

Abstract
Background
Alcoholism may result in severe neurological deficits and cognitive impairments. Many of central effects of ethanol (EtOH) can be explained by upregulation of NMDA and downregulation of GABA(A) receptors in response to a long-term EtOH consumption. Abrupt ethanol withdrawal (EW) may result in neuronal hyperexcitability leading to hallucinations, seizures, neurodegeneration and sometimes death.
Methods
Using a multidisciplinary approach in wild-type and genetically modified mice we have examined the contribution of the tissue-plasminogen activator, plasminogen and laminin to EW-induced cell death.
Results
Here we show that EW-induced neurodegeneration is mediated by the tissue plasminogen activator (tPA)/plasmin system. During EW, tPA is upregulated in the hippocampus, converts plasminogen to plasmin, which in turn degrades an extracellular matrix component laminin, leading to caspase-3-dependent cell death. Consequently, mice in which the tPA or plasminogen genes have been deleted do not show EW-induced laminin degradation, mitochondial dysfunction, and neurodegeneration. Finally we demonstrate, that disruption of the hippocampal laminin γ-1 renders the mice resistant to neurotoxic effects of EW.
Conclusions
Our data identify laminin γ-1 as a novel target to combat neurodegeneration.
Introduction
Alcoholism is a devastating condition which, beyond the personal and family impact, generates a cost of $185 billion annually in the United States alone (1). Ethanol produces a wide variety of physiological and behavioral such as initial hyperactivity and euphoria followed by sedation, hypothermia, motor and sensory incoordination, and loss of righting reflex (2, 3). Beyond its toxic effects on various tissues and organs, chronic ethanol abuse often leads to a number of neurological deficits, such as cortico/hippocampal atrophy with the resulting cognitive impairments (4-6).
Most of ethanol's effect can be explained by its neuromodulatory actions at several neurotransmitter-gated ion channels (2, 7, 8). Ethanol blocks the activity of NMDA receptors, and enhances GABA(A)- receptor-mediated signaling, therefore elevating neuronal excitability threshold (2, 7, 8). In contrast, chronic ethanol administration causes adaptive up-regulation of NMDA receptors and down-regulation of GABA(A) receptors in neurons, aimed to counteract ethanol-induced sedation. Thus, abrupt ethanol withdrawal (EW) results in a dramatic increase in neuronal excitability, which may lead to hallucinations, seizures, neurodegeneration and eventually death (8-10).
Beyond its effect on ion channels, ethanol modulates a variety of neurotransmitter systems. We have recently shown that a serine protease tissue plasminogen-activator (tPA) is induced by EW and facilitates EW-induced seizures (11). This effect of tPA is mediated through its interaction with ethanol-sensitive, NR2B-containing NMDA receptors (11).
Whether tPA contributes to ethanol-induced neurodegeneration has not been investigated. In this study we show that tPA dose-dependently facilitates EW-induced neuronal death both in vitro and in vivo. This effect is plasminogen-dependent, and can be prevented by deletion of either the tPA or plasminogen gene. EW-induced neurodegeneration correlates with the tPA/plasmin-mediated degradation of laminin in the hippocampus. Finally, we show that conditional disruption of the laminin γ-1 in the CA1 region of the hippocampus renders the mice resistant to neurotoxic effects of EW.
Methods and Materials
Animals
Experiments were performed on male, three-month old wild-type C57/Bl6, tPA -/- (12) or plasminogen -/- (13, 14) and mice in which laminin γ-1 was deleted from the forebrain (fLAMγ1 × CaMKIIα-Cre)(15). tPA-/- and plasminogen-/- mice have been backcrossed to Jackson Lab C57/BL6J for at least 9 generations. Mice homozygous for the knockout allele were used to set up breeding colonies. We used Cre(-) littermates as controls for fLAMγ1 × CaMKIIα-Cre animals.
Induction of physical dependence
The mice were housed individually and given a measured amount of liquid diet (Bio-Serv, USA) containing 2.3 - 10% v/v ethanol and vitamin supplement as their sole nutrient source. The mice were gradually introduced to the ethanol diet as follows: days 1-3, 2.3%; days 4-6, 4.7%; days 7-10, 7%; days 11-14, 10% ethanol. The pair-fed control mice were given the same volume of isocaloric ethanol-free liquid diet as the ethanol-exposed mice had consumed the previous day (11, 16). Every 24 hours the mice were rated for behavioral signs of ethanol intoxication, by an observer who was unaware of the kind or amount of diet consumed as well as genotype of the animals, as previously described (11, 16). Ethanol withdrawal (EW) was initiated on day 15 at 8 AM by removing the ethanol-containing diet and replacing it with ethanol-free diet.
In situ zymography
In situ zymography was performed as previously described (17). The slides were incubated at 37°C in a humid chamber for 2–4 h, and the developed zymograms were photographed under dark-field illumination.
Quantification of ethanol and EW-induced cell death in the hippocampus
To assess the extent of cell death animals were sacrificed after 14 days of ethanol administration (without withdrawal) or up to two days following EW. Their brains were embedded in paraffin and 4 μm-thick sections were cut at 1.7-2.2 mm posterior to bregma. To estimate the extent of cell loss sections were deparaffinized, rehydrated and stained with DAPI to visualize cell nuclei. At least four sections per animal were analyzed. The cells were counted in three consecutive fields of 300 μm of the CA1 or the CA3 region and averaged.
To confirm cell death (either necrotic or apoptotic) the above sections were processed for detection of broken DNA strands with TdT-mediated dUTP nick-end labeling (TUNEL) method (Boehringer Mannheim, FITC-based kit), performed according to manufacturers instructions.
Fluoro-Jade B staining was performed as previously described (18).
Determination of the lineage of dying cells in the hippocampus following EW
Animals were sacrificed 2 days following EW and 4μm paraffin sections were prepared (see above). TUNEL staining was performed in conjunction with neuron- (NeuN) or astrocyte-specific (glial fibrillary acidic protein; GFAP) markers (see Supplementary Material and Methods).
To distinguish the type of neurons undergoing degeneration we performed immunohistochemistry for a glutamate transporter VGLUT1 (a marker for excitatory neurons), glutamic acid decarboxylase-67 (GAD67; a marker for inhibitory interneurons) or active caspase-3 in conjunction with TUNEL staining (see Supplementary Material and Methods).
Laminin immunohistochemistry
Laminin staining was performed on 15 μm thick paraformaldehyde fixed sections as previously described (19).
Brain ethanol levels
Hippocampal amples were deproteinized and ethanol concentration was measured using Ethanol UV-method Kit (R-Biopharm, USA) according to the manufacturer's instructions (see Supplementary Material and Methods).
Cell culture experiments
Hippocampal primary neuronal cultures were prepared from embryonic day 18.5 WT and tPA-/- mice (see Supplementary Material and Methods). On day 10 after plating, the cultures were exposed to culture media containing 50 or 100 mM ethanol for 3 days. EW was induced on day 4 by replacing ethanol-containing media with regular growth media. Neuronal death was determined on day 6.
LDH measurement
LDH released into the culture media was determined using the LDH assay kit (Sigma, USA) according to manufacturer's instructions.
Determination of mitochondrial permeability
tPA+/+ or tPA-/- neurons (with or without prior administration / withdrawal of ethanol) were stained for the presence of mitochondrial J-aggregates using MitoPT kit (Immunochemistry Technologies, Bloomington, USA) according to the manufacturers instructions and observed under Zeiss LSM5 Exciter confocal microscope.
Statistical Analysis
Mann-Whitney U-test was used for statistical evaluation and p<0.05 was considered statistically significant. Diet consumption and intoxication levels were compared between genotypes using Kruskall-Wallis nonparametric ANOVA.
Results
Ethanol withdrawal causes hippocampal cell death in a chronic consumption model
It has been widely accepted that ethanol abuse causes cell death in the brain (4-6) which depends on the genetic background of the animals used, as well as the schedule and route of EtOH administration (20-22). The above parameters not only determine the extent but also the time when cell death is observed (23-25). To investigate the occurrence and severity of degeneration in our model we determined the cell number in the CA1 and CA3 regions of the hippocampus after 14 days of EtOH administration with of without withdrawal. We found that chronic administration of ethanol did not change the number of cells in the CA1 or CA3 region as compared to EtOH-naïve animals (Fig 1; p>0.05). Similarly, the cell number was not affected six hours following ethanol withdrawal (Fig 1; p>0.05). However, a 14-17% drop was observed in the CA1 two days after EtOH diet has been replaced by the control one (Fig 1; p<0.05 and Fig. 2; p<0.01). This result indicates that in our model ethanol withdrawal (EW) is necessary for cell death to occur in the CA1 region, and implicates delayed cell death mechanisms in its pathogenesis.
Figure 1. Ethanol withdrawal causes upregulation of tPA activity and triggers cell death in the hippocampus.
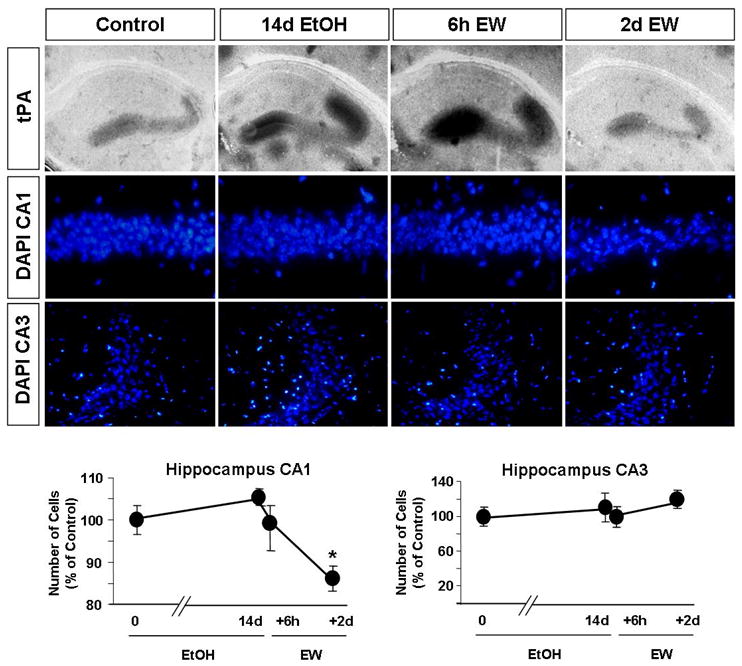
Wild-type mice were sacrificed after 14 days of EtOH administration or six hours to two days after ethanol withdrawal (EW). EtOH-naïve mice served as controls. The brains were collected and in situ zymography was performed. Ethanol administration caused an upregulation of tPA activity in the hippocampus (dark lytic zones in the upper panels), which was even more dramatic after EW. The correlation between tPA levels and cell loss was determined by counting cell number in the CA1 and CA3 region of the hippocampus using DAPI-stained sections (middle panels). 14 days of EtOH administration (14d) or six hours of EW (6h) did not affect the number of cells in the CA1 or CA3 as compared to EtOH-naïve animals (shown as 0 days). However, a 14% decrease was observed two days after EW in the CA1 region (lower panels). * p<0.05; n=3-4 per time-point. The results are presented as mean ± SEM.
Figure 2. Ethanol withdrawal-induced neurodegeneration is tPA- and plasminogen-dependent.
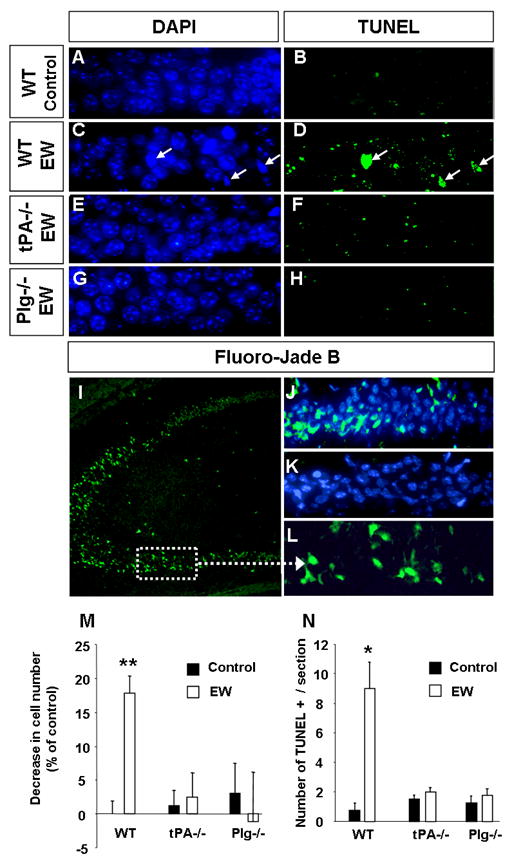
Wild-type, tPA -/- or plasminogen -/- mice were sacrificed two days following EW. EtOH-naïve mice of the same genotype served as controls. Cell loss was determined by counting cell number in the CA1 region of the hippocampus using DAPI-stained sections. To confirm cell death the above sections were processed for detection of broken DNA strands with TdT-mediated dUTP nick-end labeling (TUNEL) method. Analysis of the CA1 region revealed that the number of cells decreased in the wild-type mice (A, C, M) but not in tPA -/- (E, M) or plasminogen -/- animals (G, M) two days after EW. Similarly, we did not observe TUNEL-positive cells in CA1 region of tPA- or plasminogen-deficient mice (F, H, N) typically seen in wild-type mice after EW (B, D, N; arrows in D). Staining using another marker of neurodegeneration, Fluoro-Jade B, confirmed the presence of bright green damaged cells (as seen in the kainic acid-injected positive control; I, L) in the CA1 region in wild-type (J) but not tPA-/- mice (K) two days after EW. Altogether these results demonstrate that EW-mediated neurodegeneration is tPA- and plasminogen-dependent. Sections derived from EtOH-naïve tPA-/- or plasminogen -/- mice did not show any signs of neurodegeneration and are not shown, but are included in quantification in M and N. * p<0.05; ** p<0.01; n=4-5 per group. The results are presented as mean ± SEM.
To confirm the presence of degenerating neurons in the CA1 after EW we performed Fluoro-Jade B staining. Ethanol withdrawal resulted in an appearance of bright green, Fluoro-Jade B-positive cells in the CA1, but not CA3 region of the hippocampus (Fig. 2), which was consistent with neuronal damage we have observed in that area.
To further analyze cell death two days after EW in the hippocampus we performed TUNEL staining. The number of TUNEL-positive cells increased 12-fold in the CA1 region as compared to mice fed ethanol-free diet (Fig. 2; p<0.05). These cells were characterized by condensed chromatin in DAPI staining (Fig. 2) which is consistent with apoptotic nucleus morphology (26). Again, cell death was specific to CA1 as we did not observe TUNEL-positive cells in other regions of the hippocampus (not shown).
To investigate which cell type is predominantly affected by EW we analyzed the phenotype of TUNEL-positive cells in wild-type animals by multiple immunohistochemistry. TUNEL-positive cells were co-labeled using antibodies against neuron-specific antigen NeuN and astrocyte-specific antigen glial fibrillary acidic protein (GFAP). We found that 74.4±9 % of dying cells were NeuN-positive and therefore of a neuronal lineage (Fig 3). 14.6±4 % of TUNEL-positive cells colocalized with GFAP and were identified as astrocytes (Fig 3). The remaining 11±6 % did not express either of the cell-type specific markers and remained unidentified. These results suggest that neurons are predominantly affected by EW in the hippocampus (p<0.001).
Figure 3. Glutamatergic neurons are predominant cell type affected by ethanol withdrawal in the hippocampus.
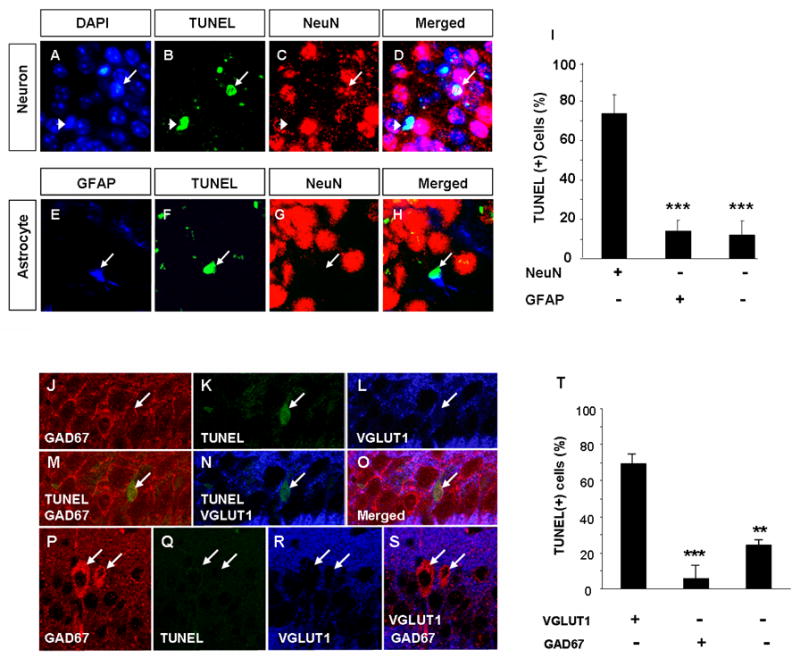
To investigate which cell type is primarily affected by EW we analyzed the phenotype of TUNEL-positive cells in the CA1 region of wild-type mice two days after withdrawal. To this end we performed co-labeling using antibodies against neuron-specific antigen NeuN and astrocyte-specific antigen glial fibrillary acidic protein (GFAP). Panels A-D show two TUNEL-positive cells with chromatin condensation visualized by the DAPI staining. One of these cells (arrowhead) colocalized with NeuN and therefore was identified as a neuron. The second cell (arrow) was NeuN-negative. Panels E-H show a GFAP-positive, damaged astrocyte (arrow). We found that 74.4% of dying cells were neurons (NeuN-positive) and 14.6% were astrocytes (GFAP-positive; quantified in I). Immunohistochemistry for VGLUT1 (a marker of excitatory neurons) and glutamic acid decarboxylase-67 (GAD67; a marker for inhibitory interneurons) in conjunction with TUNEL staining revealed that the majority of dying cells were VGLUT1-positive and GAD67-negative excitatory neurons (J-O). GAD67-positive cells (bright red in P as opposed to punctuate red GAD67-positive synaptic buttons found on most CA1 pyramidal neurons; see J) were TUNEL-negative (P-S). Quantification of the above results shown in T. *** p<0.001. The above estimation was performed using paraffin sections containing CA1 regions of four mice. The results are presented as mean ± SEM.
To determine the lineage of degenerating neurons we performed immunohistochemistry for VGLUT1 (a marker of excitatory neurons) and glutamic acid decarboxylase-67 (GAD67; a marker for inhibitory interneurons) in conjunction with TUNEL; Fig 3). The majority of GAD67-positive cells (distingished as bright red in contrast to punctuate red GAD67-positive synaptic buttons, found on most CA1 pyramidal neurons; Fig 3) were localized in stratum radiatum and were always TUNEL-negative. We found that only 5±6% of TUNEL-positive cells were GAD67-positive in stratum pyramidale of the CA1. In contrast, 70±5% of degenerating cells were VGLUT1-positive (p<0.001), which points to excitatory neurons as most vulnerable to EW.
To investigate the relationship between EW-induced neuronal death and activation of caspase-3, a critical effector protease promoting neurodegeneration, we performed immunohistochemistry (Fig 4). We found that TUNEL staining correlated well with high levels of active caspase-3 in the tissue after EW (96±6% of TUNEL-positive cells showed high caspase-3 levels; p<0.001), indicating an important role for caspases in promoting cell death in our model.
Figure 4. Ethanol withdrawal-induced neuronal death is caspase-dependent and accompanied by mitochondrial dysfunction.
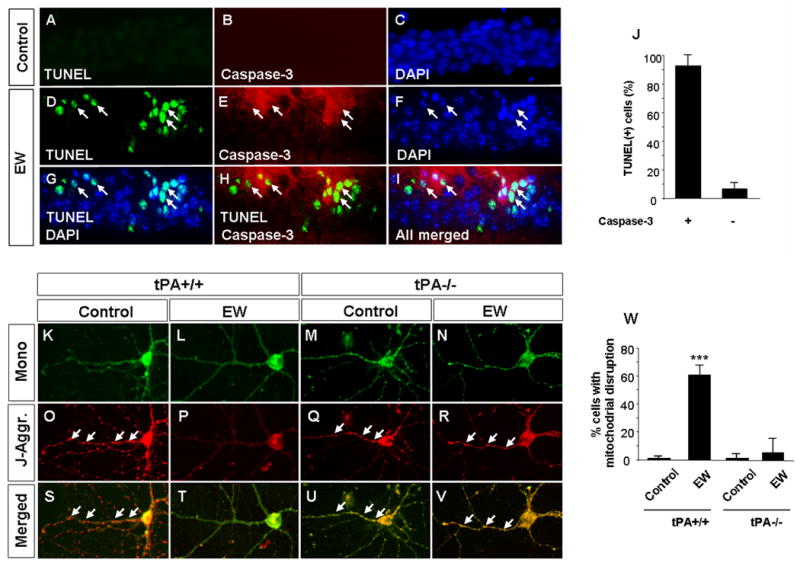
Upper panels Wild-type mice received either ethanol free diet (A-C) or ethanol containing diet followed by EW (D-I). Ethanol withdrawal resulted in neurodegeneration in the CA1 region of the hippocampus as indicated by the presence of TUNEL-positive cells (D, G, H, I). Double immunohistochemistry revealed that the majority of dying cells spatially and temporarily coincided with activation of caspase-3 (arrows in E, H, I and quantified in J). Lower panels. Wild-type (K, L, O, P, S, T) and tPA-/- (M, N, Q, R, U, V) hippocampal neurons were subjected to ethanol withdrawal and mitochondrial integrity was investigated by J-aggregate staining. EW resulted in mitochondrial disruption in wild-type neurons as evidenced by disappearance of red J-aggregates (P, T). In contrast, tPA-/- neurons were protected against EW-induced injury (arrows in R, V; quantified in W). *** p<0.001. The above experiments were repeated at least four times with similar results. The results are presented as mean ± SEM.
Neuronal death is often preceded by mitochondrial dysfunction leading to depolarization and protein leakage from these structures. To examine mitochondrial integrity after EW we subjected hippocampal neurons in culture to EW and visualized J-aggregates (Fig 4). EW resulted in a dramatic increase in the percentage of cells displaying signs of mitochondrial disruption (2±1% vs. 61±7% in control and EW groups, respectively; p<0.001) as evidenced by disappearance of J-aggregates.
EW-mediated increase in tPA activity in the hippocampus precedes neurodegeneration
High tPA activity has previously been associated with neuronal death (27, 28). To investigate whether hippocampal tPA activity correlated with EW-induced neurodegeneration we performed in situ zymography using brain sections of wild-type mice collected at various time-points during ethanol intoxication and/or EW. In line with our previous observations (11) extracellular tPA activity increased after 14 days of ethanol treatment, and even more dramatically during EW (Fig 1). This increased caseinolytic activity was due to tPA and not uPA, since it was not seen in tPA-/- mice (not shown).
EW-induced neurodegeneration is tPA- and plasmin-dependent
Since high tPA activity and seizures often correlate with cell death (10, 19, 27, 29, 30) we investigated if EW-induced neurodegeneration is tPA-dependent. To this end we induced EW in tPA-/- mice and analyzed their hippocampi two days later. Histological analysis of the CA1 region revealed that, unlike wild-type mice (Fig. 2; p<0.01), the number of cells in tPA -/- mice was not affected by EW (p>0.05). Similarly, we did not observe TUNEL-positive cells in CA1 region (Fig. 2; p>0.05) typically seen in wild-type mice after EW (p<0.05) or disruption of mitochondrial J-aggregates in tPA-deficient hippocampal neurons subjected to EW (Fig 4).
To investigate if plasminogen is necessary for EW-mediated neuronal death we subjected plasminogen -/- mice to EW and determined the number of cells in CA1 region. Plasminogen -/- mice that were fed ethanol-free diet served as control. Similarly to tPA-/- animals, we did not observe any decrease in the number of cells in the CA1 region as the result of EW in plasminogen -/- mice (Fig. 2; p>0.05). Moreover, TUNEL-positive cells were not seen in these animals (Fig. 2; p>0.05), which is consistent with the role of plasmin in EW-mediated neuronal death.
tPA and plasminogen do not affect ethanol levels, consumption or behavioral signs of intoxication
To investigate whether attenuation of EW-induced neuronal death in tPA-/- and plasminogen-/- mice can be attributed to reduced ethanol drinking we measured the volume of the EtOH diet consumed during the course of the 14-day intoxication. We did not find any differences in this parameter between wild-type animals and mice in which the tPA or plasminogen genes have been disrupted (Supplemental Fig 1; p>0.05). The amount of ethanol consumed on day 14 was 26.7±2 g/kg/d, 25.5±7 g/kg/d and 27.7±3 g/kg/d for wild-type mice, tPA-/- and plasminogen-/- mice, respectively and did not differ between the genotypes. Weight loss was similar during the course of ethanol intoxication between groups (Supplemental Fig 1) and reached the maximum of ∼20% at day 14.
To check if tPA deletion affects EtOH metabolism we measured hippocampal ethanol levels in wild-type and tPA-/- mice before EtOH diet was introduced, after 14 days of its consumption and 6 hours following its withdrawal. Hippocampal ethanol levels on day 14 amounted to 43±11 and 42±9 mM in wild-type and tPA-/- mice, respectively (Supplemental Fig 1; p>0.05 between the genotypes), and were consistent with those known to produce sedation (2, 8). These levels dropped sharply 6 hours after the ethanol has been withdrawn, and were similar in the wild-type and tPA-deficient animals (Supplemental Fig 1; p>0.05).
In spite of lack of differences in brain ethanol metabolism tPA-/- mice could still have subtle receptor/circuit anomalies that would render them resistant to behavioral effects of ethanol. To determine the effect of tPA and plasmin on EtOH-induced sedation we measured behavioral signs of ethanol intoxication in mice deficient in the above proteases and compared them with wild-type controls. We did not observe any differences in this parameter between wild-type tPA-/- or plasminogen-/- mice during the course of 14 days of ethanol administration (Supplemental Fig 1; p>0.05).
EW-induced neurodegeneration and its tPA-dependence can be mimicked in vitro
Many of the effects of EtOH in the central nervous system are mediated at the systems level and ethanol dependence is often considered an aberrant form of neuronal plasticity. This form of plasticity requires the presence of well-defined synaptic circuits that are modified by EtOH (31, 32). To investigate if the facilitation of EW-induced neurodegeneration by tPA was due to its effects at the circuit level or rather at the cellular level we subjected dissociated forebrain neurons to EtOH and induced EW. In wild-type neurons EW resulted in a 10-fold increase in the release of LDH, an indicator of degeneration, to the medium (Fig 5 lower panel) followed by a dramatic decrease in the cell number (Fig 5 upper panels). In contrast, neurons from mice deficient in tPA were resistant to EW-induced degeneration, as measured by either mitochondrial disruption (Fig 4), LDH release or cell loss (Fig 5).
Figure 5. tPA-/- neurons in culture are resistant to EW-induced neurodegeneration.
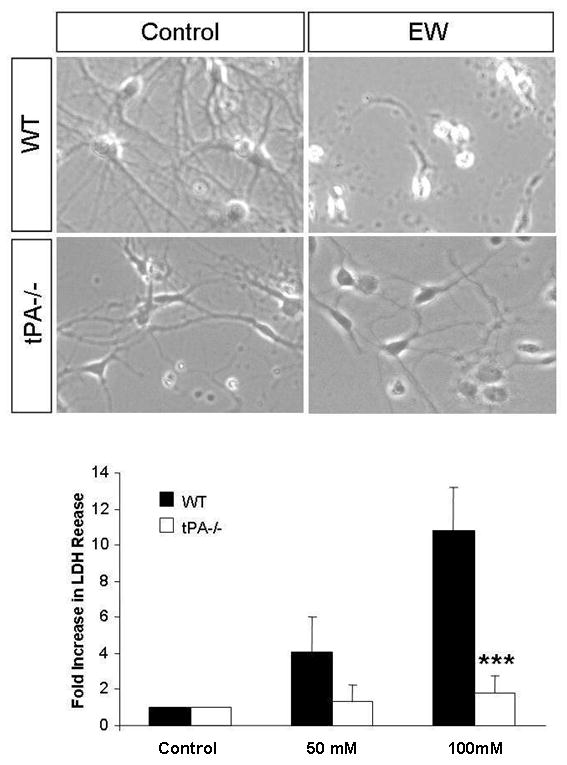
To determine whether ethanol mediates its' effects at the cellular or rather circuit level, we investigated the effects of EW on the survival of dissociated wild-type and tPA-/- hippocampal neurons in culture. Neurons were exposed to 50 or 100 mM ethanol for 3 days and subject to withdrawal. Bright field images of neurons exposed to 100mM ethanol revealed signs of cell death (rounded cell bodies, short neurites) in wild-type neurons undergoing EW, where as tPA-/- neurons were resistant to EW-induced neurodegeneration (upper panels). EW-induced neurodegeneration was quantified using the LDH assay (lower panel). Withdrawal from 50, and 100 mM ethanol resulted in a dose-dependent increase in LDH release from wild-type neurons (n=4) whereas tPA-/- neurons did not show a significant increase in LDH release (n=5). ***p<0.001. The results are presented as mean ± SEM.
EW-induced neurodegeneration correlates with tPA-dependent degradation of laminin
Chen et al. demonstrated that neurodegeneration after injection of an excitotoxin kainic acid is promoted by plasmin-catalyzed degradation of laminin (19). To investigate if EW-induced neurodegeneration correlates with laminin degradation we assessed laminin levels by immunohistochemistry in the hippocampal CA1 region of wild-type mice six hours after EW, when seizures were most pronounced. We observed a marked decrease in laminin levels surrounding neuronal bodies six hours after EW in the CA1 (Fig 6), the region that typically undergoes degeneration two days later (Fig 1 and 3). This result suggests that degradation of laminin could be responsible for EW-induced neuronal death.
Figure 6. EW-induced neurodegeneration correlates with tPA-dependent degradation of laminin.
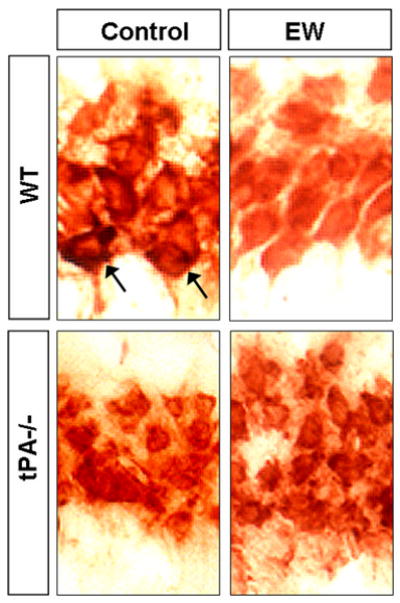
To investigate if EW-induced neurodegeneration correlates with laminin degradation we assessed laminin levels by immunohistochemistry in the hippocampal CA1 region of wild-type and tPA -/- mice during EW. EtOH-naïve mice of the same genotype served as controls. Laminin was localized mainly around cell bodies. In wild-type mice a marked decrease in laminin levels was observed six hours after EW in the CA1 (strong pericellular staining indicated by arrows), the region that degenerates two days later. Laminin degradation was not observed in tPA-/- mice, which were also resistant to neurodegeneration. These results suggest that degradation of laminin is tPA-dependent and could be responsible for EW-induced neuronal death. This experiment was repeated four times with similar results.
To investigate if EW-induced degradation of laminin was tPA-dependent we performed analysis of laminin levels in the hippocampus of tPA-/- mice with or without EW. We did not observe laminin degradation typically seen in the wild-type mice at the same time point after EW (Fig 6).
CA1-specific deletion of laminin γ-1 protects neurons from EW-induced degeneration
Degradation of laminin γ-1 chain, has previously been implicated in tPA/plasmin system-mediated neuronal death after kainic acid injection (33, 34). To investigate if laminin γ-1 was involved in EW-mediated cell death we generated mice in which the gene encoding laminin γ-1 chain has been floxed (F/F) and conditionally deleted in the CA1 region of the hippocampus (15). We treated these F/F Cre+ and their F/F Cre- control littermates with ethanol and two days after EW cell number was assessed. We found 12±2% decrease in the cell number in the CA1 region of F/F Cre- control mice two days after EW (Fig 7). On the other hand, the animals in which laminin γ-1 has been deleted were protected from EW-induced cell loss (p<0.05).
Figure 7. Conditional deletion of the laminin γ-1 gene from the CA1 region of the hippocampus protects neurons from EW-induced degeneration.
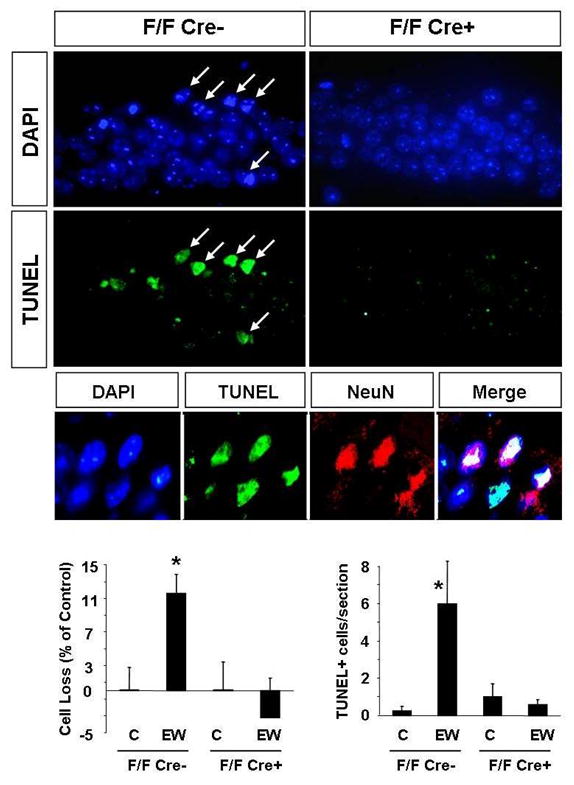
To delete laminin γ-1 in the CA1 region mice in which both copies of the laminin γ-1 gene has been floxed (F/F) were crossed with CaMKIIα-Cre transgenic mice (offspring referred to as F/F Cre+). Immunohistochemistry confirmed a complete absence of laminin γ-1 protein in the CA1 region of F/F Cre+ mice (Chen et al. manuscript in preparation). The F/F Cre- littermates served as controls. The mice were administered EtOH for 14 days and were sacrificed two days following EW. EtOH-naïve mice of the same genotype served as controls. Cell loss was determined by counting cell number in the CA1 region of the hippocampus using DAPI-stained sections. Cell death was further confirmed by visualizing broken DNA strands with the TUNEL method. Analysis of the CA1 region two days after EW revealed that the number of cells decreased in F/F Cre- mice but not in F/F Cre+ animals, in which laminin γ-1 has been deleted (upper panels and quantification in the lower panels). Similarly, we did not observe TUNEL-positive cells in CA1 region of F/F Cre+ mice, typically seen in F/F Cre- (middle panels and quantification in the lower panels) as well as C57/Bl6 animals (see Fig 2 and Fig 3) after EW. Most TUNEL-positive cells colocalized with the neuronal marker NeuN (small middle panels). Sections derived from EtOH-naïve F/F Cre+ and F/F Cre- mice did not show any signs of neurodegeneration and therefore are not shown, but are included in the quantification. * p<0.05; n=4-10 per group for each experiment. The results are presented as mean ± SEM.
To confirm that deletion of laminin γ-1 chain rendered neuroprotection we performed TUNEL staining using coronal brain sections collected two days after EW. We observed a dramatic increase in the number of TUNEL-positive cells in the CA1 region in F/F Cre- animals (to 6±2 cells per section; p<0.05; Fig 7), while hippocampi of the F/F Cre+ mice subjected to EW were TUNEL-negative. Similarly to C57/Bl6 mice (Fig 3) the majority of these TUNEL-positive cells were NeuN-positive, and were therefore identified as neurons (Fig 7).
These results indicate that the presence of laminin γ-1 during EW is necessary for cell death in the CA1 region of the hippocampus, and that it's degradation by plasmin may constitute an important mechanism mediating neurodegeneration in this region.
Discussion
One of the most common complications of alcoholism are degenerative changes observed in the course of ethanol dependence (4-6). EtOH-related degenerative changes are characterized by cell death, damage of neurites and synapses and of the myelin sheath in the cortex, hippocampus, thalamus and pons (4-6). However, depending on the dose of ethanol, route of its administration, the strain and genetic background of the animals used the above changes can be observed early after EtOH treatment or only after EW (20-24, 35-40). In our study EW was necessary to trigger cell death, which may reflect lower susceptibility of C57/BL6 mice to EtOH toxicity as compared to other strains (41) and it was limited to the CA1 region of the hippocampus. We found that EW predominantly affects excitatory neurons, as 74.4% of TUNEL-positive cells co-express neuronal marker NeuN and 70±5% of them were also VGLUT1-positive. These findings are in agreement with previous studies demonstrating neuronal death following EW both in animal models and in human alcoholics (4-6).
What is the mechanism of EW-induced neurodegeneration? Ethanol acts as an antagonist at excitatory NMDA receptors and at the same time stimulates inhibitory GABA(A) binding sites (8). Because of its pharmacological profile chronic administration of EtOH causes an adaptive upregulation of NMDA- and downregulation of GABA(A)-receptors. This creates a potential state of hyperexcitability temporarily inhibited by high doses of EtOH. However, upon cessation of drinking this predominance of excitatory transmission is unmasked, which results in EW seizures and cell death. There are numerous factors that can facilitate excitotoxic neuronal death further. One critical molecule is a serine protease tissue plasminogen activator (tPA). tPA is induced and released from neurons by various forms of neuronal activity (30, 42-44) including EW (11). We have previously demonstrated that tPA -/- mice are resistant to seizures and cell death after injection of kainic acid (19, 27, 45). We have also found that tPA interacts with NR2B, the ethanol-sensitive subunit of NMDA receptor, and promotes EW seizures (11). To investigate if tPA similarly promotes EW-induced neurodegeneration we administered EtOH to tPA-/- mice and determined cell death in the hippocampus upon its withdrawal. Mice or neurons deficient in tPA were resistant to EW-induced mitochondrial disruption and neurodegeneration as indicated by the presence of J-aggregates, cell number and lack of TUNEL-positive neurons. This finding is in line with the role of tPA in facilitating physical dependence to EtOH and promoting neuronal death in different animal models.
High seizure activity during EW can deteriorate brain damage and facilitate cell death (46-48). Considerable evidence suggest that EW-induced epileptic activity emanate from either brain stem or forebrain and are propagated and augmented within specific neuronal circuits (31). To investigate if the tPA-dependent element of EW-induced neurodegeneration is secondary to convulsions or whether the two phenomena can be separated we eliminated the neuronal network component. Dissociated mixed forebrain neuronal/astrocytic cultures were treated with EtOH and EW was initiated. Faster development of ethanol dependence in vitro compared to the in vivo model may be due to continuous EtOH exposure (no circadian variations in ethanol intake which is typical for mouse models) and limited potential for compensatory negative feedback from inhibitory interneurons. We have found that in the wild-type cultures neuronal death was still observed, suggesting that EW-induced neurodegeneration has a cellular component independent of the epileptic circuitry. Deletion of the tPA gene completely prevented cell death in vitro. This confirmed our in vivo results pointing to the role of tPA as a critical factor facilitating EW-induced neuronal death.
tPA can use several mechanisms to exert its effects in the nervous system. These involve direct or indirect interactions with NMDA receptors (11, 29, 49), LRP receptors (50), activation of BDNF (51) and MMP-9 (52). One well established substrate for tPA is plasminogen which it activates to a broad spectrum protease, plasmin. To investigate whether tPA-mediated neurodegeneration following EW is plasmin-dependent we assessed cell death in plasminogen -/- mice. We found that these mice were resistant to neuronal death after EW suggesting that the effect of tPA is mediated by activation of plasminogen. This result is in line with our previous findings pointing to the role of plasmin in the central nervous system injury after kainic acid injection (19, 27, 45). However, in our previous paper we found that the facilitating effect of tPA on EW seizures was plasminogen-independent (11). Our results collectively suggest that the mechanism of EW-seizures and EW-induced neurodegeneration can be genetically dissected by the deletion of the plasminogen gene and that both plasminogen-dependent (cleavage of laminin) and plasminogen-independent (interaction with NR2B subunit of NMDA receptor) play different albeit complementary roles in promoting EW-induced seizures and neuronal death.
What could be the mechanism that the tPA/plasmin system uses to promote EW-induced cell death? One well-characterized substrate for plasmin is an extracellular matrix protein, laminin (19, 33). Through its interactions with membrane-bound integrins laminin provides means of communication and attachment between the cell and its outside surroundings. We have previously shown that degradation of laminin by plasmin during excitotoxic insult triggers cell death (19). We found here that, similarly to kainic acid injection, laminin is degraded in hippocampal regions that eventually undergo degeneration. Laminin degradation occurred six hours after EW was initiated, when tPA activity was the highest and seizures were most pronounced. Both laminin degradation and neurodegeneration were prevented by the deletion of the tPA gene. This result strongly suggests a correlation between the tPA/plasmin-mediated loss of laminin and EW-induced neuronal death.
If EW-induced neurodegeneration was caused by a loss of laminin support, then deletion of laminin should sensitize mice towards EW-mediated insult. That was not the case. Deletion of laminin γ-1, a primary substrate for plasmin, rendered CA1 neurons resistant to degeneration. What could be the reason for such an effect? One possibility is that the presence of laminin γ-1 could ensure proper functioning of some cellular mechanisms involved in neurodegeneration, such as correct membrane placement, trafficking or anchoring of excitatory ion channels. Another possibility is that laminin degradation products, released after EW, could facilitate neurodegeneration. We have recently reported that laminin degradation products are essential for upregulation of kainate receptor subunit KA1 in the hippocampus in response to excitotoxic injury (53). Whether similar mechanisms operate after EW requires further investigation.
In summary, we have found that plasmin-mediated degradation of laminin is critical for neurodegeneration after EW, a common complication seen in alcoholics (see Fig 8 for summary). Consequently, deletion of the tPA, plasminogen or laminin γ-1 genes prevents EW-induced neuronal loss in the hippocampus. These findings add to our knowledge of the mechanism of EW and may contribute towards developing better therapies against central nervous system injury.
Figure 8. A proposed model of EW-induced neurodegeneration.

In basal conditions tPA is stored intracellularly, and is released into the extracellular space following EtOH administration (see Fig 1). During intoxication, EtOH (gray circles) blocks NMDA receptors (black circles) and together with extracellular tPA (see (11) is responsible for their upregulation. Abrupt withdrawal of EtOH leaves NMDA receptors unblocked, and the resulting hyperexcitability causes further release of tPA from neurons (see Fig 1). High extracellular levels of tPA activate plasminogen to plasmin, which in turn degrades a component of extracellular matrix, laminin (see Fig 6). Laminin γ-1 degradation renders neurons more sensitive to damaging effects of EW, which results in caspase-3-dependent cell death, while the absence of laminin γ-1 is protective (see Fig 7).
Supplementary Material
Acknowledgments
This work was supported by 5R01AA014630-03 and 5R01NS038472-09 grants from NIAAA and NINDS to S.S, MRC Project Grant (G0500231/73852) and a Marie Curie Excellence Grant (MEXT-CT-2006-042265 from European Commission) to R.P. The authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.NIAAA. Updating Estimates of the Economic Costs of Alcohol Abuse in the United States. National Institute on Alcohol Abuse and Alcoholism 2000 [Google Scholar]
- 2.Tabakoff B, Hoffman PL. Alcohol addiction: an enigma among us. Neuron. 1996;16:909–912. doi: 10.1016/s0896-6273(00)80113-0. [DOI] [PubMed] [Google Scholar]
- 3.Diamond I, Gordon AS. Cellular and molecular neuroscience of alcoholism. Physiol Rev. 1997;77:1–20. doi: 10.1152/physrev.1997.77.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Charness ME. Brain lesions in alcoholics. Alcohol Clin Exp Res. 1993;17:2–11. doi: 10.1111/j.1530-0277.1993.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 5.Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5:73–78. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Lovinger DM. Excitotoxicity and alcohol-related brain damage. Alcohol Clin Exp Res. 1993;17:19–27. doi: 10.1111/j.1530-0277.1993.tb00720.x. [DOI] [PubMed] [Google Scholar]
- 7.Grant KA. Emerging neurochemical concepts in the actions of ethanol at ligand-gated ion channels. Behav Pharmacol. 1994;5:383–404. doi: 10.1097/00008877-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Lovinger DM. Alcohols and neurotransmitter gated ion channels: past, present and future. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:267–282. doi: 10.1007/pl00005051. [DOI] [PubMed] [Google Scholar]
- 9.Hall W, Zador D. The alcohol withdrawal syndrome. Lancet. 1997;349:1897–1900. doi: 10.1016/S0140-6736(97)04572-8. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman PL, Grant KA, Snell LD, Reinlib L, Iorio K, Tabakoff B. NMDA receptors: role in ethanol withdrawal seizures. Ann N Y Acad Sci. 1992;654:52–60. doi: 10.1111/j.1749-6632.1992.tb25955.x. [DOI] [PubMed] [Google Scholar]
- 11.Pawlak R, Melchor JP, Matys T, Skrzypiec AE, Strickland S. Ethanol-withdrawal seizures are controlled by tissue plasminogen activator via modulation of NR2B-containing NMDA receptors. Proc Natl Acad Sci U S A. 2005;102:443–448. doi: 10.1073/pnas.0406454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 13.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. doi: 10.1101/gad.9.7.794. [DOI] [PubMed] [Google Scholar]
- 14.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995;92:2585–2593. doi: 10.1161/01.cir.92.9.2585. [DOI] [PubMed] [Google Scholar]
- 15.Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163:889–899. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malinowska B, Napiorkowska-Pawlak D, Pawlak R, Buczko W, Gothert M. Ifenprodil influences changes in mouse behaviour related to acute and chronic ethanol administration. Eur J Pharmacol. 1999;377:13–19. doi: 10.1016/s0014-2999(99)00393-3. [DOI] [PubMed] [Google Scholar]
- 17.Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 20.Arendt T, Allen Y, Sinden J, Schugens MM, Marchbanks RM, Lantos PL, Gray JA. Cholinergic-rich brain transplants reverse alcohol-induced memory deficits. Nature. 1988;332:448–450. doi: 10.1038/332448a0. [DOI] [PubMed] [Google Scholar]
- 21.Riley JN, Walker DW. Morphological alterations in hippocampus after long-term alcohol consumption in mice. Science. 1978;201:646–648. doi: 10.1126/science.566953. [DOI] [PubMed] [Google Scholar]
- 22.Walker DW, Barnes DE, Zornetzer SF, Hunter BE, Kubanis P. Neuronal loss in hippocampus induced by prolonged ethanol consumption in rats. Science. 1980;209:711–713. doi: 10.1126/science.7394532. [DOI] [PubMed] [Google Scholar]
- 23.Cadete-Leite A, Tavares MA, Paula-Barbosa MM. Alcohol withdrawal does not impede hippocampal granule cell progressive loss in chronic alcohol-fed rats. Neurosci Lett. 1988;86:45–50. doi: 10.1016/0304-3940(88)90180-2. [DOI] [PubMed] [Google Scholar]
- 24.Pawlak R, Skrzypiec A, Sulkowski S, Buczko W. Ethanol-induced neurotoxicity is counterbalanced by increased cell proliferation in mouse dentate gyrus. Neurosci Lett. 2002;327:83–86. doi: 10.1016/s0304-3940(02)00369-5. [DOI] [PubMed] [Google Scholar]
- 25.Phillips SC, Cragg BG. Chronic consumption of alcohol by adult mice: effect on hippocampal cells and synapses. Exp Neurol. 1983;80:218–226. doi: 10.1016/0014-4886(83)90018-3. [DOI] [PubMed] [Google Scholar]
- 26.Saraste A. Morphologic criteria and detection of apoptosis. Herz. 1999;24:189–195. doi: 10.1007/BF03044961. [DOI] [PubMed] [Google Scholar]
- 27.Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 28.Tsirka SE, Rogove AD, Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- 29.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 30.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 31.Faingold CL, N'Gouemo P, Riaz A. Ethanol and neurotransmitter interactions--from molecular to integrative effects. Prog Neurobiol. 1998;55:509–535. doi: 10.1016/s0301-0082(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 32.Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, Merlo-Pich E, Weiss F. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- 33.Chen ZL, Indyk JA, Strickland S. The hippocampal laminin matrix is dynamic and critical for neuronal survival. Mol Biol Cell. 2003;14:2665–2676. doi: 10.1091/mbc.E02-12-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Indyk JA, Chen ZL, Tsirka SE, Strickland S. Laminin chain expression suggests that laminin-10 is a major isoform in the mouse hippocampus and is degraded by the tissue plasminogen activator/plasmin protease cascade during excitotoxic injury. Neuroscience. 2003;116:359–371. doi: 10.1016/s0306-4522(02)00704-2. [DOI] [PubMed] [Google Scholar]
- 35.Arendt T, Schugens MM, Bigl V. The cholinergic system and memory: amelioration of ethanol-induced memory deficiency by physostigmine in rat. Acta Neurobiol Exp (Wars) 1990;50:251–261. [PubMed] [Google Scholar]
- 36.Bengoechea O, Gonzalo LM. Effects of alcoholization on the rat hippocampus. Neurosci Lett. 1991;123:112–114. doi: 10.1016/0304-3940(91)90170-x. [DOI] [PubMed] [Google Scholar]
- 37.Beracochea D, Lescaudron L, Verna A, Jaffard R. Neuroanatomical effects of chronic ethanol consumption on dorsomedial and anterior thalamic nuclei and on substantia innominata in mice. Neurosci Lett. 1987;73:81–84. doi: 10.1016/0304-3940(87)90035-8. [DOI] [PubMed] [Google Scholar]
- 38.Cadete-Leite A, Tavares MA, Pacheco MM, Volk B, Paula-Barbosa MM. Hippocampal mossy fiber-CA3 synapses after chronic alcohol consumption and withdrawal. Alcohol. 1989;6:303–310. doi: 10.1016/0741-8329(89)90087-6. [DOI] [PubMed] [Google Scholar]
- 39.Collins MA, Corso TD, Neafsey EJ. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcohol Clin Exp Res. 1996;20:284–292. doi: 10.1111/j.1530-0277.1996.tb01641.x. [DOI] [PubMed] [Google Scholar]
- 40.Lescaudron L, Seguela P, Geffard M, Verna A. Effects of long-term ethanol consumption on GABAergic neurons in the mouse hippocampus: a quantitative immunocytochemical study. Drug Alcohol Depend. 1986;18:377–384. doi: 10.1016/0376-8716(86)90102-x. [DOI] [PubMed] [Google Scholar]
- 41.Schauwecker PE. Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol. 2002;178:219–235. doi: 10.1006/exnr.2002.8038. [DOI] [PubMed] [Google Scholar]
- 42.Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 43.Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci. 1996;16:2220–2225. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- 45.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukoyanov NV, Sa MJ, Madeira MD, Paula-Barbosa MM. Selective loss of hilar neurons and impairment of initial learning in rats after repeated administration of electroconvulsive shock seizures. Exp Brain Res. 2004;154:192–200. doi: 10.1007/s00221-003-1658-3. [DOI] [PubMed] [Google Scholar]
- 47.Park JH, Cho H, Kim H, Kim K. Repeated brief epileptic seizures by pentylenetetrazole cause neurodegeneration and promote neurogenesis in discrete brain regions of freely moving adult rats. Neuroscience. 2006;140:673–684. doi: 10.1016/j.neuroscience.2006.02.076. [DOI] [PubMed] [Google Scholar]
- 48.Rao MS, Hattiangady B, Reddy DS, Shetty AK. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J Neurosci Res. 2006;83:1088–1105. doi: 10.1002/jnr.20802. [DOI] [PubMed] [Google Scholar]
- 49.Matys T, Strickland S. Tissue plasminogen activator and NMDA receptor cleavage. Nat Med. 2003;9:371–372. 372–373. doi: 10.1038/nm0403-371. author reply. [DOI] [PubMed] [Google Scholar]
- 50.Zhuo M, Holtzman DM, Li Y, Osaka H, DeMaro J, Jacquin M, Bu G. Role of tissue plasminogen activator receptor LRP in hippocampal long- term potentiation. J Neurosci. 2000;20:542–549. doi: 10.1523/JNEUROSCI.20-02-00542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Lee SR, Arai K, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003 doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 53.Yu H, Chen Z, Yu WM, Barker-Carlson K, Strickland S. Laminin-dependent up-regulation of KA1 receptor expression promotes excitotoxic neuronal death. Society for Neuroscience Abstracts. 2007 604.6/EE16. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.