

Abstract
Integrin-linked kinase (ILK) is a serine/threonine protein kinase that plays an important role in integrin signaling and cell proliferation. We used Cre recombinase (Cre)-loxP technology to study CNS restricted knock-out of the ilk gene by either Nestin-driven or gfap-driven Cre-mediated recombination. Developmental changes in ilk-excised brain regions are similar to those observed in mice lacking the integrin β1 subunit in the CNS, including defective laminin deposition, abnormal glial morphology, and alterations in granule cell migration. Decreases in 6-bromodeoxyuridine (BrdU) pulse labeling and proliferating cell nuclear antigen expression in the external granule cell layer of the cerebellum demonstrated that proliferation is disrupted in granule cells lacking ILK. Previous studies have shown that laminin-sonic hedgehog (Shh)-induced granule cell precursor (GCP) proliferation is dependent on β1 integrins, several of which bind laminin and interact with ILK through the β1 cytoplasmic domain. Both ex vivo deletion of ilk and a small molecule inhibitor of ILK kinase activity decreased laminin-Shh-induced BrdU labeling in cultured GCPs. Together, these results implicate ILK as a critical effector in a signaling pathway necessary for granule cell proliferation and cerebellar development.
Keywords: granule cell precursor, integrin, integrin-linked kinase, laminin, proliferation, sonic hedgehog
Introduction
In the developing early postnatal cerebellum, β1 integrins are important in supporting the proliferation of granule cells, thereby promoting the postnatal expansion of the cerebellum. During development, granule cell precursors (GCPs) form the external granule layer (EGL), a postnatal proliferative zone that lies between the meningeal layer and the Purkinje cell layer (PCL) (Goldowitz and Hamre, 1998; Wang and Zoghbi, 2001). GCPs express laminin receptors within the β1 integrin family (α6β1 and α7β1), and proliferating cells are in close association with laminin isoforms contained within the basement membrane (Blaess et al., 2004). Within the EGL, laminin strongly enhances the mitogenic effects of sonic hedgehog (Shh), a potent inducer of GCP proliferation (Pons et al., 2001; Lewis et al., 2004). In vitro, β1 integrins appear to act cell autonomously to regulate laminin-Shh-induced GCP proliferation. Presently, the downstream effectors in this proliferative pathway remain essentially unknown.
Functions of integrins are mediated through the recruitment of cytoplasmic proteins that connect integrins to the actin cytoskeleton (Grashoff et al., 2004; Hannigan et al., 2005). Integrin-linked kinase (ILK) is one of these proteins, first discovered as a protein that interacts with the cytoplasmic tails of the β1- and β3-integrin subunits (Hannigan et al., 1996). ILK is a key effector of integrin function, regulating cell adhesion and anchorage-dependent growth (Grashoff et al., 2004; Hannigan et al., 2005). ILK functions as both an adaptor protein and serine/threonine kinase: the former important for the adhesive functions of integrins and the latter critical for cell cycle progression (Grashoff et al., 2004; Hannigan et al., 2005). Loss-of-function studies in Caenorhabditis elegans and Drosophila have revealed an important role for ILK as a scaffold protein, recruiting integrins and stabilizing actin at adhesion sites (Zervas et al., 2001; Mackinnon et al., 2002). In addition, genetic or pharmacological inhibition of ILK suppresses proliferation in vitro and in vivo (D'Amico et al., 2000; Persad et al., 2000; Grashoff et al., 2003; Terpstra et al., 2003; Tan et al., 2004). In the majority of cases, these effects can be explained by inhibition of ILK kinase activity (Grashoff et al., 2004; Hannigan et al., 2005). ILK phosphorylates glycogen synthase kinase 3β (GSK-3β) and protein kinase B (PKB)/AKT, two enzymes known to be involved in cell proliferation (Delcommenne et al., 1998; Persad et al., 2000, 2001a,b). These phosphorylation events ultimately induce the expression of cell-cycle-promoting genes such as cyclins and c-myc (Grashoff et al., 2004; Hannigan et al., 2005).
Defects in integrin function have profound effects on development and may underlie some of the genetic diseases that lead to cortical malformations (Georges-Labouesse et al., 1998; Miner et al., 1998; Walsh, 1999; Halfter et al., 2002; Moore et al., 2002; Beggs et al., 2003). Lack of integrin β1 gene expression in mice leads to peri-implantation lethality (Fassler and Meyer, 1995). In nervous system-restricted integrin β1 knock-out mice, aberrant formation of lamina and folia in the cerebral and cerebellar cortices have been observed (Graus-Porta et al., 2001). These developmental abnormalities are primarily attributable to altered glia and basement membrane interactions, which result in marked instability of the basal lamina. In the cerebellum, the absence of β1 integrins also results in reduced GCP proliferation (Blaess et al., 2004). Similar to the absence of β1 integrins, the absence of ILK expression in mice also leads to peri-implantation lethality (Sakai et al., 2003). Therefore, to investigate ILK function in the brain, we selectively knocked-out ilk using Cre recombinase (Cre)-loxP technology. In the cerebellum, we found that ILK deficiency caused phenotypic changes similar to those observed in mice lacking β1 integrins, including granule cell ectopia, defective laminin deposition, glial network changes, and reduced GCP proliferation.
Materials and Methods
Transgenic mouse strains
We used previously described mouse lines, namely, nestin-Cre (Tronche et al., 1999), glial fibrillary acidic protein (gfap)-Cre (Kwon et al., 2001), and ilkloxP (Terpstra et al., 2003). IlkloxP/loxP;gfap-Cre and ilkloxP/loxP; nestin-Cre mice were generated by crossing IlkloxP/loxP mice with IlkloxP/+; gfap-Cre and ilkloxP/+;nestin-Cre, respectively. IlkloxP/loxP;gfap-Cre and ilkloxP/loxP;nestin-Cre were compared with littermates. The animals examined were of mixed background. Specifically, the background of the nestin-Cre, gfap-Cre, and ilkloxP mice were C57BL/6/ICR, FVB/C57BL/6/129 and C57BL/6, respectively.
Cerebellar cultures and drug exposure
Cultures of cerebellar granule cells were prepared from postnatal day 4 (P4) to P5 mice and plated onto coverslips coated with poly-d-lysine (100 μg/ml) and laminin (5 μg/ml) as described previously (Cohen-Cory et al., 1991; Mills et al., 2003). Primary cerebellar neuronal cultures from ilkloxP/loxP or wild-type mice were infected with Cre recombinase-expressing adenovirus (AdCre) at 2 d in vitro as described previously (Troussard et al., 2003). Conditioned media was removed, and cells were incubated with virus in OptiMEM for 4 h at 37°C. The conditioned media was then added back, and cells were incubated for 4 d. For proliferation assays, Shh (1–3 μg/ml; R&D Systems, Minneapolis, MN) was added to Neurobasal media at 6 d in vitro, and cultures were incubated for 3 d. For experiments using the ILK kinase inhibitor KP-392 [formerly known as KP-SD-1 (Persad et al., 2001b)] Shh (1 μg/ml) was added to cultures at 6 d in vitro with or without KP-392 (50 or 100 μm) or vehicle control. Four hours before fixation, cultures were treated with 20 μm 6-bromodeoxyuridine (BrdU). Cells were fixed with methanol, treated with 2 m HCl for 1 h, neutralized in 0.1 m sodium borate buffer, pH 8.5, and washed with PBS. BrdU-labeled cells were visualized by a fluorescein-conjugated anti-BrdU antibody. The total number of BrdU-positive cells represents the number of cells that were counted in four randomly chosen fields of view per treatment group.
Western blotting
Lysates from primary cultures were prepared in Tris-Cl buffer, pH 7.6, containing 1% NP-40, 150 mm NaCl, 1 mm EDTA, 3.8 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm PMSF, 2 mm NaF, and 1 mm Na3VO4 as described previously (Mills et al., 2003). ILK was detected using a monoclonal anti-ILK antibody (BD Transduction Laboratories, Lexington, KY). All other antibodies are as described previously (Mills et al., 2003).
Histology
For BrdU labeling, mice were injected intraperitoneally with 100 μg of BrdU per gram of body weight and perfused 2 h later. Mice were perfused transcardially with 4% paraformaldehyde in PBS. Brains were either processed for paraffin sectioning or submerged in 30% sucrose, embedded, and sectioned using a cryostat. Ten micrometer sections were treated with 4N HCl for 10 min before the addition of primary antibody (see below). The presence of iron was detected in paraffin sections using Perl's Prussian blue method (Carleton, 1980). For Nissl staining, brains were submerged in 30% sucrose overnight at 4°C and sectioned directly at 40 μm using a frozen sliding microtome. Sections were then dehydrated overnight in 70% EtOH, stained with 0.1% cresyl violet/0.5% acetic acid, rinsed in 70% dH2O/95% ethanol and chloroform, and differentiated with 1.7% acetic acid in 95% EtOH. Frozen and paraffin-embedded sections were used for immunohistochemistry (see below).
Immunohistochemistry
Sections were blocked in 3% milk or 5% horse serum and solubilized in 0.2% Triton X-100 in PBS. We performed immunohistochemistry with primary antibodies to BrdU (Roche Diagnostics, Indianapolis, IN), proliferating cell nuclear antigen (PCNA) (Santa Cruz Biotechnology, Santa Cruz, CA), Englebreth Holm-Swarm laminin (Sigma, St. Louis, MO), monoclonal ILK (Santa Cruz Biotechnology), polyclonal ILK (Cell Signaling Technology, Beverly, MA), neuronal-specific nuclear protein (NeuN) (Chemicon, Temecula, CA), and GFAP (Chemicon or DakoCytomation, Carpenteria, CA). Sections were incubated in primary antibodies for 2 hat room temperature or overnight at 4°C. For immunofluorescence microscopy of nestin-Cre adult animals, we performed detection of primary antibodies with Alexa Fluor 488 or 594 secondary antibodies (Invitrogen, Carlsbad, CA). For all other immunofluorescence staining, unconjugated primary antibodies were detected with biotinylated secondary antibodies, followed by amplification with streptavidin–FITC or streptavidin–Texas Red. For costaining by immunofluorescence, ILK was visualized using a polyclonal ILK antibody, whereas BrdU and GFAP were visualized using conjugated monoclonal antibodies anti-BrdU–fluorescein (Roche Diagnostics), and anti-GFAP–cyanine 3 (Sigma), respectively. For light microscopy, we performed detection of primary antibodies with biotinylated secondary antibodies, followed by amplification with a streptavidin–peroxidase conjugate, treatment with Nova Red substrate, and hematoxylin counterstain (when stated).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling and DAPI staining. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) of cerebellar neuronal cells was measured using the In Situ Cell Death Detection kit (fluorescein; Boehringer Mannheim, Indianapolis, IN) according to the methods of Miller et al. (1997). The number of TUNEL-positive cells was calculated as a percentage of total cell number (DAPI-stained cells). TUNEL of cerebellar slices was measured with the same In Situ Cell Death Detection kit according to the instructions of the manufacturer.
Statistical analysis
Data depicted in the graphs represent the mean ± SEM of results. The minimum level of statistical significance was set at α = 0.05. Intergroup comparisons were made with a one-way ANOVA, followed by either Tukey's multiple comparison post hoc test or Scheffé's correction unless otherwise stated.
Results
Cre recombinase activity and ILK expression in brain
To generate mice with postnatal, CNS-restricted knock-out of the ILK gene, we crossed gfap-Cre mice or nestin-Cre mice with ilkloxP mice. ilkloxP/loxP;gfap-Cre mice were born at the expected Mendelian ratios and were indistinguishable from littermate controls. However, death increased at 21 weeks in ilkloxP/loxP;gfap-Cre mice (4%), increasing to 50% mortality at 30 weeks of age. Premature death did not occur in ILK+/+;gfap-Cre or ILK+/loxP; gfap-Cre littermate controls. Locomotor abilities revealed that step length, gait width, and alternation coefficient were similar to littermate controls at 13 weeks of age (data not shown). However, naive ilkloxP/loxP;gfap-Cre mice at 13 weeks of age exhibited impaired performance on the rotating rod, indicating that a preexisting motor performance deficit was present at this time. Likewise, ilkloxP/loxP;nestin-Cre mice were born at the expected Mendelian ratios. As with IlkloxP/loxP;gfap-Cre mice, increased death was not observed in young adult nestin-Cre mutant mice. Although the life expectancy, beyond early adulthood, was not formally studied, an increase in death was not observed in older nestin-Cre mice colonies housed for 24 weeks. Locomotor abilities were not studied in ilkloxP/loxP;nestin-Cre mice.
The pattern of Cre recombination in the nestin-Cre mouse line has been characterized previously by crossing nestin-Cre mice with ROSA R29R reporter mice in which Cre-mediated recombination is required for expression of the lacZ reporter gene. Nestin is an intermediate neurofilament expressed in neural precursors in the developing embryo (Lendahl et al., 1990). In the nestin-Cre mouse line, Cre-mediated recombination occurs in the precursors of neurons and glia in the CNS. Therefore, LacZ expression was evident in embryonic day 15.5 (E15.5) embryos throughout the cerebellar anlage (Blaess et al., 2004). At P7, all cells in the EGL, internal granule cell layer (IGL), and PCL expressed LacZ. A similar breeding strategy was used to characterize the pattern of Cre-mediated recombination in a gfap-Cre transgenic mouse model in which Cre recombination was under the control of a modified glial fibrillary acidic protein (GFAP) promoter (Kwon et al., 2001). Although traditionally considered a glial promoter, Cre recombination in neuronal cells has been reported for a number of gfap-Cre transgenic mice and is likely attributable to Cre being transiently expressed in neuronal precursors during development (Zhuo et al., 2001). In the cerebellum of these gfap-Cre reporter mice, Cre recombination had occurred in the majority of granule neurons in the EGL and IGL at P14 (Kwon et al., 2001). Cre-mediated recombination was variable in granule cells in the EGL at earlier developmental time points (at P5, Cre-mediated recombination had occurred in ∼20% of granule cells in some folia, whereas in other folia this number was less). Surprisingly, Cre activity in cells immunopositive for S100β (a glial marker) was only occasionally observed (Kwon et al., 2001).
We analyzed the expression of ILK in gfap-Cre and nestin-Cre mice immunohistochemically using two different ILK antibodies. At P10, ILK immunoreactivity was relatively strong in the EGL of gfap-Cre littermate control mice (Fig. 1a). Although a sizeable reduction in ILK immunoreactivity was observed in some folia of ilkloxP/loxP;gfap-Cre mice at P10, ILK excision was not complete at this developmental time (Fig. 1b). A more dramatic reduction in ILK expression was observed in adult ilkloxP/loxP;gfap-Cre mice (Fig. 1d,f) compared with littermate controls (Fig. 1c,e). Loss of ILK expression was evident in the granule cell layer (gcl; containing granule cell somas) and molecular layer (ml; containing granule cell axons) of the cerebellum (Fig. 1d,f). The reduction of ILK staining was consistent with a loss of ILK expression in both neuronal as well as glial cells (Fig. 1d,f). In contrast to gfap-Cre mutant mice, a dramatic loss of ILK immunoreactivity was observed in nestin-Cre mutant mice at P10. Loss of ILK staining was visible at low magnification throughout the cerebellum, consistent with a dramatic reduction of ILK expression in neuronal and glial cells (Fig. 1h).
Figure 1.
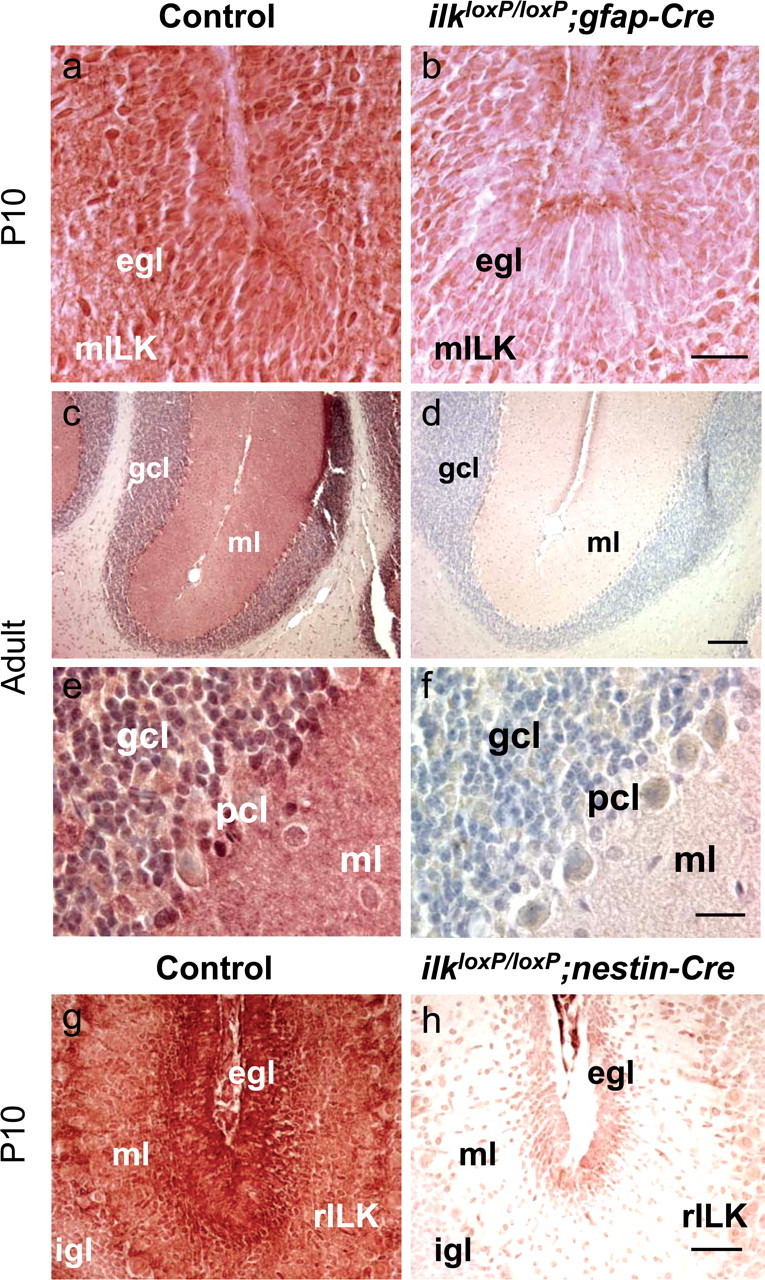
ILK expression in the mouse brain. a–f, Sagittal sections of the cerebellum in gfap-Cre mice. Sections were stained with a monoclonal antibody to ILK. ILK is expressed in the EGL of P10 mice. At this developmental time, incomplete loss of ILK immunoreactivity occurs in the EGL of ilkloxP/loxP; gfap-Cre mice compared with littermate controls (a, b). Loss of ILK immunoreactivity is apparent in the granule cell neurons of the adult mutant mouse cerebellum, as shown in low-magnification (d) and high-magnification (f) panels. Specifically, ILK immunoreactivity is decreased in the granule cell layer (gcl; containing granule cell somas) and molecular layer (ml; containing granule cell axons) of the cerebellum. g, h, Sagittal sections of the cerebellum in nestin-Cre mice. Sections were stained with a polyclonal antibody to ILK. ILK is strongly expressed throughout the EGL and molecular layer of the P10 littermate control mice (g). A dramatic reduction of ILK immunoreactivity is present in ilkloxP/loxP; nestin-Cre mice (h) compared with littermate controls (g). Scale bars: b, f, 20 μm; d, 1 mm; h, 100 μm.
We have previously characterized ILK expression in various tissues and found ILK to be relatively highly expressed in the adult mammalian brain in neuronal cells (Mills et al., 2003). To further characterize the cell type in which ILK is normally found (and thereby exerting its effect), we costained cerebellar sections at P10 with antibodies to ILK and GFAP. Costaining revealed a pattern of ILK immunoreactivity that only partially colocalized with GFAP-positive cells (Fig. 2a–c). ILK staining that was GFAP negative was consistent with GCP staining (Fig. 2b,c). Costaining of sagittal sections of P10 mice (that had been previously injected with BrdU) with antibodies to ILK and BrdU revealed ILK-positive cells within the proliferative zone of the EGL (Fig. 2d–f), suggesting that ILK is expressed in GCPs.
Figure 2.

ILK expression occurs in granule precursor cells and Bergmann glial cells of the EGL. Sagittal sections of the cerebellum of P10 mice were stained with antibodies to ILK (green) and GFAP (red) (a–c). Although ILK immunoreactivity (b) appears to colocalize with some GFAP-positive cells (c), a large number of ILK-positive cells are GFAP negative, appearing as a honeycomb staining pattern within the cerebellar folium (b). c, Inset, A high-magnification view of glia end feet is shown, illustrating costaining for both ILK and GFAP. ILK immunoreactivity colocalizes with the proliferation zone of the EGL (d–f). Sagittal sections of P10 wild-type mice (that were previously injected with BrdU) were costained with antibodies to ILK (red) and BrdU (green). ILK-immunoreactive cells (d) occurring near the basement membrane were also BrdU positive (e). f, Inset, A high-magnification view of cells within the proliferating zone (BrdU-positive cells) that costain for ILK. Scale bars, 50 μm.
Developmental changes in the mutant mouse brain
The cerebellar architecture of CNS-restricted ilk knock-out mice was markedly different between ilkloxP/loxP;gfap-Cre and ilkloxP/loxP;nestin-Cre. The cerebellar architecture of ilkloxP/loxP; nestin-Cre was similar to that seen for β1loxP/loxP;nestin-Cre mice (Graus-Porta et al., 2001; Blaess et al., 2004). At P10, nestin-Cre mutant mice exhibited an irregular IGL, fused folia, and granule cell ectopia (Fig. 3b,d,f) compared with littermate controls (Fig. 3a,c,e). Granule cell ectopias in nestin-Cre mutant mice occurred within folia and on the surface of the cerebellum. Fused folia are evident in higher-magnification figures (Fig. 3, compare control in c with mutant in d) of areas marked by asterisks (Fig. 3a,b). NeuN (a marker of postmitotic neuronal cells) staining revealed that ectopic granule cells were no longer proliferating (Fig. 3f). Similar cerebellar perturbations were observed in adult nestin-Cre mutant animals (Fig. 4b,d). In contrast, the cerebellar cortex of ilkloxP/loxP;gfap-Cre mice showed a regular foliation pattern at P10 (data not shown) and in the adult (Fig. 4f) compared with littermate controls (Fig. 4e).
Figure 3.
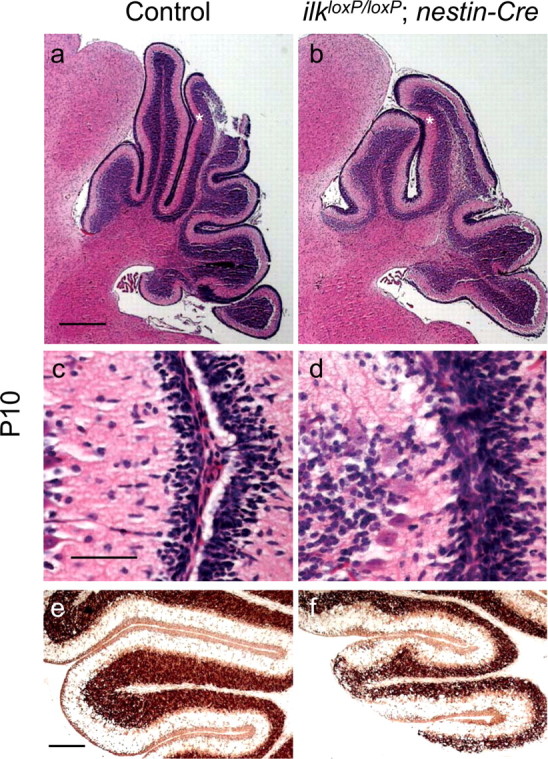
Cerebellar architecture of CNS-restricted ILK knock-out mice during postnatal development. a–f, Sagittal sections of control (a, c, e) or ilkloxP/loxP; nestin-Cre (b, d, f) P10 mice. a–d, Sagittal sections of P10 animals were stained for hematoxylin–eosin. The folia in the mutant mice lacked fissures, and adjacent EGLs were fused (b, d). Similar areas of littermate controls are provided for comparison (a, c). Areas of the folia labeled by asterisks in a and b are shown at higher magnification in c and d, respectively. e, f, Sagittal sections of P10 animals were stained for the postmitotic neuronal marker NeuN. Ectopic granule neurons staining positively for NeuN invade the molecular layer of nestin-Cre mutant mice, indicating that these cells are no longer dividing. Scalloping of the IGL of mutant mice was also observed at this developmental time. Scale bars: a, 500 μm; c, 50 μm; e, 200 μm.
Figure 4.
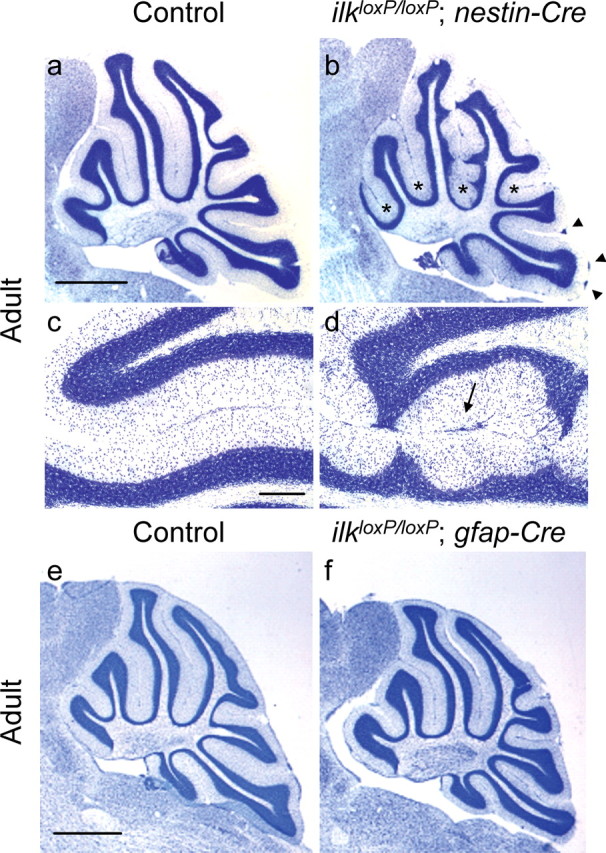
Cerebellar architecture of CNS-restricted ILK knock-out adult mice. a–d, Sagittal sections of control (a, c) or ilkloxP/loxP; nestin-Cre (b, d) adult mice stained for cresyl violet. As occurred in younger animals, scalloping of the IGL and granule cell ectopias (b, d) were observed in the cerebellum of adult nestin-Cre mutant mice compared with control (a, c). Asterisks label folia containing granule cell ectopia between folia and arrowheads label granule cell ectopia found on the cerebellar surface below the pial surface (b). d, A high-magnification view illustrating inappropriate positioning of granule cells within the molecular layer and ectopias between folia (arrow). e, f, Sagittal sections of control (e) and ilkloxP/loxP; gfap-Cre adult (f) mice stained for cresyl violet. In contrast to nestin-Cre mutant mice, a regular foliation pattern occurred in the cerebellum of these adult mice. Scale bars: a, e, 1 mm; c, 200 μm.
In nervous system-restricted β1 integrin knock-out mice, development defects in the basement membrane between the surface of the cerebellum and the pia–meninges results in abnormal foliation (Graus-Porta et al., 2001). In particular, laminin in mutant mice was severely reduced at the cerebellar surface and was almost absent in the folia. Similarly, altered laminin staining was observed in both CNS-restricted ilk knock-out mice lines, indicative of defects in basement membrane assembly (Figs. 5, 6). A marked reduction of laminin was observed at the cerebellar surface [compare laminin staining of gfap-Cre mutant mice with littermate controls (Fig. 5, f with e and h with g, respectively)] and within the folium [compare laminin staining of gfap-Cre mutant mice with littermate controls (Fig. 5, c with a and d with b, respectively)] of 10-d-old ilkloxP/loxP;gfap-Cre mice. Laminin deficits were more pronounced in nestin-Cre mutant mice than gfap-Cre mutant mice. In littermate controls of P10 nestin-Cre mice, laminin staining was continuous along the cerebellar surface, penetrating deep into the cerebellar folia (Fig. 6a,c). In P10 nestin-Cre mutant mice, laminin was either fragmented (Fig. 6d) or entirely absent from the cerebellar surface (Fig. 6b, see area within arrows) and did not always penetrate into the cerebellar folia (Fig. 6b, see folia with arrowhead). Even in areas of the cerebellum in which laminin was present, assembly may have been altered because staining appeared thicker and disorganized (Fig. 6d). In the adult, laminin deficits were observed in the meningeal basement membrane of both ilkloxP/loxP;nestin-Cre (Fig. 6e–h) and ilkloxP/loxP;gfap-Cre mice (data not shown). Laminin staining was either markedly reduced or absent from the cerebellar surface (Fig. 6, compare h with g, a littermate control) of ilkloxP/loxP;nestin-Cre mice and from between the folia (Fig. 6, compare f with e, a littermate control), especially when there was fusion of folia. Loss of basal lamina matrix integrity has been reported to result in microvascular permeability and erythrocyte leakage. Microvascular abnormalities were also observed in CNS-restricted ILK knock-out mice (Figs. 5i,j, 6i,j). Laminin staining appeared fragmented in large blood vessels within the molecular layer of ilkloxP/loxP;nestin-Cre mice (Fig. 6j). In P15 ilkloxP/loxP;gfap-Cre mice, remote (Fig. 5j, black arrow) and recent hemorrhage (Fig. 5j, white arrow) in the form of focal parenchymal and perivascular collections of erythrocytes, serum proteins, and blood breakdown products were observed. Distinct clusters of cells resembling erythrocytes appeared within the granule cell layer and molecular layer (data not shown). Positive iron staining associated with these cells in ilkloxP/loxP;gfap-Cre mutant mice was also evident (Fig. 5j) but was absent from littermate controls (Fig. 5i).
Figure 5.
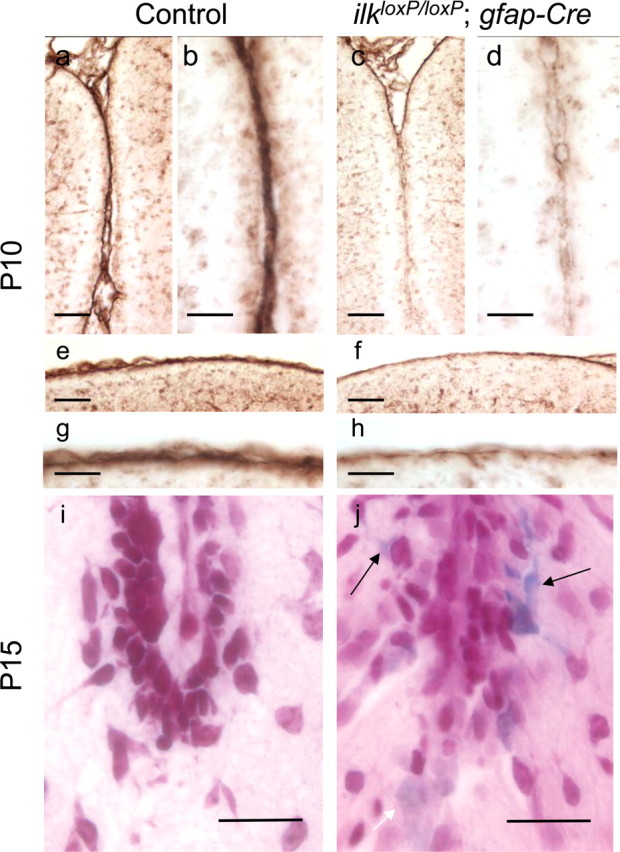
Altered laminin staining in the cerebellum of ilkloxP/loxP; gfap-Cre mutant mice. a–h, Laminin staining in control (a, b, e, g) and ilkloxP/loxP; gfap-Cre (c, d, f, h) mice at P10. Laminin staining deficits were evident between the folia (c, d) and at the cerebellar surface (f, h) compared with wild-type littermate controls. A low-magnification (a, c) and high-magnification (b, d) view of laminin staining indicate that laminin was incorporated into the folium in the cerebellum of the wild-type mouse (a, b) but was markedly reduced in the mutant mouse (c, d). Similar changes were observed at the cerebellar surface (e–h). A reduction of laminin staining at the cerebellar surface in gfap-Cre mutant mice is evident at both low (f) and high (h) magnification compared with littermate controls (e, g). i, j, Altered microvasculature in 15-d-old ilkloxP/loxP; gfap-Cre mice. The cerebellum of mutant mice showed evidence of remote and recent hemorrhage in the form of focal parenchymal and perivascular collections of erythrocytes, serum proteins, and blood breakdown products. Faint (white arrow) and intense (black arrows) iron labeling associated with cells resembling erythrocytes (j) indicate that microhemorrhages were present within the cerebellum of mutant mice but were absent from littermate controls (i). Scale bars: a, c, e, f, 50 μm; b, d, g, h, i, j, 20 μm.
Figure 6.
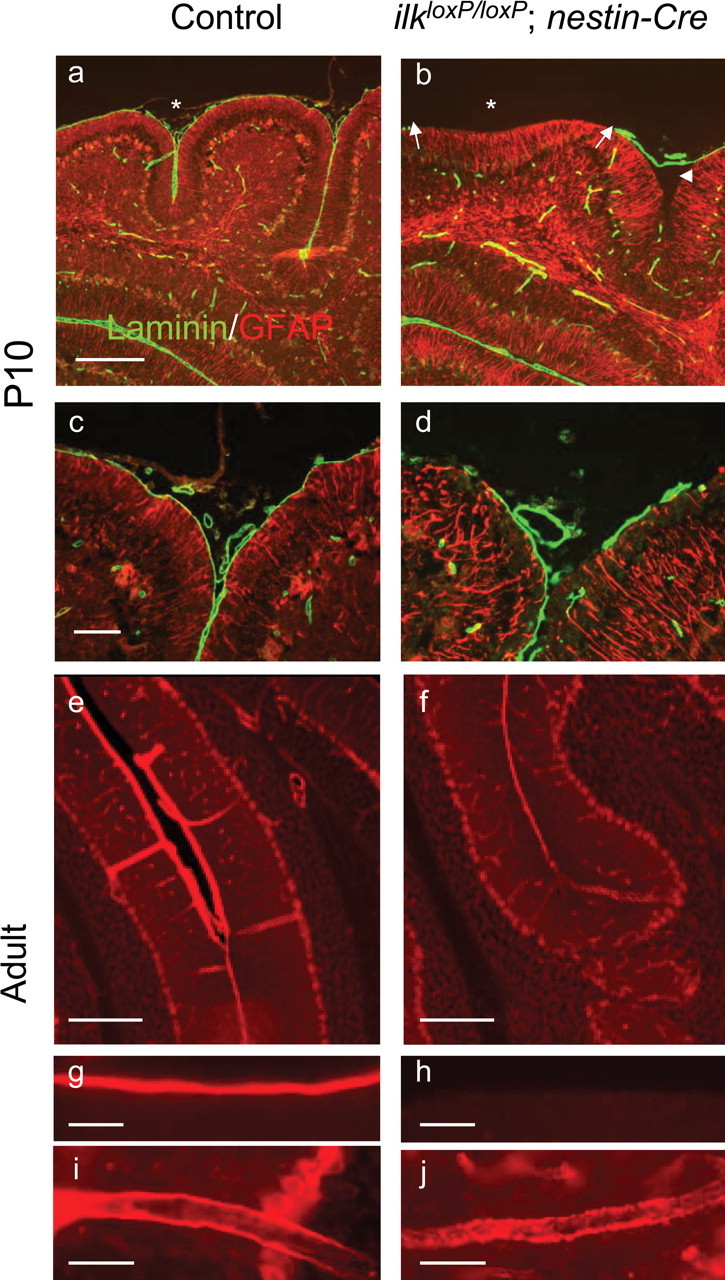
Altered laminin staining in the cerebellum of ilkloxP/loxP; nestin-Cre mutant mice. a–d, Laminin staining in control (a, c) and ilkloxP/loxP; nestin-Cre (b, d) P10 mice. Low (a, b) and high (c, d) magnification of two different cerebellar folia costained for laminin (green) and GFAP (red). In nestin-Cre mutant mice, laminin staining was absent from regions of the cerebellar surface (b, d), appearing either entirely absent (b, arrowhead) or fragmented (d). Loss of laminin correlated with the loss of fissures (b, asterisk) or folia depth (b, arrowhead). e–j, Laminin staining in control (e, g, i) and adult ilkloxP/loxP; nestin-Cre (f, h, j) mice. Laminin staining was markedly reduced between fused folia of mutant mice (f) compared with littermate controls (e). Laminin appeared stripped from the cerebellar surface of mutant mice (h) compared with littermate controls (g). Laminin staining appeared fragmented (note punctuate staining) in the large blood vessel walls within the molecular layer of the mutant mice (j) compared with littermate controls (i). Scale bars: a, e, f, 200 μm; c, g–j, 50 μm.
Abnormal glial fibers resulting from altered glia–basement membrane interactions have been observed in nervous system-specific integrin β1-deficient mice (Graus-Porta et al., 2001). To investigate the integrity of the glial network in ILK knock-out mice, glial fibers were stained for GFAP. In wild-type mice, glial processes formed a regular network, extending across the molecular layer (Fig. 7a,c). Processes terminated with end feet at the basement membrane, forming a continuous layer (Fig. 7a,c). In ilkloxP/loxP;gfap-Cre 15-d-old mice, glial processes extended across the molecular layer, but end-feet formation was disrupted at the cerebellar surface and between the folia (Fig. 7b,d). Glial network defects were much more pronounced in nestin-Cre mutant mice at P10 and occurred throughout the cerebellum, even in areas in which the basal lamina appeared intact. Glial processes in ilkloxP/loxP;nestin-Cre mice were highly irregular, meandering within the molecular layer, and lacked end feet (Fig. 7, compare GFAP staining in nestin-Cre mutant mice in f with littermate controls in e). nestin-Cre mice at P10 were pulse labeled with BrdU and costained for GFAP (red) and BrdU (green). Compared with the controls, BrdU immunoreactivity indicate that a proliferative zone in the EGL was still present but reduced within the folia, despite severe glial network abnormalities (see g and h depicting the same folia, except with both BrdU and GFAP costaining). In adult nestin-Cre mutant mice, glial processes also lacked end feet at the cerebellar outer surface (Fig. 7j) and between the folia (data not shown).
Figure 7.
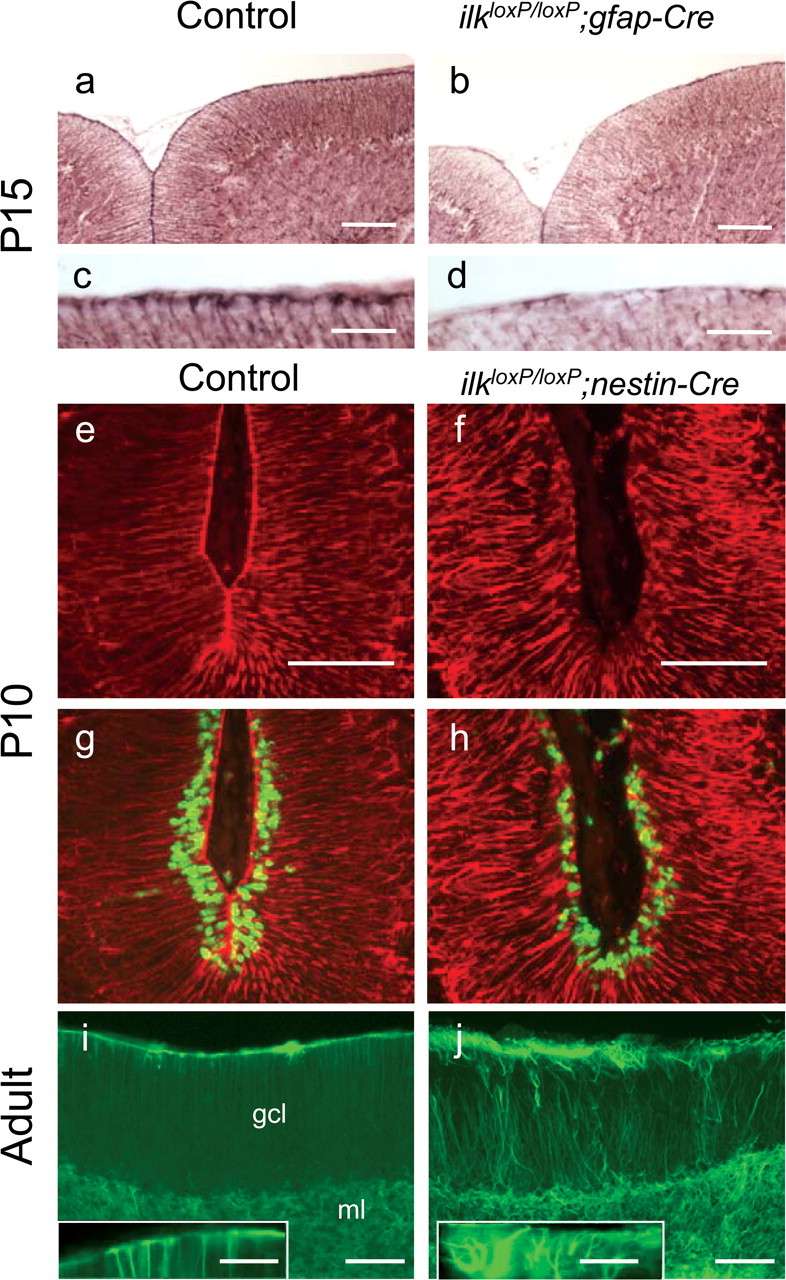
Defects in the glial network of CNS restricted ilk knock-out mice. a–d, GFAP staining in control (a, c) or ilkloxP/loxP;gfap-Cre (b, d) mice at P15. A reduction in the number of glial fibers having end feet was observed in mutant mice at the cerebellar outer surface (b, d) compared with littermate controls (a, c). e–h, nestin-Cre mice were pulsed with BrdU 10 d after birth, and sagittal sections of the cerebellum were costained for GFAP (red) and BrdU (green). GFAP staining indicates that the glial fiber network is severely disrupted in ilkloxP/loxP; nestin-Cre mouse (f). Glial fibers are seen to meander, and end feet are absent from the basement membrane. BrdU immunoreactivity indicates that a proliferative zone in the EGL (albeit reduced) was still present within the folia despite severe glial network abnormalities (g, h, depicting the same folia except with both BrdU and GFAP costaining). Glial fibers in the adult cerebellum of control (i) and ilkloxP/loxP; nestin-Cre mutant (j) mice were stained for GFAP (green). In wild-type mice (i), glial fibers formed a regular network and were anchored at the basement membrane with end feet (i, inset, a high magnification of the wild-type mouse cerebellar surface). As occurred postnatally, in ilkloxP/loxP; nestin-Cre adult mice (j), directional abnormalities of glial fibers were observed. Processes projected in abnormal directions through the molecular layer and lacked end feet at the cerebellar outer surface (j, inset, a high-magnification view of the mutant cerebellar surface). Scale bars: a, b, e, f, i, j, 100 μm; c, d, 30 μm; inset, 50 μm.
ILK deletion causes abnormalities in proliferation
In integrin β1 CNS-restricted knock-out mice, defects in postnatal proliferation of GCPs were observed. BrdU pulse labeling revealed that, at P0, the EGL contained normal numbers of granule cells (indicating that generation of the initial GCP pool was normal in these mutant mice). However, a proliferative defect in the EGL by P2 was observed, particularly in developing folia (Blaess et al., 2004). In addition, survival of cells within the EGL was not different from that in controls (Blaess et al., 2004). Because ILK is a downstream effector of β1, we wanted to determine whether loss of ILK expression also inhibits the postnatal proliferation of GCPs. To examine this possibility, we analyzed BrdU pulse labeling and the expression of PCNA in ilkloxP/loxP;gfap-Cre mice using immunohistochemical techniques. The number of progenitor cells expressing PCNA in the outer EGL at P14 was decreased in ilkloxP/loxP;gfap-Cre mice compared with littermate controls (Fig. 8, compare gfap-Cre mutant mice in b and d with littermate controls a and c). Similarly, BrdU pulse labeling demonstrated a proliferative defect in GCPs in the EGL of gfap-Cre mutant mice at P14. Compare representative folia 1 (F1) and folia 2 (F2) in mutant mice (Fig. 8f, h) with littermate controls (Fig. 8e,g). To determine whether or not the survival of the GCP pool was normal at P14–P15 in gfap-Cre mutant mice, TUNEL was performed and did not appear different in mutants (Fig. 8p) from littermate controls (Fig. 8o). Together, these results indicate that, in postnatal ilkloxP/loxP;gfap-Cre mice, the GCP pool failed to expand because of a proliferative defect rather than a survival defect.
Figure 8.
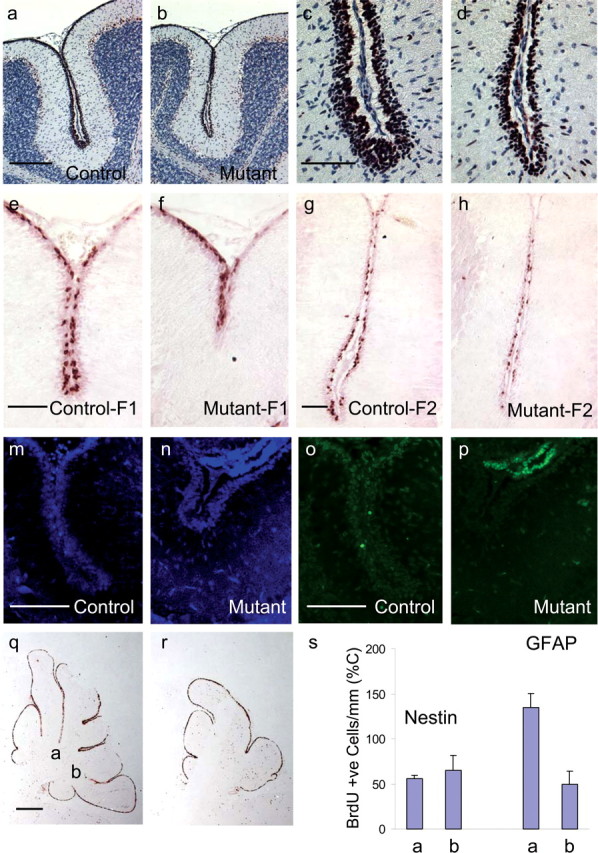
Granule cell precursor proliferation is impaired in the ilkloxP/loxP; gfap-Cre and ilkloxP/loxP; nestin-Cre mouse brain. a–d, Immunodetection of PCNA is shown in sagittal cerebellar sections of P14 mice. Anti-PCNA antibody was detected by a peroxidase-labeled secondary antibody in combination with Nova Red substrate (reddish brown). Sections have been counterstained with hematoxylin (blue). Proliferative defects occur in the EGL of the folia of mutant mice (b) compared with littermate controls (a). A higher-magnification view of these same folia are represented in c and d. e–h, Representative BrdU labeling at P14 from two different folia labeled F1 and F2 in mutant mice (f, h) and their littermate controls (e, g) are provided. BrdU labeling revealed strong proliferative defects within the cerebellar folia of mutant mice (f, h) compared with littermate controls (e, g). m–p, TUNEL of the EGL of gfap-Cre mice at P14 (o, p) and corresponding areas stained for DAPI (m, n). TUNEL of ilkloxP/loxP; gfap-Cre mice at P14 (p) did not appear different from control (o). q, r, Sagittal sections of nestin-Cre mutant mice (r) and littermate controls (q) at 10 d postnatal were pulsed with BrdU 2 h before the animals were killed and later stained for BrdU (reddish brown). A significant reduction in the number of proliferating cells in the EGL was observed, partly attributable to the loss of folia depth (r). s, gfap-Cre and nestin-Cre mice were injected with BrdU at 10 d postnatal and were killed 2 h later. The number of proliferating GCPs were measured in sagittal cerebellar sections by counting the number of cells having incorporated BrdU in the folia indicated (q). Two or more sections were counted per animal (2–3 wild-type and mutant mice). The number of BrdU-labeled cells in ilkloxP/loxP; gfap-Cre mice (GFAP) and ilkloxP/loxP; nestin-Cre mice (Nestin) along the cerebellar surface adjacent to the primary and secondary fissures are quantified per millimeter of cerebellar surface. This value is expressed as a percentage of the number of BrdU-labeled cells per millimeter cerebellar surface in the littermate control animals. Scale bars: a, 200 μm; c, e, g, m, o, 50 μm; q, 500 μm.
Compared with controls (Fig. 8q), loss of BrdU pulse labeling in ilkloxP/loxP;nestin-Cre mice at P10 (Fig. 8r) was much more profound than that observed for ilkloxP/loxP;gfap-Cre mice. This was in part attributable to the loss of folia. Although some fissures were often absent in nestin-Cre mutant mice, the primary and secondary fissures were consistently present. Therefore, GCP proliferation was measured along the cerebellar surface adjacent to these fissures by counting the number of cells having incorporated BrdU. Because depths of folia differ between Cre mutant animals and littermate controls, the length of the cerebellar surface along each fissure was measured. Proliferation was then quantified per millimeter of cerebellar surface, and this was expressed as a percentage of littermate control animals (Fig. 8s). BrdU pulse labeling in ilkloxP/loxP;nestin-Cre mice indicate that proliferation was <65% (of that in littermate controls) within the cerebellar surface adjacent to the secondary fissure of both nestin-Cre and gfap-Cre mutant animals. A similar reduction in BrdU labeling was seen within the cerebellar surface adjacent to the primary fissure of nestin-Cre mutant mice. In contrast, in gfap-Cre mutant mice, proliferation was not reduced (Fig. 8s). The length of the cerebellar surface adjacent to the primary and secondary fissures (expressed as a percentage of control) for nestin-Cre mutant animals was 63.9 ± 3.4 and 75.2 ± 6.8%, respectively, whereas the length of the cerebellar surface adjacent to the primary fissure and secondary fissures for gfap-Cre mutant animals was 59.3 ± 2.5 and 84.8 ± 2.1%, respectively.
Serial sections comparing hematoxylin–eosin staining (Fig. 9b) with BrdU staining (Fig. 9d) revealed that, in nestin-Cre mice at P10, both the width of the external granule cell layer and BrdU labeling were reduced in areas of the cerebellum lacking fissures (Fig. 9, compare regions marked by white arrows in b and d with comparable regions in control animals marked in a and c). In P10 nestin-Cre littermate control mice, laminin staining is continuous and intimately associated with the proliferative zone in the EGL (Fig. 9c,e). Although in nestin-Cre mutant mice at P10, dual staining for BrdU and laminin revealed that, even in areas in which laminin is present, it was dissociated from proliferating GCPs (Fig. 9d,f). Moreover, areas of the cerebellum in which laminin is dissociated from the proliferative zone corresponded to areas of reduced BrdU labeling and fissure depth (Fig. 9c–f). Together, these findings suggest that proliferative defects were associated with areas of the cerebellum in which the basement membrane was disrupted.
Figure 9.
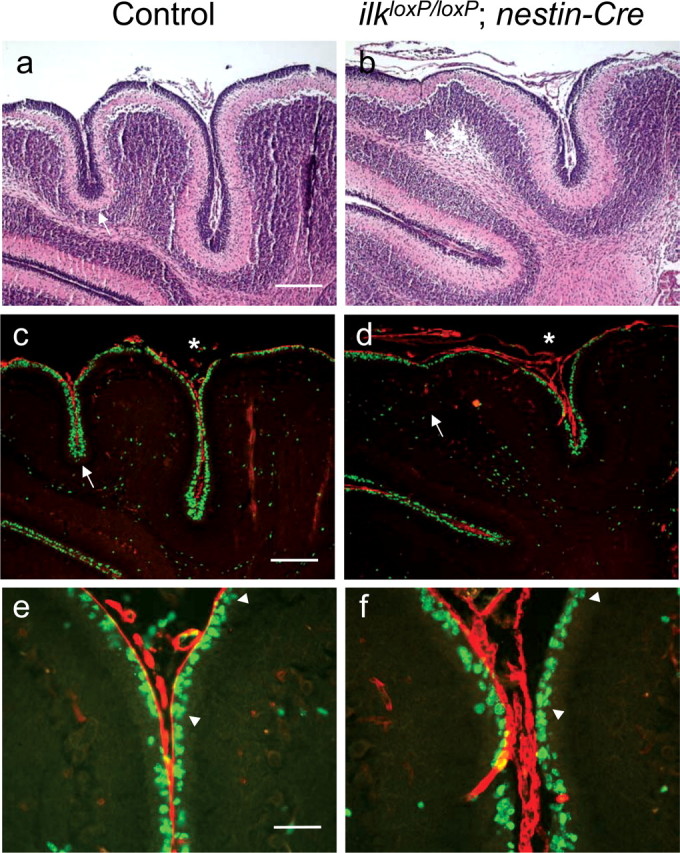
Dual staining for BrdU and laminin in nestin-Cre postnatal mice. Costaining for BrdU (green) and laminin (red) in nestin-Cre mice at 10 d postnatal (c, d) and corresponding area stained for hematoxylin–eosin to reveal general cytoarchitecture (a, b). A reduction in the thickness of the external granule cell layer is observed in areas of the cerebellum lacking fissures (white arrows, a–d). Dual staining for BrdU and laminin in an adjacent section revealed that laminin staining is intimately associated with the proliferating zone in the EGL of wild-type animals, whereas laminin is dissociated from BrdU-positive cells in areas of the nestin-Cre mutant mouse cerebellum. Areas of the cerebellum in which laminin is dissociated from the proliferative zone correspond to areas having reduced BrdU labeling and folia depth (white asterisk, d). e, f, High-magnification view of fissures labeled by asterisks (c, d), indicating that, although laminin staining appears continuous, it no longer resides flush against proliferating GCPs in nestin-Cre mutant mice: note the area flanked by arrowheads (f) compared with littermate control (e). Scale bars: a, c, 200 μm; e, 50 μm.
Shh-induced granule cell proliferation requires ILK
Studies using mice having a CNS-restricted knock-out of the integrin β1 subunit gene indicate that β1 integrin expression is required to recruit the laminin-Shh complex to the surface of GCP and subsequently promote proliferation (Blaess et al., 2004). Although these results are consistent with a model in which β1 integrins and Shh cooperate together to regulate the activity of second-messenger systems, the downstream effectors in this pathway have not been studied. To determine whether ILK is involved in integrin-stimulated GCP proliferation, ILK was excised ex vivo using an AdCre virus. Western blot analyses indicate that, 4 d after infection of ilkloxP/loxP cerebellar cultures, ILK expression was markedly reduced compared with control cultures (Fig. 10b). GCPs derived from wild-type or ilkloxP/loxP mice were plated on laminin and infected with AdCre virus. After AdCre infection, the number of BrdU-incorporating cells after a 3 d exposure to Shh (1 and 3 μg/ml) was significantly reduced in ilkloxP/loxP cultures compared with ILK wild-type cultures (Fig. 10a). Granule cell survival after AdCre infection was determined by the percentage of TUNEL-negative cells. Survival of AdCre-infected cells was not different from mock-infected sister cultures, indicating that viral infection did not reduce proliferation by inhibiting survival (Fig. 10c). Proliferation was also quantitated in GCP cultures plated onto laminin and exposed to Shh (1 μg/ml) in the presence or absence of KP-392, a small molecule inhibitor of ILK kinase activity. In a previous study, we have shown that, although KP-392 (at concentrations of 50 and 100 μm) inhibited phosphorylation of the ILK substrates AKT and GSK-3, survival of neuronal cell lines in the presence of trophic support was not compromised (Mills et al., 2003). Similar results were found in a subsequent study using cultured primary neurons (Zhou et al., 2004). The number of BrdU-incorporating granule cells after exposure to Shh was markedly decreased by KP-392 (50 and 100 μm) compared with numbers in cultures treated with vehicle control (Fig. 10d).
Figure 10.

Ex vivo excision of ilk and ILK inhibition decreased granule cell precursor proliferation. a, Cerebellar granule neurons from ilk loxP/loxP or WT mice were plated on poly-d-lysine–laminin and maintained in Neurobasal media for 2 d. At that time, cells were infected with AdCre virus. Four days after infection, media was changed, and cultures were maintained in the presence or absence of Shh (1 and 3 μg/ml) for 3 d. Before fixation, cells were pulsed with BrdU for 4 h and labeled with a fluorescein-conjugated anti-BrdU antibody. Quantitation of BrdU-positive wild-type or floxed cells was performed. Data represent the average ± SEM of three to seven separate culture platings. b, Representative Western blot of cell lysates from GCP cultures generated from ilkloxP/loxP mice, infected with an AdCre virus or mock control, and probed with a monoclonal antibody to ILK. c, Granule cell survival was measured in ilkloxP/loxP cultures by the number of TUNEL-negative cells in culture. TUNEL-negative cells were expressed as the percentage of total cell number (DAPI-positive cells). Survival of AdCre-infected cultures was not different from mock-infected sister cultures treated with the same concentration of Shh. Comparisons were made using a paired t test (data are mean±SEM and represent 3 experiments; α = 0.05). d, At 6 d in vitro, 1 μg/ml Shh was added to GCP cultures with or without the ILK inhibitor KP-392 at a concentration of 50 or 100 μm (50 KP and 100 KP, respectively). C represents vehicle control.
Discussion
We have selectively excised the ilk gene from the CNS by Cre-loxP technology and have shown abnormal brain development and early lethality in mutant mice. Some developmental changes in ilk-excised brain regions are reminiscent of integrin β1 CNS knock-out mice, including granule cell ectopia, defective laminin deposition, and glial network changes (Graus-Porta et al., 2001). A follow-up study on integrin β1 CNS knock-out mice indicated that Shh induced GCP proliferation is severely reduced (Blaess et al., 2004). In the present paper, we show that ILK is also required for normal GCP proliferation. In vivo, deletion of ilk from the cerebellar anlage decreased laminin deposition and proliferation of granule cells in the EGL. Ex vivo deletion of ilk from purified cerebellar granule cells decreased laminin-Shh-induced BrdU labeling as did a small molecule inhibitor of ILK kinase activity. Overall, our data demonstrate that ILK is a critical effector in a signaling pathway necessary for granule cell proliferation and cerebellar development.
In the developing CNS, the initial deposition of extracellular matrix components into the basement membrane is dependent on meningeal cells, whereas subsequent remodeling is dependent on glial cells. Specifically, meningeal fibroblasts secrete components of the basal lamina that bind receptors on glial end feet of the glia limitans, allowing assembly of a functional basal lamina (Sievers et al., 1994; Shearer and Fawcett, 2001; Beggs et al., 2003; Blaess et al., 2004). Results from β1loxP/loxP;nestin-Cre knock-out mice support the idea that interactions between β1-class integrins in glial cells with the ECM are important for proper glial network development and maintenance of the basement membrane (Graus-Porta et al., 2001; Blaess et al., 2004). If β1 and ILK cooperate to regulate glia cell process outgrowth and differentiation, glial network defects in ilk mutant mice would in turn lead to abnormalities in basement membrane assembly. Glial cells may be the primary cell type responsible for the basal lamina defects seen in CNS-restricted ILK knock-out mice because excision of ILK from postmitotic neurons did not result in lamination defects (Niewmierzycka et al., 2005). Indeed, glial network defects in gfap-Cre mutant mice were much less dramatic than those observed in nestin-Cre mutant mice. These glial network changes correlated with the severity of laminin loss. Together, our data support the hypothesis that ILK is an important effector in glia development, mediating integrin-dependent assembly of the meningeal basement membrane.
Proliferative defects observed in β1loxP/loxP;nestin-Cre mice are essentially recapitulated in ilk mutant models. In β1loxP/loxP;nestin-Cre mice, defects in the external basal lamina correlated with both changes in proliferation of GCPs and an overall reduction in size of the cerebral and cerebellar cortices (Blaess et al., 2004). Similarly, in both ILK mutant mice models, decreased BrdU labeling in the proliferative zone of the EGL correlated with defects in the basal lamina. Shh is a powerful mitogen of postnatal GCP proliferation in the cerebellum (Dahmane and Ruiz-i-Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999). A previous study has shown that Shh-induced granule cell proliferation is potentiated by the integrin ligand laminin (Blaess et al., 2004). In cerebellar granule cells cultured from β1loxP/loxP;nestin-Cre mice and plated onto laminin, Shh-induced proliferation was significantly reduced (Blaess et al., 2004). Similarly, both ex vivo deletion of ilk and a small molecule inhibitor of ILK kinase activity decreased laminin-Shh-induced BrdU labeling in cultured GCPs. Therefore, these data support the hypothesis that ILK is an important effector in a β1-integrin-dependent signaling pathway that regulates proliferation. Although ILK may also mediate integrin-dependent assembly of the meningeal basement membrane, thereby affecting GCP proliferation indirectly, ILK also likely regulates GCP proliferation cell autonomously. Evidence for the direct role of ILK on GCP proliferation is provided by our in vitro studies because inhibition of ILK decreased GCP proliferation in relatively pure cultures of granule cells in the presence of laminin.
Although both ilk mutant mouse models exhibit defects in proliferation and the basal lamina, clear differences exist between the two mouse models. The ilkloxP/loxP;nestin-Cre mice were most similar to β1loxP/loxP;nestin-Cre mice, exhibiting gross changes in cerebellar architecture (fused and irregular folia) and extreme disorganization of the superficial glial limitans (changes that were not observed in ilkloxP/loxP;gfap-Cre mutant mice). These differences are likely attributable to the pattern of Cre-mediated ilk excision. For example, in nestin-Cre mice, it has been reported that Cre recombinase activity is present in E15.5 embryos throughout the cerebellar anlage. In contrast to nestin-Cre mice, Cre activity in gfap-Cre transgenic mice has been reported to occur postnatally in granule neurons of the cerebellum: only by P14 had Cre recombination occurred in the majority of granule cells in the EGL and IGL (Kwon et al., 2001). Although both nestin-Cre and gfap-Cre mouse lines are suitable in vivo models to study a role for ILK in neuronal cell precursor proliferation, each come with limitations. For example, in the nestin-Cre mouse model, migration defects and resulting ectopias confound the proliferative defect. Specifically, it is difficult to discern whether or not a proliferation or migration defect underlies ectopic granule cells that are no longer proliferating (but rather express NeuN, a marker associated with differentiated neurons). Furthermore, early excision of ilk might cause secondary defects that exacerbate cerebellar changes. For example, the fused folia that were observed in nestin-Cre mutant mice would prevent meningeal cell penetration into fissures. This would likely contribute, in part, to cerebellar foliation defects because meningeal cells have been shown to be important in neuronal cell migration and radial glial cell differentiation (Hartmann et al., 1998). Conversely, although changes in cerebellar architecture were not observed in gfap-Cre mutants, Cre recombinase activity occurs in the majority of granule cells in the EGL at P14, a developmental time near the end of the EGL proliferative period. Indeed, at P10, ILK immunoreactivity was observed in some cells of the EGL, and pulse labeling revealed that proliferative defects occurred in only some of the folia (likely reflecting incomplete and somewhat variable ILK excision at this developmental time point). Furthermore, folia depth was reduced in both Cre mutants (which might lead to a decrease in BrdU pulse labeling because of a decrease in the number of precursor cells). Therefore, BrdU pulse labeling was expressed per millimeter of cerebellar surface in both Cre mutant models. Controlling for the decrease in cerebellar surface, proliferative defects were observed in both CNS-restricted ilk knock-out models, providing evidence that ILK is an important regulator of neuronal precursor proliferation. Indeed, in vitro results indicate that Shh-induced proliferation in GCP cells is reduced in the absence of ILK (in which the initial number of precursor cells is the same), further supporting a role for ILK in neuronal precursor proliferation.
ILK phosphorylates GSK-3β and PKB/AKT, two enzymes known to be involved in cell proliferation (Delcommenne et al., 1998; Persad et al., 2000, 2001a,b). Phosphorylation of these targets had been shown to occur in vivo (Edwards et al., 2005; Younes et al., 2005) and in vitro (Delcommenne et al., 1998; Persad et al., 2000, 2001a,b). In a recent publication characterizing the role of ILK in the dorsal forebrain, no difference in phosphorylation levels of GSK-3β or AKT was observed in E14.5 dorsal forebrain extracts of Emx1-Cre; ILKfl/fl mutants (Niewmierzycka et al., 2005). ILK was not deleted in the cerebellum in this study. To determine whether GSK-3β phosphorylation was important in GCP proliferation in the cerebellum, we stained sagittal sections of wild-type mice using a phosphospecific antibody to GSK-3β. We could not detect phosphospecific GSK-3β immunoreactivity in the EGL, although the molecular layer stained modestly for this phosphorylated protein (J.M., unpublished observations). Nevertheless, a role for ILK kinase activity in GCP proliferation is suggested by the fact that the ILK kinase inhibitor KP-392 reduced Shh-induced proliferation in vitro. Previously, ILK has been shown to act downstream of phosphatidylinositol 3 (PI3)-kinase in mouse DRG neurons to regulate localized GSK-3β phosphorylation and axon elongation (Mills et al., 2003; Zhou et al., 2004). Therefore, assessment of ILK kinase activity by this substrate may be more relevant in other in vivo neuronal mouse models.
The precise mechanism of ILK-mediated regulation of GCP proliferation remains unknown. At least two possibilities exist, involving its signaling or scaffolding functions. If Shh-β1 complexes bind to laminin, then ILK, by binding to the integrin β1-cytoplasmic tail, may link the Shh-β1 complex to the actin cytoskeleton, thereby regulating the cytoskeletal changes required for proliferation. However, the kinase activity of ILK also plays an important role in integrin-mediated cell–matrix interactions and cell proliferation. In vitro, constitutive ILK activation or overexpression has been shown to stimulate cyclin D1 expression (Radeva et al., 1997; D'Amico et al., 2000; Persad et al., 2001a). Proliferative defects were observed in mice having ILK-null growth-plate chondrocytes, whereas ex vivo excision of ILK resulted in reduced cyclin D1 expression (Grashoff et al., 2003; Terpstra et al., 2003). In GCPs, Shh-induced proliferation and regulation of D-type cyclin expression occurs via N-myc (Kenney et al., 2003, 2004). Moreover, Shh and PI3-kinase signaling pathways converge on N-myc to regulate neuronal cell-cycle progression (Kenney et al., 2003, 2004). Therefore, ILK may be a key effector in a PI3-kinase-dependent proliferative pathway in neuronal precursor cells. Of course, ILK scaffolding function and kinase activity are not entirely distinct because the kinase and adapter properties of ILK appear to function together in a PI3-kinase-dependent manner, regulating integrin-mediated cell attachment and signal transduction (Attwell et al., 2003). Regardless of the precise mechanism, the fact that ILK regulates proliferation in a variety of tissues and is an important effector in Shh signaling suggests that ILK plays a fundamental role in regulating the proliferative capacity of precursor cells throughout the body.
Footnotes
This work was supported by Canadian Institutes of Health Research and National Cancer Institute of Canada. We thank Dr. Suzy Baker for generously providing the gfap-cre mice. We thank Dr. Jane Roskams and members of her laboratory for helpful discussions and Dr. Jeremy Mills for his help with the statistical analysis. We also thank Howard Tearle for his invaluable technical assistance.
Correspondence should be addressed to Shoukat Dedhar, British Columbia Research Centre, 675 West 10th Avenue, Vancouver, British Columbia, Canada V5Z 1L3. E-mail: sdedhar@interchange.ubc.ca.
B. Rico's present address: Institute of Neuroscience, University Miguel Hernandez–Consejo Superior de Investigaciones Científicas, 03550 San Juan, Alicante, Spain.
DOI:10.1523/JNEUROSCI.1852-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260830-11$15.00/0
References
- Attwell S, Mills J, Troussard A, Wu C, Dedhar S (2003) Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN. Mol Biol Cell 14: 4813–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40: 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S, Graus-Porta D, Belvindrah R, Radakovits R, Pons S, Littlewood-Evans A, Senften M, Guo H, Li Y, Miner JH, Reichardt LF, Muller U (2004) β1-Integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci 24: 3402–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleton H (1980) Carleton's histological technique, Ed 5. New York: Oxford UP.
- Cohen-Cory S, Dreyfus CF, Black IB (1991) NGF and excitatory neurotransmitters regulate survival and morphogenesis of cultured cerebellar Purkinje cells. J Neurosci 11: 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico M, Hulit J, Amanatullah DF, Zafonte BT, Albanese C, Bouzahzah B, Fu M, Augenlicht LH, Donehower LA, Takemaru K, Moon RT, Davis R, Lisanti MP, Shtutman M, Zhurinsky J, Ben-Ze'ev A, Troussard AA, Dedhar S, Pestell RG (2000) The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J Biol Chem 275: 32649–32657. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz-i-Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126: 3089–3100. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA 95: 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LA, Thiessen B, Dragowska WH, Daynard T, Bally MB, Dedhar S (2005) Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene 24: 3596–3605. [DOI] [PubMed] [Google Scholar]
- Fassler R, Meyer M (1995) Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev 9: 1896–1908. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A (1998) Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol 8: 983–986. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K (1998) The cells and molecules that make a cerebellum. Trends Neurosci 21: 375–382. [DOI] [PubMed] [Google Scholar]
- Grashoff C, Aszodi A, Sakai T, Hunziker EB, Fassler R (2003) Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep 4: 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Thievessen I, Lorenz K, Ussar S, Fassler R (2004) Integrin-linked kinase: integrin's mysterious partner. Curr Opin Cell Biol 16: 565–571. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367–379. [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U (2002) A critical function of the pial basement membrane in cortical histogenesis. J Neurosci 22: 6029–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, Dedhar S (2005) Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer 5: 51–63. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S (1996) Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379: 91–96. [DOI] [PubMed] [Google Scholar]
- Hartmann D, Ziegenhagen MW, Sievers J (1998) Meningeal cells stimulate neuronal migration and the formation of radial glial fascicles from the cerebellar external granular layer. Neurosci Lett 244: 129–132. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH (2003) Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 130: 15–28. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Widlund HR, Rowitch DH (2004) Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131: 217–228. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29: 404–411. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD (1990) CNS stem cells express a new class of intermediate filament protein. Cell 60: 585–595. [DOI] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP (2004) Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol 270: 393–410. [DOI] [PubMed] [Google Scholar]
- Mackinnon AC, Qadota H, Norman KR, Moerman DG, Williams BD (2002) C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr Biol 12: 787–797. [DOI] [PubMed] [Google Scholar]
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson Jr EM (1997) Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol 139: 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O'Connor TP, Dedhar S (2003) Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci 23: 1638–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR (1998) Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol 143: 1713–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418: 422–425. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF (2005) Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci 25: 7022–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S (2000) Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci USA 97: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S (2001a) Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol 153: 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S (2001b) Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem 276: 27462–27469. [DOI] [PubMed] [Google Scholar]
- Pons S, Trejo JL, Martinez-Morales JR, Marti E (2001) Vitronectin regulates Sonic hedgehog activity during cerebellum development through CREB phosphorylation. Development 128: 1481–1492. [DOI] [PubMed] [Google Scholar]
- Radeva G, Petrocelli T, Behrend E, Leung-Hagesteijn C, Filmus J, Slingerland J, Dedhar S (1997) Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J Biol Chem 272: 13937–13944. [DOI] [PubMed] [Google Scholar]
- Sakai T, Li S, Docheva D, Grashoff C, Sakai K, Kostka G, Braun A, Pfeifer A, Yurchenco PD, Fassler R (2003) Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev 17: 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer MC, Fawcett JW (2001) The astrocyte/meningeal cell interface—a barrier to successful nerve regeneration? Cell Tissue Res 305: 267–273. [DOI] [PubMed] [Google Scholar]
- Sievers J, Pehlemann FW, Gude S, Berry M (1994) Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol 23: 135–149. [DOI] [PubMed] [Google Scholar]
- Tan C, Cruet-Hennequart S, Troussard A, Fazli L, Costello P, Sutton K, Wheeler J, Gleave M, Sanghera J, Dedhar S (2004) Regulation of tumor angiogenesis by integrin-linked kinase (ILK). Cancer Cell 5: 79–90. [DOI] [PubMed] [Google Scholar]
- Terpstra L, Prud'homme J, Arabian A, Takeda S, Karsenty G, Dedhar S, St-Arnaud R (2003) Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol 162: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103. [DOI] [PubMed] [Google Scholar]
- Troussard AA, Mawji NM, Ong C, Mui A, St-Arnaud R, Dedhar S (2003) Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem 278: 22374–22378. [DOI] [PubMed] [Google Scholar]
- Wallace VA (1999) Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9: 445–448. [DOI] [PubMed] [Google Scholar]
- Walsh CA (1999) Genetic malformations of the human cerebral cortex. Neuron 23: 19–29. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY (2001) Genetic regulation of cerebellar development. Nat Rev Neurosci 2: 484–491. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22: 103–114. [DOI] [PubMed] [Google Scholar]
- Younes MN, Kim S, Yigitbasi OG, Mandal M, Jasser SA, Dakak Yazici Y, Schiff BA, El-Naggar A, Bekele BN, Mills GB, Myers JN (2005) Integrin-linked kinase is a potential therapeutic target for anaplastic thyroid cancer. Mol Cancer Ther 4: 1146–1156. [DOI] [PubMed] [Google Scholar]
- Zervas CG, Gregory SL, Brown NH (2001) Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J Cell Biol 152: 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD (2004) NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron 42: 897–912. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A (2001) hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31: 85–94. [DOI] [PubMed] [Google Scholar]