retinal neuromodulation: the role of dopamine. Vis Neurosci. 1991;7:87–97. doi: 10.1017/s0952523800010968. [DOI] [PubMed] [Google Scholar]
- 15.Dowling JE, Sidman RL. Inherited retinal dystrophy in the rat. J Cell Biol. 1962;14:73–109. doi: 10.1083/jcb.14.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.el Azazi M, Wachtmeister L. The postnatal development of the oscillatory potentials of the electroretinogram V. Relation to the double peaked a-wave. Acta Ophthalmol. 1993;71:32–38. doi: 10.1111/j.1755-3768.1993.tb04956.x. [DOI] [PubMed] [Google Scholar]
- 17.Eder DJ, Williams TP. A method of isorhodopsin analysis and the photoreversal of rhodopsin intermediates. Am J Optom Arch Am Acad Optom. 1973;50:765–776. doi: 10.1097/00006324-197310000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Euler T, Wässle H. Immunocytochemical identification of cone bipolar cells in the rat retina. J Comp Neurol. 1995;361:461–478. doi: 10.1002/cne.903610310. [DOI] [PubMed] [Google Scholar]
- 19.Ezzeddine ZD, Yang X, DeChiara T, Yancopoulos G, Cepko CL. Postmitotic cells fated to become rod photoreceptors can be respecified by CNTF treatment of the retina. Development. 1997;124:1055–1067. doi: 10.1242/dev.124.5.1055. [DOI] [PubMed] [Google Scholar]
- 20.Faktorovich EG, Steinberg RH, Yasumura D, Matthes MT, LaVail MM. Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J Neurosci. 1992;12:3554–3567. doi: 10.1523/JNEUROSCI.12-09-03554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuhrmann S, Kirsch M, Hofmann HD. Ciliary neurotrophic factor promotes chick photoreceptor development in vitro. Development. 1995;121:2695–2706. doi: 10.1242/dev.121.8.2695. [DOI] [PubMed] [Google Scholar]
- 22.Fulton AB, Hansen RM, Findl O. The development of the rod photoresponse from dark-adapted rats. Invest Ophthalmol Vis Sci. 1995;36:1038–1045. [PubMed] [Google Scholar]
- 23.Grünert U, Martin PR, Wässle H. Immunocytochemical analysis of bipolar cells in the macaque monkey retina. J Comp Neurol. 1994;348:607–627. doi: 10.1002/cne.903480410. [DOI] [PubMed] [Google Scholar]
- 24.Hackett SF, Schoenfeld CL, Freund J, Gottsch JD, Bhargave S, Campochiaro PA. Neurotrophic factors, cytokines and stress increase expression of basic fibroblast growth factor in retinal pigmented epithelial cells. Exp Eye Res. 1997;64:865–873. doi: 10.1006/exer.1996.0256. [DOI] [PubMed] [Google Scholar]
- 25.Hetling JR, Pepperberg DR. Sensitivity and kinetics of mouse flash responses determined in vivo from paired-flash electroretinograms. J Physiol (Lond) 1999;516:593–609. doi: 10.1111/j.1469-7793.1999.0593v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonasaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, K (AB)-2(Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett. 1996;388:11–15. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 27.Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan MW, Iwata RT, Sterrett CB. Retinal detachment prevents normal assembly of disk membranes in vitro. Invest Ophthalmol Vis Sci. 1990;31:1–8. [PubMed] [Google Scholar]
- 30.Kirsch M, Fuhrmann S, Wiese A, Hofmann HD. CNTF exerts opposite effects on in vitro development of rat and chick photoreceptors. NeuroReport. 1996;7:697–700. doi: 10.1097/00001756-199602290-00004. [DOI] [PubMed] [Google Scholar]
- 31.Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the TrkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- 32.Knapp AG, Schiller PH. The contribution of ON-cone bipolar cells to the electroretinogram of rabbits and monkeys. Vision Res. 1984;24:1841–1846. doi: 10.1016/0042-6989(84)90016-6. [DOI] [PubMed] [Google Scholar]
- 33.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamb TD. Stochastic simulation of activation in the G-protein cascade of phototransduction. Biophys J. 1994;67:1439–1454. doi: 10.1016/S0006-3495(94)80617-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamb TD. Photoreceptor spectral sensitivities: common shape in the long-wavelength region. Vision Res. 1995;35:3083–3091. doi: 10.1016/0042-6989(95)00114-f. [DOI] [PubMed] [Google Scholar]
- 36.Lamb TD, Pugh EN., Jr G-protein cascades: gain and kinetics. Trends Neurosci. 1992;15:291–298. doi: 10.1016/0166-2236(92)90079-n. [DOI] [PubMed] [Google Scholar]
- 37.LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol. 1973;58:650–661. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, Steinberg RH. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- 39.Levine EM, Roelink H, Turner J, Reh TA. Sonic hedgehog promotes rod photoreceptor differentiation in mammalian retinal cells in vitro. J Neurosci. 1997;17:6277–6288. doi: 10.1523/JNEUROSCI.17-16-06277.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Libby R, Hunter DD, Brunken WJ. Developmental expression of laminin beta 2 in rat retina. Further support for a role in rod morphogenesis. Invest Ophthalmol Vis Sci. 1996;37:1651–1661. [PubMed] [Google Scholar]
- 41.Linberg KA, Fisher SK. Ultrastructural evidence that horizontal cell axon terminals are presynaptic in the human retina. J Comp Neurol. 1988;268:281–297. doi: 10.1002/cne.902680211. [DOI] [PubMed] [Google Scholar]
- 42.Liu C, Peng M, Laties AM, Wen R. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344. doi: 10.1523/JNEUROSCI.18-04-01337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu ZZ, Zhu LQ, Eide FF. Critical role of TrkB and brain-derived neurotrophic factor in the differentiation and survival of retinal pigment epithelium. J Neurosci. 1997;17:8749–8755. doi: 10.1523/JNEUROSCI.17-22-08749.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martínez A, Alcantara S, Borrell V, Del Río JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mascarelli F, Raulais D, Courtois Y. Fibroblast growth factor phosphorylation and receptors in rod outer segments. EMBO J. 1989;8:2265–2273. doi: 10.1002/j.1460-2075.1989.tb08351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milam AH, Dacey DM, Dizhoor AM. Recoverin immunoreactivity in mammalian cone bipolar cells. Vis Neurosci. 1993;10:1–12. doi: 10.1017/s0952523800003175. [DOI] [PubMed] [Google Scholar]
- 49.Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for l-2-amino-4-phosphonobutyrate. J Biol Chem. 1993;268:11868–11873. [PubMed] [Google Scholar]
- 50.Neophytou C, Vernallis AB, Smith A, Raff MC. Müller-cell-derived leukaemia inhibitory factor arrests rod photoreceptor differentiation at a postmitotic pre-rod stage of development. Development. 1997;124:2345–2354. doi: 10.1242/dev.124.12.2345. [DOI] [PubMed] [Google Scholar]
- 51.Nomura A, Shigemoto R, Nakamura Y, Okamoto N, Mizuno N, Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77:361–369. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 52.Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- 53.Pugh EN, Jr, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram, and the effect of age and light-rearing history on the magnitude of these components. In: Williams, Thistle, editors. Phoytostasis and related phenomena. Plenum; New York: 1997. pp. 93–128. [Google Scholar]
- 54.Ratto GM, Robinson DW, Yan B, McNaughton PA. Development of the light response in neonatal mammalian rods. Nature. 1991;351:654–657. doi: 10.1038/351654a0. [DOI] [PubMed] [Google Scholar]
- 55.Reichardt LF, Fariñas I. Neurotrophic factors and their receptors: roles in neuronal development and function. Oxford UP; New York: 1997. [Google Scholar]
- 56.Rickman DW, Brecha NC. Expression of the proto-oncogene, trk, receptors in the developing rat retina. Vis Neurosci. 1995;12:215–222. doi: 10.1017/s0952523800007896. [DOI] [PubMed] [Google Scholar]
- 57.Rickman DW, Rickman CB. Suppression of TrkB expression by antisense oligonucleotides alters a neuronal phenotype in the rod pathway of the developing rat retina. Proc Natl Acad Sci USA. 1996;93:12564–12569. doi: 10.1073/pnas.93.22.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rickman DW, Orris A, DeChiara T, Yancopoulos G. Retinal development in the BDNF knockout mouse. Soc Neurosci Abstr. 1998;24:295. [Google Scholar]
- 59.Rohrer B, Iuvone PM, Stell WK. Stimulation of dopaminergic amacrine cells by stroboscopic illumination or fibroblast growth factor (bFGF, FGF-2) injections: possible roles in prevention of form-deprivation myopia in the chick. Brain Res. 1995;686:169–181. doi: 10.1016/0006-8993(95)00370-6. [DOI] [PubMed] [Google Scholar]
- 60.Rothe T, Bähring R, Carroll P, Thoenen H, Grantyn R. Repetitive firing deficits in brain-derived neurotrophic factor knockout mice. Soc Neurosci Abstr. 1996;22:2009. [Google Scholar]
- 61.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 62.Udovichenko IP, Newton AC, Williams DS. Regulation of the phosphorylation state of rhodopsin by dopamine. J Biol Chem. 1998;273:7181–7184. doi: 10.1074/jbc.273.13.7181. [DOI] [PubMed] [Google Scholar]
- 63.Ugolini G, Cremisi F, Maffei L. TrkA, TrkB and p75 mRNA expression is developmentally regulated in the rat retina. Brain Res. 1995;704:121–124. doi: 10.1016/0006-8993(95)01191-9. [DOI] [PubMed] [Google Scholar]
- 64.Veruki ML, Wässle H. Immunohistochemistry localization of dopamine D1 receptors in rat retina. Eur J Neurosci. 1996;8:2286–2297. doi: 10.1111/j.1460-9568.1996.tb01192.x. [DOI] [PubMed] [Google Scholar]
- 65.Wässle H, Boycott BB. Functional architecture of the mammalian retina. Physiol Rev. 1991;71:447–480. doi: 10.1152/physrev.1991.71.2.447. [DOI] [PubMed] [Google Scholar]
- 66.Wässle H, Grünert U, Röhrenbeck J. Immunocytochemical staining of AII-amacrine cells in the rat retina with antibodies against parvalbumin. J Comp Neurol. 1993;332:407–420. doi: 10.1002/cne.903320403. [DOI] [PubMed] [Google Scholar]
- 67.Wakabayashi K, Gieser J, Sieving PA. Aspartate separation of the scotopic threshold response (STR) from the photoreceptor a-wave of the cat and monkey ERG. Invest Ophthalmol Vis Sci. 1988;29:1615–1622. [PubMed] [Google Scholar]
- 68.Wen R, Song Y, Cheng T, Matthes MT, Yasumura D, LaVail MM, Steinberg RH. Injury-induced upregulation of bFGF and CNTF mRNAS in the rat retina. J Neurosci. 1995;15:7377–7385. doi: 10.1523/JNEUROSCI.15-11-07377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wexler EM, Berkovich O, Nawy S. Role of the low-affinity NGF receptor (p75) in survival of retinal bipolar cells. Vis Neurosci. 1998;15:211–218. doi: 10.1017/s095252389815201x. [DOI] [PubMed] [Google Scholar]
- 70.Yoshida K, Imaki J, Fujisawa H, Harada T, Ohki K, Matsuda H, Hagiwara M. Differential distribution of CaM kinases and induction of c-fos expression by flashing and sustained light in rat retinal cells. Invest Ophthalmol Vis Sci. 1996;37:174–179. [PubMed] [Google Scholar]
- 71.Zhang DR, Yeh HH. Protein kinase C-like immunoreactivity in rod bipolar cells of the rat retina: a developmental study. Vis Neurosci. 1991;6:429–437. doi: 10.1017/s0952523800001292. [DOI] [PubMed] [Google Scholar]
- 72.Zhong DZ, Pei C, Xiu-Qin L. Neurobehavioral study of prenatal exposure to hypothermia combined with irradiation. Neurotoxicol Teratol. 1996;18:703–709. doi: 10.1016/s0892-0362(96)00127-4. [DOI] [PubMed] [Google Scholar]