

Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder that is caused by a pathological expansion of CAG repeats within the gene encoding for a 350 kD protein called huntingtin. This polyglutamine expansion within huntingtin is the causative factor in the pathogenesis of HD, however the underlying mechanisms have not been fully elucidated. Nonetheless, it is becoming increasingly clear that alterations in mitochondrial function play key roles in the pathogenic processes in HD. The net result of these events is compromised energy metabolism and increased oxidative damage, which eventually contribute to neuronal dysfunction and death. Mitochondria from striatal cells of a genetically accurate model of HD take up less calcium and at a slower rate than mitochondria from striatal cells derived from normal mice. Further, respiration in mitochondria from these mutant huntingtin-expressing cells is inhibited at significantly lower calcium concentrations compared to mitochondria from wild type cells. Considering these and other findings this review explores the evidence suggesting that mutant huntingtin, directly or indirectly impairs mitochondrial function, which compromises cytosolic and mitochondrial calcium homeostasis, and contributes to neuronal dysfunction and death in HD.
Introduction
Huntington disease (HD) is a neurodegenerative disease that is caused by the pathological elongation of the CAG repeats in exon one of the huntingtin protein gene (27, 30, 81), although the resulting pathogenic processes have not been fully elucidated (27). However transcriptional deregulation (3, 15) and mitochondrial dysfunction (49, 56, and 63) have been strongly implicated in the pathogenesis of HD. In this review, we explore the role of mitochondrial dysfunction in the pathogenesis of HD and the contribution of transcriptional dysregulation, and discuss possible therapeutic interventions based on these findings.
1. Huntington’s Disease
1.1 Clinical and Pathological Aspects
HD is an autosomal dominant neurodegenerative disorder, which inevitably leads to the death of affected individuals. The clinical features of HD classically involve progressive motor dysfunction and psychiatric disturbances with gradual dementia (32). The clinical progression of HD is paralleled by a selective pattern of neuronal degeneration initially in the caudate and striatum and at later stages of the disease in the cerebral cortex (81). In the striatum the neuronal loss is associated with reactive fibrillary astrocytosis, and projection neurons in the striatum and cortex appear to be more vulnerable than interneurons (81). Intraneuronal aggregates which are immunoreactive for huntingtin and ubiquitin also characterize HD brain (16). Although initially it was suggested that the aggregates contributed significantly to neuronal cell death, more recent studies indicate that the aggregates may not be toxic entities per se (1).
A fundamental step in understanding the cellular and molecular mechanisms associated with HD occurred with the localization (30) and the identification of the gene that contained the disease-causing mutation (27). The HD gene is located on the short arm of the chromosome 4, (locus 4q16.3), and encodes for a 350 kD protein named huntingtin (27). Translation of the mutated gene results in an abnormally expanded stretch of glutamine (Q) residues near the N-terminal domain of huntingtin (27) (begins at residue 18). In the non-affected population this CAG/Q domain ranges from 6 to 39 repeats, whereas subjects with more than 39 CAG/Q repeats will almost invariably develop HD (2, 30). In general, the number of the CAG/Q repeats is inversely correlated with the age of onset of the disease (7).
1.2 Transcriptional Dysregulation in HD
Despite the fact that the mutated gene responsible for HD was identified more than 15 years ago (27) and the effects of mutant huntingtin have been studied extensively, the mechanisms by which the mutant huntingtin protein causes neurodegeneration have not yet been fully elucidated. However, it is becoming apparent that transcriptional dysregulation and mitochondrial dysfunction (for review see 10) contribute to the pathogenesis of HD, and that the two processes are likely linked (3, 26, 61). Below we highlight several of the studies which demonstrate that mutant huntingtin disrupts transcriptional processes, with a focus on targets that impact mitochondrial function.
In HD, as well as other polyglutamine diseases, there is clear evidence of transcriptional dysregulation (3, 33, 60). Early studies demonstrated that mutant huntingtin interacts with CREB binding protein (CBP) and attenuates CBP-dependent gene expression (60). Further, mutant huntingtin interacts with the histone acetyltransferase domain (HAT) and inhibits activity (75). Increased expression of CBP or treatment with histone deacetylase (HDAC) inhibitors reversed polyglutamine toxicity both in cultured mammalian cells and in flies (75). A dysregulation of p53 has been proposed to play a role in the mitochondria-associated cellular dysfunction and behavioral abnormalities of HD (3). Mutant huntingtin binds to p53 and upregulates the levels of nuclear p53 as well as p53 transcriptional activity in neuronal cultures. p53 levels are increased in HD patient’s lymphoblast, and ablation of p53 prevents mitochondrial membrane depolarization and cytotoxicity in HD cells (3).
PGC-1α is a member of a family of transcriptional co-activators that regulate the expression of proteins involved in mitochondrial function and the maintenance of glucose, lipid and energy homeostasis (15, 42). PGC-1α interacts with a number of transcription factors including NRF-1 and NRF-2 which regulate the expression of mitochondrial respiratory genes (65), as well as members of the PPAR family including PPARγ (42). Indeed, PGC-1α plays a central and crucial role in modulating the expression of genes that impact mitochondrial function, as PGC-1α knockout mice exhibit defects in energy metabolism (43). Recent studies have provided evidence that the expression of PGC-1α is repressed by mutant huntingtin, due in part to the fact that mutant huntingtin interferes with the TAF4/CREB signaling pathway (15). In addition, it can be speculated that because cAMP levels and hence CREB phosphorylation and CRE signaling are significantly decreased in mutant huntingtin expressing striatal cells (23), this may also contribute to the downregulation of PGC-1α expression.
PGC-1α plays a central role in regulating the expression of mitochondrial genes and recent findings have implicated this co-activator in neurodegenerative processes. PGC-1α is required for the expression of numerous genes that detoxify ROS (74) and increased expression of PGC-1α protects cultured neuronal-like cells against oxidative stress (74). PGC-1α target genes are decreased in HD human brain and an HD mouse model, and striatal neurons from the HD mouse model expressing exogenous PGC-1α were resistant to the toxic effects of the succinate dehydrogenase inhibitor 3-nitropropionic acid (3-NP) (82). Moreover, PGC-1α mRNA and protein levels are significantly decreased in mutant huntingtin knockin mice as well in the STHdhQ111/Q111 cell line (15). When PGC-1α knockout mice were crossed with HD knock-in mice this resulted in increased neurodegeneration of striatal neurons and motor abnormalities in the HD mice. Additionally, expression of PGC-1α partially protects against the toxic effects of mutant huntingtin in cultured striatal neurons (15). Overall, these data indicate that in HD there is defective PGC-1α functioning and therefore downstream events are likely impaired.
1.3. Mitochondrial Dysfunction in HD
It was first speculated that there was energetic impairment in HD because HD patients exhibit profound weight loss despite sustained caloric intake (10, 11). Further, PET scans revealed marked reductions in glucose utilization in the striatum of HD patients in early stages prior to pronounced striatal atrophy. In addition, these studies revealed that energy dysfunction precedes the onset of clinical symptoms of the disorder, suggesting that an energy failure may play a primary role in the pathogenesis of HD (5, 35, 36, 37, 39, 46, 83). Early ultrastructural studies of cortical biopsies obtained from patients with either juvenile or adult onset HD showed abnormal mitochondria morphology (25, 79). Mitochondrial functional abnormalities were also observed in early studies. In 1974 a defect in succinate dehydrogenase, a component of both the Krebs cycle and the complex II of the electron transport chain, in the caudate and to a lesser extent in the cortex of postmortem HD brains was reported (73). Subsequent studies confirmed that there was a significant decrease in complex II activity in the caudate nucleus of HD brains (an approximately 50% decrease) relative to the levels in matched control brains. In addition to decreases in complex II activity, decreases in complex III activity in the caudate and putamen, and of complex IV in the putamen have been observed (12, 28, 45). However the majority of these cases showed advanced neuropathology including dramatic striatal atrophy (pathological grades 3 and 4 of HD), and therefore alterations in the source (i.e., glial, neuronal, etc) of the mitochondria is likely to have been affected. Interestingly, in presymptomatic and grade 1 HD cases no impairment of mitochondrial complex activities was observed (29).
Animal and cell models of HD have provided compelling evidence that mitochondrial function is impaired in HD, and that this occurs early in the disease process and is likely fundamental to the pathogenesis of HD. 3-NP is an irreversible inhibitor of succinate dehydrogenase that inhibits both the TCA cycle and complex II activity, and in animal models, administration of 3-NP results in selective lesioning of the striatum (9). Low doses of 3-NP administered chronically to both rodents and non-human primates resulted in pathology and symptomatology resembling HD (specific striatal lesions with selective vulnerability of medium-sized spiny neurons) (4, 8, 18). It is intriguing to note that striatal mitochondria contain more cyclophilin D than cortical mitochondria and are more sensitive to calcium-induced mitochondrial permeability transition pore (mPTP) opening (13). Early studies in rats exposed to intrastriatal injection of malonate (a reversible inhibitor of succinate dehydrogenase) (4) further support the hypothesis that impairment of mitochondrial function plays an important role in the pathogenesis of HD. Intrastriatal injection of malonate produced age-dependent striatal lesions, with medium-sized spiny neurons being selectively affected (4). These observations have lead to the hypothesis that the expression of mutant huntingtin results in impaired mitochondrial energy metabolism and calcium handling and therefore decreases in energy levels of the cells, increases in oxidative damage, and potentially secondary excitotoxic death (for a review see 10 and 11).
Mitochondrial dysfunction is evident in two well established HD mice models; the 150/150Q mutant huntingtin knock-in mice (41), and the R6/2 mice (44). Mitochondria isolated from 150/150Q mutant huntingtin knock-in mice show an increased sensitivity to calcium-induced mPTP opening (14) and striatal neurons from heterozygous 150/150Q mutant huntingtin knock-in mice were more prone to undergo “deregulation” in response to NMDA compared to neurons from wild type mice (55). The R6/2 HD mouse model express exon 1 of the huntingtin gene with 155 CAG expansion (44). In these mice a significant reduction in aconitase, an enzyme involved in the Krebs cycle has been reported. The activities of complex IV in the striatum and cerebral cortex were also reported to be significantly decreased in the R6/2 mice (78). Moreover, these results suggest that the deficiency in complex IV precedes neuronal death in the R6/2 mice and thus contribute to the pathogenesis (78). A decreased stability of mitochondria from the HD R6/2 mouse muscle against calcium-induced mPTP opening has been detected. Further, complex I-dependent respiration of R6/2 mitochondria was more sensitive to calcium-induced inhibition than wild-type mitochondria (24). In addition, significant alterations in mitochondrial ultrastructure were seen, consistent with metabolic stress in the heart of R6/2 mice (48). Overall these mouse models exhibit mitochondrial and metabolic defects that are consistent with the defects that occur in HD pathology.
More recent findings have provided additional evidence of mitochondrial dysfunction in HD. Lymphoblasts derived from HD patients manifest a much greater increase in mitochondrial depolarization than control samples when treated with toxins that target complexes II and IV (64). When ATP/ADP ratios were evaluated in 40 human lymphoblastic cell lines an inverse between CAG repeat length in the HD gene and the ATP/ADP ratio was observed (69). Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin (49). This cell line is considered a genetically accurate model of HD (80). Further the mutant huntingtin-expressing cells exhibit a significant increase in sensitivity to 3-NP (23, 63). Taken together there is clear and compelling evidence that mitochondrial dysfunction is a significant contributor to the HD pathogenesis.
1.4 Mitochondria Calcium-Handling Defects in HD
It is becoming increasingly apparent that mitochondrial calcium handling defects are associated with the pathogenesis of HD. Mitochondria from lymphoblasts of HD patients have a lower Δψm and depolarize at lower calcium loads than do mitochondria from controls (56). Similar defects were noted in brain mitochondria from transgenic mice expressing full-length mutant huntingtin, and this defect preceded the onset of pathological or behavioral abnormalities (56). In addition, Gizatullina et al showed that skeletal muscle of transgenic HD R6/2 mice is characterized by increased vulnerability of HD mitochondria to calcium stress, leading to energetic depression and muscle atrophy (24). Furthermore, mitochondria from HD rats that expressed 51 glutamine repeats (htt51Q) exhibited a decreased Δψm stability in response to calcium, lower capacities and rates of mitochondrial calcium transport, and a decreased calcium threshold for mPTP opening (21). Moreover, the presence of full-length mutant huntingtin at physiological levels in clonal striatal cells has been clearly demonstrated to result in deficits in mitochondrial-dependent calcium handling (49, 54).
When subjected to increasing calcium concentrations, mitochondria from mutant huntingtin expressing cells were significantly more sensitive to calcium-induced decreases in state 3 respiration and Δψm than mitochondria from wild type cells (49). Further, mutant huntingtin expressing cells had a reduced mitochondrial calcium uptake capacity in comparison with wild-type cells (49, 59). Decreases in state 3 respiration were associated with increased mitochondrial membrane permeability. The Δψm defect was attenuated in the presence of ADP and the decreases in calcium uptake capacity were abolished in the presence of mPTP opening inhibitors (49). Treatment of the mutant huntingtin expressing cells with HDAC inhibitors (trichostatin A or sodium butyrate) ameliorated the mitochondrial calcium handling defects, suggesting the involvement of transcriptional dysregulation (54). These findings clearly indicate that mutant huntingtin expressing cells have mitochondrial calcium handling defects and that the increased sensitivity to calcium-induced mitochondrial depolarization maybe a contributing mechanism to the mitochondrial dysfunction in HD.
Although mutant huntingtin induced transcriptional dysregulation likely contributes to the mitochondrial dysfunction in HD, direct effects cannot be ruled out. Choo et al showed that huntingtin was present in a purified mitochondrial fraction in association with the outer mitochondrial membrane in clonal striatal cells established from wild-type and mutant huntingtin knock-in mice (14). Further, a recombinant truncated mutant huntingtin construct, but not a wild-type, directly induced mPTP opening in isolated mouse liver mitochondria, an effect that was prevented completely by cyclosporin A (CsA) and ATP (14). These data suggest that mutant huntingtin, in addition to modifying protein expression by affecting transcriptional processes, could be acting directly on mitochondria and modifying their function.
2. Mechanisms to Ameliorate Mitochondrial Dysfunction in HD
2.1 Mitochondrial Permeability Transition Pore (mPTP) Opening Inhibitors
It has been suggested that the neuroprotective properties of CsA are due in part to its ability to prevent mPTP opening in response to high levels of calcium or oxidative stress (52, 58). Exposure to high levels of calcium or oxidative stress results in the mPTP opening of the inner mitochondrial membrane, causing disruption of Δψm, and swelling of mitochondria (40, 47, 58). In vitro CsA attenuates apoptosis induced by the mitochondrial complex 1 inhibitor rotenone (68), and also the calcium ionophore A23187 (58). CsA also prevents Δψm loss resulting from exposure to NMDA in cortical neurons (52). Additionally, CsA and bongkrekic acid significantly attenuated NMDA-induced calcium peak and Δψm loss in YAC128 medium-size spiny neurons (MSNs) (17). The YAC128 mouse model express full-length human huntingtin with 128 glutamine repeats and exhibits selective striatal neurodegeneration and large increases in apoptosis after NMDA receptor activation (70, 72). Also, CsA has been demonstrated to be neuroprotective in vivo. Using procedures which facilitate molecule penetration of blood brain barrier, CsA has reduced neuronal death in ischemia-reperfusion (71), hypoglycemia (19), and traumatic brain injury (53). In addition, Leventhal and colleges demonstrated that treatment with CsA protected striatal neurons toxicity induced by 3-NP in vitro and in vivo (40). Interestingly, CsA prevented ultrastructural mitochondrial alterations and decreased apoptosis in myoblasts obtained from Ullrich congenital muscular dystrophy patients (47). Therefore, CsA or new mPTP opening inhibitors may be of potential therapeutic benefit by protecting vulnerable neurons populations affected in HD.
2.2 PPARγ Activators
PGC-1α plays a central role in regulating the expression of mitochondrial genes and recent findings have implicated this co-activator in neurodegenerative processes. Another key regulator of PGC-1α function is the NAD+-dependent deacetylase SIRT1 (22, 31). SIRTs catalyze both deacetylation and ADP-ribosylation reactions which are coupled to the cleavage of NAD+ and result in deacetylated lysine, O-acetyl-ADP-ribose and nicotinamide (31). PGC-1α is a substrate of a SIRT1 and deacetylation of PGC-1α results in the upregulation of mitochondrial metabolic genes (22). Treatment with resveratrol (a well known antioxidant and sirtuin activator) specifically rescued early neuronal dysfunction phenotypes induced by mutant polyglutamines expression in Caenorhabditis elegans (57). In others studies, treatment of mice with resveratrol significantly increased their aerobic capacity, as evidenced by their increased running time and consumption of oxygen in muscle fibers (38). These effects were explained by the fact that in addition to being an antioxidant, resveratrol activates SIRT1 resulting in subsequent deacetylation and activation of PGC-1α, and thus induction of OX/PHOS and mitochondrial biogenesis genes which improved mitochondrial function (38). These and other findings suggest that an increase in SIRT1 activity in HD could facilitate activation of the PGC-1α-PPARγ signaling pathway and thus improve mitochondrial function.
PGC-1α is a potent co-activator of the type II nuclear receptor PPARγ. A variety of endogenous compounds activate PPARγ including 15-deoxy-Δ12,14-prostaglandin J2 (15Δ-PGJ2) and nitrolinoleic acid (LNO2) (66). Further, there are numerous exogenous agents including the thiazolidinediones (TZDs) (rosiglitazone, pioglitazone, troglitazone) that are PPARγ agonists (6, 77). PPARγ agonists have been shown to be neuroprotective and improve mitochondrial function (20, 34, 59, 67). It was also demonstrated that when rosiglitazone was administered orally to mice substantial amounts were found in the brain and after 7 days of treatment there was clear evidence of mitochondrial biogenesis in the brain (76). In our studies pretreatment of mutant striatal cells with the PPAR-γ agonist rosiglitazone prevented the loss of Δψm, mitochondrial calcium deregulation, and oxidative stress overproduction in response to thapsigargin (59). Additionally, the PPARγ signaling pathway was significantly impaired in the mutant huntingtin striatal cells with decreases in PPARγ expression and reduced PPARγ transcriptional activity (59). Also, treatment with rosiglitazone increased mitochondrial mass levels, further suggesting a role for the PPARγ pathway in mitochondrial function in striatal cells (59). These findings suggest that activation of the PPARγ signaling pathway could ameliorate the mitochondrial deficits in HD. Therefore PPARγ agonists could represent a potential tool to consider in the treatment of neurodegenerative disorders, including HD.
3. Conclusions and Working Hypothesis
Figure 1 illustrates our hypothesis of how mutant huntingtin may compromise mitochondrial function and possible therapeutic targets. We hypothesize that the mutant huntingtin expression induced inhibition of CREB/TAF4 as well as CBP results in a downregulation and decrease in the activity of PGC-1α, and this subsequently results in a decrease in the activity of transcription factors such as PPARγ and hence a decrease in the expression of mitochondrial genes which results in compromised mitochondrial function which contributes to striatal cell dysfunction and death. We also hypothesize that this mutant huntingtin induced pathogenic cascade of events can be attenuated by increasing the activity and level of PGC-1α. We hypothesize that SIRT1 is a good point for therapeutic intervention as SIRT1 activates PGC-1α and potential SIRT1 activating drugs are available. This would result in increased expression of mitochondrial genes, improved mitochondrial function and hence increased striatal cell function and survival. In conjunction with increasing PGC-1α, increasing the activity of the target nuclear receptor PPARγ (e.g., with TZDs) would also contribute to the striatal cell function and survival in HD by increasing the activity of PPARγ which would increase the expression of mitochondrial genes resulting improved function and biogenesis. Also, the use of CsA derivatives or new design inhibitors of mPTP opening could be an important factor to consider ameliorating mitochondrial dysfunction in HD pathology.
Figure 1. Mutant huntingtin expression compromises mitochondrial function.
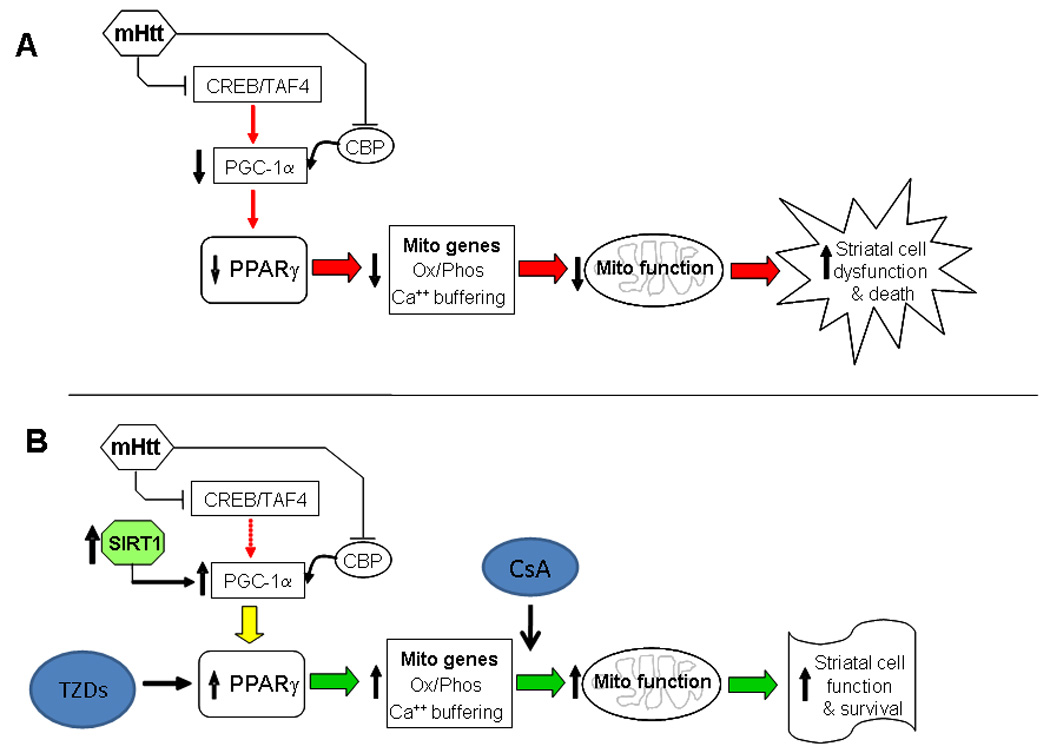
Diagram illustrating possible involvement of mutant huntingtin (mHtt) in compromising mitochondrial function and thus contributing to a loss of neuronal viability in HD. In A the disease state is shown. mHtt has been shown to interact with and/or attenuate the activity of CREB/TAF4 (15) and CBP (75), resulting in decreased expression (15) and activity of PGC-1α which in turn down regulates the activity of transcriptional activity by PPARγ and other PGC-1α dependent genes (15, 74). This results in a down regulation of mitochondrial genes impaired mitochondrial function and increased cell death and dysfunction in HD. In B the mechanisms of possible intervention therapies are shown. Increasing SIRT1 activity would enhance the activity of PGC-1α and result in appropriate activation of downstream genes. Increasing the activity of PPARγ alone (using TDZs drugs) (59, 77) or the use of new mPTP opening inhibitors (47, 53) would enhance mitochondrial gene expression and/or function and ameliorate neuronal loss dysfunction and loss in HD.
Abbreviations
- HD
Huntington’s Disease
- mPTP
mitochondrial Permeability Transition Pore
- CsA
Cyclosporin A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 2.Ashizawa T, Wong LJ, Richards CS, Caskey CT, Jankovic J. CAG repeat size and clinical presentation in Huntington's disease. Neurology. 1994;44:1137–1143. doi: 10.1212/wnl.44.6.1137. [DOI] [PubMed] [Google Scholar]
- 3.Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral in Huntington's disease. Neuron. 2005;47:1–3. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berent S, Giordani B, Lehtinen S, Markel D, Penney JB, Buchtel HA, Starosta-Rubinstein S, Hichwa R, Young AB. Positron emission tomographic scan investigations of Huntington's disease: cerebral metabolic correlates of cognitive function. Ann Neurol. 1988;23:541–546. doi: 10.1002/ana.410230603. [DOI] [PubMed] [Google Scholar]
- 6.Berger J, Moller DE. The mechanisms of action of PPARs. Annu. Rev. Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 7.Brandt J, Bylsma FW, Gross R, Stine OC, Ranen N, Ross CA. Trinucleotide repeat length and clinical progression in Huntington's disease. Neurology. 1996;46:527–531. doi: 10.1212/wnl.46.2.527. [DOI] [PubMed] [Google Scholar]
- 8.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 10.Browne SE. Mitochondria and Huntington’s disease pathogenesis. Insight from Genetic and Chemical Models. Ann NY Acad Sci. 2008;1147:358–382. doi: 10.1196/annals.1427.018. [DOI] [PubMed] [Google Scholar]
- 11.Browne SE, Beal MF. The energetics of Huntington's disease. Neurochem Res. 2004;29:531–546. doi: 10.1023/b:nere.0000014824.04728.dd. [DOI] [PubMed] [Google Scholar]
- 12.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 13.Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 15.Cui L, Jeong H, Borovecki F, Parkhurst C, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 16.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 17.Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci. 1998;18:5151–5159. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuenzalida K, Quintanilla RA, Ramos P, Piderit D, Fuentealba RA, Martinez G, Inestrosa NC, Bronfman M. PPAR gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- 21.Gellerich FN, Gizatullina Z, Nguyen HP, Trumbeckaite S, Vielhaber S, Seppet E, Zierz S, Landwehrmeyer B, Riess O, von Horsten S. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem. 283;2008:30715–30724. doi: 10.1074/jbc.M709555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gines S, Seong IS, Fossale E, Ivanova E, Trettel F, Gusella JF, Wheeler VC, Persichetti F, MacDonald ME. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum Mol Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 24.Gizatullina ZZ, Lindenberg KS, Harjes P, Chen Y, Kosinski CM, Landwehrmeyer BG, Ludolph AC, Striggow F, Zierz S, Gellerich FN. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol. 2006;59:407–411. doi: 10.1002/ana.20754. [DOI] [PubMed] [Google Scholar]
- 25.Goebel HH, Heipertz R, Scholz W, Iqbal K, Tellez-Nagel I. Juvenile Huntington chorea: clinical, ultrastructural, and biochemical studies. Neurology. 1978;28:23–31. doi: 10.1212/wnl.28.1.23. [DOI] [PubMed] [Google Scholar]
- 26.Greenamyre JT. Huntington's disease--making connections. N Engl J Med. 2007;356:518–520. doi: 10.1056/NEJMcibr067022. [DOI] [PubMed] [Google Scholar]
- 27.T.H. s. D.C.R. Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group [see comments] Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 28.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 29.Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- 30.Gusella JF, MacDonald ME, Ambrose CM, Duyao MP. Molecular genetics of Huntington's disease. Arch Neurol. 1983;50:1157–1163. doi: 10.1001/archneur.1993.00540110037003. [DOI] [PubMed] [Google Scholar]
- 31.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 32.Harper PS. The epidemiology of Huntington's disease. Hum Genet. 1992;89:365–376. doi: 10.1007/BF00194305. [DOI] [PubMed] [Google Scholar]
- 33.Helmlinger D, Tora L, Devys D. Transcriptional alterations and chromatin remodeling in polyglutamine diseases. Trends Genet. 2006;22:562–570. doi: 10.1016/j.tig.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem. 2007;100:1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- 35.Kuhl DE, Metter EJ, Riege WH, Markham CH. Patterns of cerebral glucose utilization in Parkinson's disease and Huntington's disease. Ann Neurol. 1984;15 Suppl:S119–S125. doi: 10.1002/ana.410150723. [DOI] [PubMed] [Google Scholar]
- 36.Kuhl DE, Phelps ME, Markham CH, Metter EJ, Riege WH, Winter J. Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann Neurol. 1982;12:425–434. doi: 10.1002/ana.410120504. [DOI] [PubMed] [Google Scholar]
- 37.Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntington's disease. Brain. 1990;113:1405–1423. doi: 10.1093/brain/113.5.1405. [DOI] [PubMed] [Google Scholar]
- 38.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 39.Leenders KL, Frackowiak RS, Quinn N, Marsden CD. Brain energy metabolism and dopaminergic function in Huntington's disease measured in vivo using positron emission tomography. Mov Disord. 1986;1:69–77. doi: 10.1002/mds.870010110. [DOI] [PubMed] [Google Scholar]
- 40.Leventhal L, Sortwell CE, Hanbury R, Collier TJ, Kordower JH, Palfi S. Cyclosporin A protects striatal neurons in vitro and in vivo from 3-nitropropionic acid toxicity. J Comp Neurol. 2000;425:471–478. doi: 10.1002/1096-9861(20001002)425:4<471::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 41.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 42.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 45.Mann VM, Cooper JM, Javoy-Agid F, Agid Y, Jenner P, Schapira AH. Mitochondrial function and parental sex effect in Huntington's disease [letter] Lancet. 1990;336:749. doi: 10.1016/0140-6736(90)92242-a. [DOI] [PubMed] [Google Scholar]
- 46.Martin WR, Clark C, Ammann W, Stoessl AJ, Shtybel W, Hayden MR. Cortical glucose metabolism in Huntington's disease. Neurology. 1992;42:223–229. doi: 10.1212/wnl.42.1.223. [DOI] [PubMed] [Google Scholar]
- 47.Merlini L, Angelin A, Tiepolo T, Braghetta P, Sabatelli P, Zamparelli A, Ferlini A, Maraldi NM, Bonaldo P, Bernardi P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci U S A. 2008;105:5225–5229. doi: 10.1073/pnas.0800962105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 50.Milakovic T, Quintanilla RA, Johnson GV. Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J Biol Chem. 2006;281:34785–34795. doi: 10.1074/jbc.M603845200. [DOI] [PubMed] [Google Scholar]
- 51.Morris M. Dementia and cognitive changes in Huntington's disease. Adv Neurol. 1995;65:187–200. [PubMed] [Google Scholar]
- 52.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 53.Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 54.Oliveira JM, Chen S, Almeida S, Riley R, Goncalves J, Oliveira CR, Hayden MR, Nicholls DG, Ellerby LM, Rego AC. Mitochondrial-dependent Ca2+ handling in Huntington's disease striatal cells: effect of histone deacetylase inhibitors. J Neurosci. 2006;26:11174–11186. doi: 10.1523/JNEUROSCI.3004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Goncalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- 56.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 57.Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genetics. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 58.Petersén A, Castilho RF, Hansson O, Wieloch T, Brundin P. Oxidative stress, mitochondrial permeability transition and activation of caspases in calcium ionophore A23187-induced death of cultured striatal neurons. Brain Res. 2000;857:20–29. doi: 10.1016/s0006-8993(99)02320-3. [DOI] [PubMed] [Google Scholar]
- 59.Quintanilla RA, Jin YN, Fuenzalida K, Bronfman M, Johnson GV. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: possible role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in the pathogenesis of Huntington disease. J Biol Chem. 2008;283:25628–25637. doi: 10.1074/jbc.M804291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riley BE, Orr HT. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev. 2006;20:2183–2192. doi: 10.1101/gad.1436506. [DOI] [PubMed] [Google Scholar]
- 61.Ross CA, Thompson LM. Transcription meets metabolism in neurodegeneration. Nat Med. 2006;12:1239–1241. doi: 10.1038/nm1106-1239. [DOI] [PubMed] [Google Scholar]
- 62.Ross CA, Margolis RL, Rosenblatt A, Ranen NG, Becher MW, Aylward E. Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA) Medicine (Baltimore) 1997;76:305–338. doi: 10.1097/00005792-199709000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Ruan Q, Lesort M, MacDonald ME, Johnson GV. Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum Mol Genet. 2004;13:669–681. doi: 10.1093/hmg/ddh082. [DOI] [PubMed] [Google Scholar]
- 64.Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr., Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 65.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 66.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, Zimmer A, Heneka MT. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998;8091:12–17. doi: 10.1016/s0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- 69.Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RH, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 70.Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 71.Shiga Y, Onodera H, Matsuo Y, Kogure K. Cyclosporin A protects against ischemia-reperfusion injury in the brain. Brain Res. 1992;595:145–148. doi: 10.1016/0006-8993(92)91465-q. [DOI] [PubMed] [Google Scholar]
- 72.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–15567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 73.Stahl WL, Swanson PD. Biochemical abnormalities in Huntington's chorea brains. Neurology. 1974;24:813–819. doi: 10.1212/wnl.24.9.813. [DOI] [PubMed] [Google Scholar]
- 74.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelmman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 75.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 76.Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, Ghosh S, Nock C, Saunders A, Roses A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J Alzheimers Dis. 2007;11:45–51. doi: 10.3233/jad-2007-11108. [DOI] [PubMed] [Google Scholar]
- 77.Sundararajan S, Jiang Q, Heneka M, Landreth G. PPARgamma as a therapeutic target in central nervous system diseases. Neurochem Int. 2006;49:136–144. doi: 10.1016/j.neuint.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 78.Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
- 79.Tellez-Nagel I, Johnson AB, Terry RD. Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol. 1974;33:308–332. doi: 10.1097/00005072-197404000-00008. [DOI] [PubMed] [Google Scholar]
- 80.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 81.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 82.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 83.Young AB, Penney JB, Starosta-Rubinstein S, Markel DS, Berent S, Giordani B, Ehrenkaufer R, Jewett D, Hichwa R. PET scan investigations of Huntington's disease: cerebral metabolic correlates of neurological features and functional decline. Ann Neurol. 1986;20:296–303. doi: 10.1002/ana.410200305. [DOI] [PubMed] [Google Scholar]