

Abstract
Background
Opioid receptors are clinically important targets for both pain and alcohol abuse. Three opioid receptors have been cloned: μ, δ, and κ, all of which effect alcohol consumption in animal models. Naltrexone is a non-selective opioid antagonist used for alcoholism, whose clinical utility is limited by poor efficacy and adverse side-effects. Here, we demonstrate that the therapeutic limitations of naltrexone may reflect its poor selectivity. Despite decades of research, several mysteries surround the pharmacology of these receptors. For example, in vivo two pharmacologically-defined subtypes of δ receptors exist.
Methods
Effects of δ subtype-selective ligands (naltrindole, naltriben, TAN-67, 7-benzylidenenaltrexone) were measured on ethanol consumption in C57BL/6 wild-type and opioid receptor knock-out mice using a limited access 2-bottle choice paradigm. Affinity and efficacy of naltriben, 7-benzylidenenaltrexone and TAN-67 was measured in vitro using radioligand binding and Ca2+-mobilizationa assays.
Results
We show that the subtypes of the δ receptor, δ1 and δ2, have opposing effects on ethanol consumption. We find that these effects are synergistic; thereby suggesting that δ1 and δ2 receptors are distinct molecular targets. Indeed, we provide both in vitro as well as in vivo evidence that the δ1 subtype is a μ-δ heterodimer and that the δ2 subtype is most likely a δ homomer.
Conclusions
Together, these data provide insight into the limited actions of the clinically-important drug naltrexone, and identify a novel target with improved specificity and efficacy for the development of new therapeutics for the treatment of alcoholism.
Keywords: GPCR, delta opioid receptor, subtypes, alcoholism, ethanol, heterodimerization
Introduction
Excessive ethanol consumption and alcohol addiction are a serious threat to society, both socially as well as economically. The involvement of opioid receptors in ethanol consumption, reward and dependence has long been known (1). The opioid family consists of three members: the μ-opioid (MOR) δ-opioid (DOR), and κ-opioid (KOR) receptors (2). It is well established that blockade of the opioid system with antagonists can reduce ethanol consumption (3). Indeed, naltrexone (Revia™), a non-selective opioid antagonist, is one of only three drugs approved by the FDA for the treatment of alcoholism. However, naltrexone is not universally effective as it predominantly benefits heavy drinkers (4; 5), shows very poor compliance, and causes significant side effects, including somnolence and vomiting. Importantly, it is not entirely clear which opioid receptor(s) mediate the beneficial effects of naltrexone on reduced drinking, and which mediate the side effects.
Each of the opioid receptors influences drinking in animals models. Mice with a disruption of either the MOR (6) or KOR (7) show a decreased preference for ethanol. However, the role of the DOR in alcohol intake is less clear. Mice with a disruption in the DOR show no difference in ethanol consumption when alcohol naïve (8); however, they do show an enhancement in ethanol consumption after being exposed to ethanol for some time (8). Whereas only one DOR gene has been cloned (9; 10), pharmacologically, two DOR receptors isoforms have been identified in vivo: DOR1s are activated by [Tyr-d-Pen-Gly-p-Chloro-Pen-d-Pen]-Enkephalin (DPDPE) and antagonized by 7-benzylidenenaltrexone (BNTX), while DOR2s are activated by deltorphin II and antagonized by naltriben (NTB) (11; 12). Naltrexone (NTX) as well as the non-selective DOR ligand naltrindole (NTI) can antagonize both DOR1 and DOR2.
Non-subtype selective antagonists at the DOR, including ICI 174,864 and NTI, as well as the DOR2-selective antagonist NTB, have been shown to decrease ethanol consumption in some studies (13-18), whereas others have found no effect of these drugs (19-23). Paradoxically, as mentioned above, disruption of the DOR gene has been shown to increase drinking in some paradigms. Together, these contradictory findings led us to hypothesize that DOR1 and DOR2s could have opposing effects on ethanol consumption. Importantly, no studies have examined either the specific role of DOR1 in ethanol consumption or the relative role of DOR1s versus DOR2s in drinking. In addition, there has been little insight into the molecular nature of the DOR1 and DOR2 subtypes, which cannot be recapitulated in systems where the DOR gene is heterologously expressed (24; 25).
Based on the observations of several groups that opioid receptors can form heterodimeric complexes with altered pharmacologies (26; 27), we postulated that heterodimerization of the DOR receptor with either the MOR or KOR could produce the pharmacologically-distinct DOR1 or DOR2 subtype. Here, we show that the DOR1 and DOR2 have opposing effects on ethanol consumption in C57BL/6 mice. Furthermore, utilizing mice disrupted for each of the opioid receptors, we demonstrate that the effects of the DOR1 on drinking require both the DOR and the MOR gene, consistent with the hypothesis that the DOR1 is a MOR/DOR heterodimer. Interestingly, functionality of DOR2 only requires DOR, suggesting these receptors are most likely DOR homomers.
Materials and methods
Limited access drinking paradigm
To study the effects of DOR ligands on voluntary ethanol or sucrose consumption a 2-bottle choice drinking paradigm was employed (see supplemental material for a detailed description of the method).
Locomotor activity assay
To determine the effect of DOR subtype-selective drugs on mouse locomotion, a locomotor activity assay was performed (see supplemental material for a detailed description of the method)
Blood alcohol concentration
Blood alcohol concentrations were measured in mice as previously described (28) immediately after the 4 hour ethanol access period had ended, or after an intragastric gavage (i.g.) or intra peritoneal (i.p.) injection to study ethanol intake, uptake and metabolism, respectively (see supplemental material for a detailed description of the method).
Receptor biotinylation and serial immunoprecipitation
To determine whether DOR and MOR interact with each other on the cell surface in vitro, a serial immunoprecipitation study was performed (see supplemental material for a detailed description of the method). Receptor heterodimers and homomers were immunoprecipitated either from lysates generated from cells co-expressing MOR and DOR (Fig. 7A, MOP/DOP co-express) or from lysates generated from cells expressing only MOR and only DOR that were mixed together after lysis. Receptors were immunoblotted for DOR expression as previously described (29).
Figure 7. HEK-293 cells co-expressing DOR and MOR display a DOR1 phenotype.
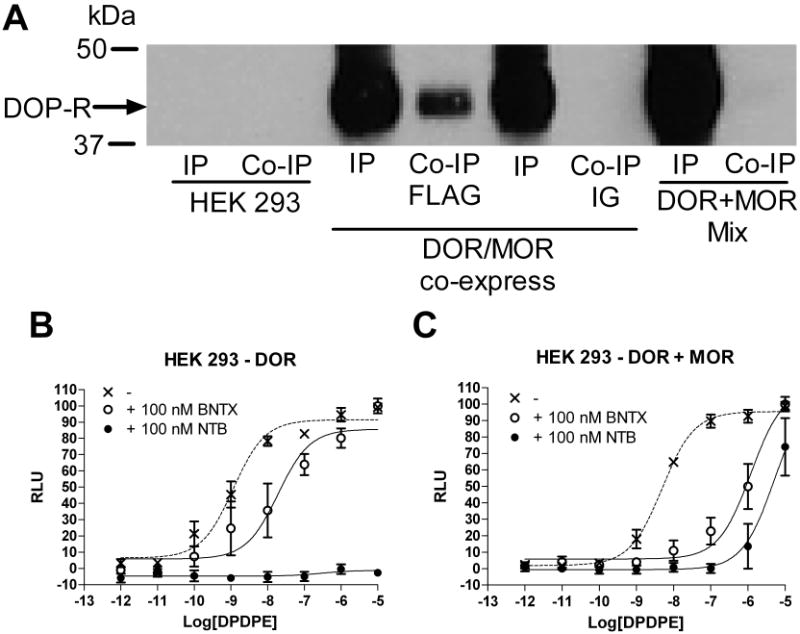
A, Biotinylated, surface DORs co-immunoprecipitate with surface MORs in HEK 293 cells co-expressing DOR and MOR (IP = immunoprecipitate = DOR homomers, Co-IP = co-immunoprecipitate = DOR-MOR heterodimers, DOP/MOP co-express). Receptors did not co-immunoprecipitate when lysates from cells expressing MOR alone were mixed with lysate from cells expressing DOR alone (DOP + MOP mix) or when non conjugated IG beads (IG) were used instead of FLAG conjugated beads (FLAG) see supplementary methods. B, C, Inhibition of DPDPE-induced calcium mobilization (dotted line) by the DOR1 antagonist BNTX (100 nM, open circles) or the DOR2 antagonist NTB (100 nM, closed circles) in HEK 923 cells expressing DOR (B) alone or co-expressing DOR and MOR (C).
[3H]DPDPE binding studies
To study the affinities of DOR subtype-selective ligands in vitro, [3H]DPDPE binding on HEK 293 cells stably expressing DOR, MOR alone or expressing DOR and MOR together was performed in triplicate as previously described (30, see supplemental material).
Calcium mobilization assay
To study the functional response of DOR subtype-selective ligands in vitro, Ca2+-mobilization was measured in triplicate in HEK-293 cells stably expressing DOR or co-expressing DOR and MOR as previously described (29, see supplemental material).
Condition place preference assay
The rewarding properties of TAN-67 were assessed using a condition placed preference paradigm (see supplemental material for a detailed description of the method).
Drugs
Ethanol solutions were prepared in tap water using 95% (vol/vol) ethanol (Gold Shield Chemical Co., Hayward, CA, USA). Naltriben mesylate (NTB), SB205607 dihydrobromide (TAN-67) and 7-benzylidenenaltrexone maleate (BNTX) were purchased from Tocris (MO, USA). Naltrindole hydrochloride (NTI), naltrexone hydrochloride (NTX), β-nicotinamide and alcohol dehydrogenase were purchased from Sigma-Aldrich (MO, USA). All compounds were dissolved in saline, with the exception of BNTX and NTB, which were dissolved in 5% DMSO. All drugs were prepared immediately prior to injection and were administered s.c. at a volume of 10 ml/kg.
Results
Non-selective opioid antagonists have a minimal effect on ethanol consumption, but naltriben, a DOR2-selective antagonist decreases ethanol consumption
Two groups of 9 male C57BL/6 mice were conditioned to prefer a 10% ethanol solution over water in a limited access drinking paradigm (see methods). After 15 training sessions (3 weeks), mice showed a stable 75% preference for ethanol over water. Whereas a dose of 1.5 mg/kg of the non-selective opioid antagonist NTX resulted in a decrease in ethanol consumption [F(3,40) = 13.39, p <0.0001], a relatively high dose (5 mg/kg), which likely antagonizes both MORs and DORs, had only moderate effects on the drinking behavior of the mice (Figure 1). The non-selective DOR antagonist NTI (10 or 15 mg/kg) did not alter either ethanol consumption or preference [F(3,40) = 1.89, p = 0.13] (Figure 1). In contrast, we found that the DOR2-selective antagonist NTB, dose dependently decreased both ethanol consumption [F(3,32) = 8.06, p = 0.0004] and ethanol preference [F3,32) = 3.476, p = 0.027] in C57BL/6 mice (Figure 1), consistent with the effects of this drug previously reported for rats (15).
Figure 1. Non-selective opioid antagonists have only a moderate effect on ethanol consumption compared to the selective DOR2 antagonist naltriben.
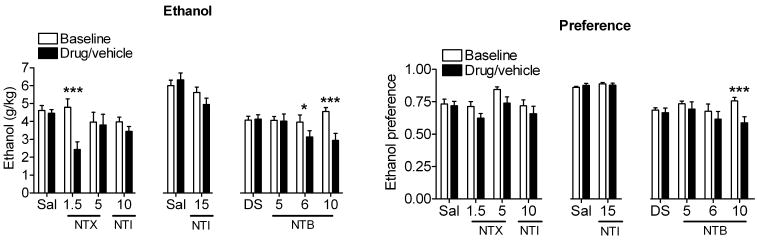
C57BL/6 mice (3× n=9) were injected s.c. with vehicle (5% DMSO or saline), 1.5 or 5 mg/kg of the non-selective opioid antagonist naltrexone (NTX), 10 or 15 mg/kg of the DOR1/R2-selective antagonist naltrindole (NTI), 5, 6 or 10 mg/kg of the DOR2-selective antagonist naltriben (NTB). Thirty minutes after injection, ethanol and water consumption were measured over a 4 hour period. Ethanol preference = ethanol consumption/(ethanol consumption + water consumption). (*p<0.05; **p<0.01).
The DOR1 agonist TAN-67 decreases ethanol consumption in C57BL/6 mice
Since antagonism of DOR2 decreased ethanol consumption but combined antagonism of DOR1 and DOR2 with NTI had no effect, we hypothesized that selective antagonism of DOR1 might promote drinking. Conversely, selective activation of DOR1 should decrease ethanol consumption. To examine this possibility, we tested the effect of the DOR1-selective antagonist BNTX and the DOR1-selective agonist TAN-67 on ethanol consumption. BNTX (5 or 10 mg/kg) did not decrease ethanol consumption [F(2,24) = 0.68, p = 0.51] or preference [F(2,24) = 0.37, p = 0.69] (Figure 2), unlike the DOR2-selective antagonist NTB (Figure 1). In contrast, administration of the DOR1 agonist TAN-67 caused a dose-dependent decrease in ethanol consumption [F(2,24) = 7.05, p = 0.0039] and preference [F(2,24) = 10.41, p = 0.0006] (Figure 2). The absence of any increase in drinking with BNTX could reflect that, in these mice, the level of ethanol consumption may have reached a maximally reinforcing or physiologically tolerable level.
Figure 2. The DOR1 agonist TAN-67 decreases ethanol consumption.
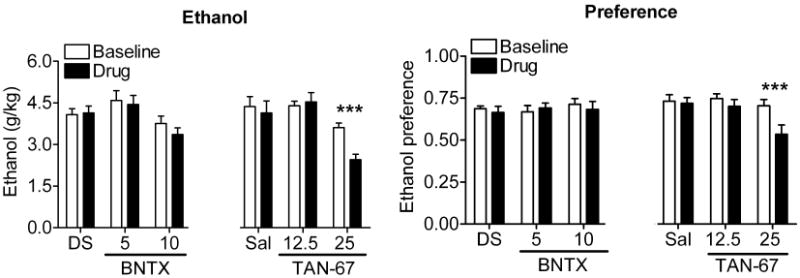
C57BL/6 mice (2× n=9) were injected s.c. with vehicle (5% DMSO or saline), 5 or 10 mg/kg of the DOR1-selective antagonist BNTX, 12.5 mg/kg or 25 mg/kg of the DOR1-selective agonist TAN-67. Thirty minutes after injection, ethanol and water consumption were measured over a 4 hour period. Ethanol preference = ethanol consumption/(ethanol consumption + water consumption). (*p<0.05; **p<0.01).
TAN-67 and NTB do not affect locomotion, sucrose consumption, ethanol uptake and ethanol metabolism and decreases blood alcohol concentration
Opioids are known to affect locomotor activity (31). Therefore, it was possible that the effects of TAN-67 and NTB on ethanol consumption were secondary to the effects on locomotion. However, mice injected with 25 mg/kg TAN-67 or 6 mg/kg NTB did not show any significant difference in locomotor activity compared to vehicle treated animals [F(3,28) = 1.15, p = 0.34] (Figure 3A), suggesting this is not the case. In addition, neither TAN-67 [F(1,16) = 0.057, p = 0.81] nor NTB [F(1,16) = 0.62, p = 0.44] had an affect on sucrose intake (Figure 3B), demonstrating that the effect on consumption was selective to ethanol but not sucrose, a “natural” reward. In both the locomotor assay [F(3,28)=1.155, p=0.40] and the sucrose intake assay [F(3,32)=0.3121, p=0.82] no significant difference between saline and DMSO treated groups was observed. To confirm that TAN-67 and NTB reduced physiologically relevant measures of ethanol intoxication, we measured the blood alcohol concentration (BAC) of mice (2 groups each n=9) treated with TAN-67 or NTB compared to controls. We observed a significant reduction in the BAC in the mice treated with 25 mg/kg TAN-67 or 6 mg/kg NTB [F(2,28) = 14.85, p <0.0001](Figure 3C). Importantly, neither TAN-67 nor NTB significantly affect ethanol uptake from the gut after oral gavage with 1.5 g/kg ethanol [F(3,26) = 2.509, p=0.084] (Supplemental Figure 4A). We next examined whether TAN-67 or NTB decreased the BAC in mice by affecting ethanol metabolism. Mice were treated s.c. with 25 mg/kg TAN-67, 6 mg/kg NTB or vehicle 30 minutes prior to an intra peritoneal (i.p.) injection of 2.5 g/kg ethanol. No differences in the metabolic rate was observed between vehicle and TAN-67 [F(1,8) = 0.087, p = 0.59] or NTB treated mice [F(1,8) = 0.087, p = 0.78] (Supplemental Figure 4B).
Figure 3. The DOR1 agonist TAN-67 does not affect locomotion or sucrose preference and decreases blood alcohol concentration without inhibiting ethanol uptake.
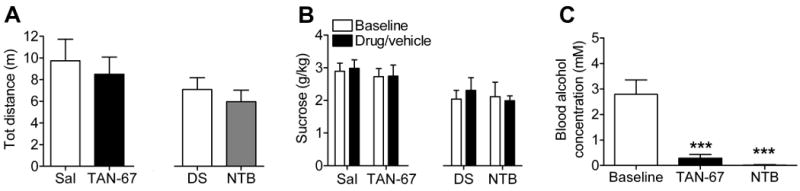
(A) C57BL/6 mice (n=8) were injected s.c. with vehicle (saline or 5% DMSO), or 25 mg/kg of the DOR1 agonist TAN-67 or 6 mg/kg of the DOR2 antagonist NTB. Thirty minutes after injection locomotor activity (distance traveled) was measured for four hours. (B) C57BL/6 mice (n=9) trained to prefer sucrose were injected s.c. with vehicle (saline or 5% DMSO), 25 mg/kg TAN-67 or 6 mg/kg NTB. Thirty minutes after injection sucrose consumption was measured over a 4 hour period. (C) C57BL/6 mice were injected s.c. with vehicle (saline or 5% DSMO) 25 mg/kg TAN-67 or 6 mg/kg NTB. Thirty minutes after injection mice (n=9) were exposed to ethanol and water consumption for a 4 hour period in a 2-bottle choice paradigm and blood alcohol levels (BAC) were measured immediately afterwards. (***p<0.001).
TAN-67 and NTB act at distinct receptor sites to reduce ethanol consumption
Because the DOR1 and DOR2 are defined solely by pharmacology, we next examined whether TAN-67 and NTB decreased ethanol consumption by acting at different receptor sites. As expected, the effect of TAN-67 (25 mg/kg) on ethanol consumption was blocked by 10 mg/kg of the DOR1 antagonist BNTX [F(4.38) = 4.89, p = 0.0028] (Figure 4A). However, co-administration of TAN-67 (25 mg/kg) together with the DOR2 antagonist NTB (10 mg/kg) caused a greater decrease in drinking than either drug alone [F(4,38) = 24.29, p < 0.0001] (Figure 4B). Furthermore, low doses of NTB (5 mg/kg) and TAN-67 (12.5 mg/kg), that by themselves had no effect on ethanol consumption, caused a significant decrease in ethanol consumption when they were co-administered [F(4,40) = 3.179, p = 0.023] (Figure 4C). Together, these data suggest that TAN-67 and NTB are acting on distinct targets to modulate ethanol intake.
Figure 4. The DOR1 antagonist BNTX blocks the effect of TAN-67, whereas the DOR2 antagonist NTB enhances the effect of TAN-67.

(A) C57BL/6 mice (n=9) were injected s.c. with vehicle (5% DMSO or saline), 25 mg/kg TAN-67, 10 mg/kg BNTX or 25 mg/kg TAN-67 + 10 mg/kg BNTX. (B) C57BL/6 mice (n=9) were injected s.c. with vehicle (5% DMSO or saline) 10 mg/kg NTB, 25 mg/kg TAN-67 or NTB + TAN-67. (C) C57BL/6 mice (n=9) were injected s.c. with vehicle (5% DMSO or saline) 5 mg/kg NTB, 12.5 mg/kg TAN-67 or NTB + TAN67. Thirty minutes after injection ethanol consumption was measured over a 4 hour period. (*p<0.05; **p<0.01).
Demonstration that DOR1 activity requires a functional MOR
We next examined the molecular mechanism underlying the distinct actions of the DOR1 and DOR2s in drinking. We favored the hypothesis that the DOR1 could be a heterodimer of the DOR and another opioid receptor (see introduction). To examine this possibility, we generated C57BL/6 mice with disruptions in the DOR, MOR or KOR gene (Supplemental Figure 1). Knock-out (KO) mice were trained to prefer ethanol using the same paradigm that was employed for the wild-type mice. As previously reported (7; 8), KOR KO mice showed a decrease in ethanol consumption, whereas mice with a disruption of the DOR displayed an increased consumption of ethanol compared to wild type (WT) mice (Supplemental Figure 5). Our MOR KO mice did not show any change in basal ethanol consumption compared to WT mice (Supplemental Figure 5). This is in contrast with earlier findings (6; 32), and may reflect differences in the genetic backgrounds of the mice. Our mice are on a purebred C57BL/6 background, whereas previous studies used mice on a mixed C57/129sv background. The C57BL/6 inbred mice are known to consume much more ethanol than 129/sv inbred mice (33). In addition, mice drink significantly more ethanol in the intermittent access paradigm utilized in our studies compared to the 24 hour access paradigm used in the initial knockout studies.
We next used this set of opioid receptor knock out mice to examine the hypothesis that one of the DOR subtypes was a heterodimer of DOR and another opioid receptor. As expected, the effects on drinking of the DOR1 selective agonist TAN-67 (25 mg/kg) were lost in mice with a disruption of the DOR1 gene (Figure 5A and 5B). Intriguingly, the effects of TAN-67 on both consumption [F(3,30) = 3.28, p = 0.03] (Figure 5A) and preference [F(3,30) = 4.01, p = 0.016] (Figure 5C) were also significantly diminished in mice with a disruption of the MOR gene, but were not affected by a disruption of the KOR gene (Figure 5A and 5D). Together, these data show that DOR1 activity requires a functional MOR, suggesting that DOR1 could be a heterodimer of the DOR and the MOR.
Figure 5. TAN-67 activity is affected by disruption of DOR and MOR.
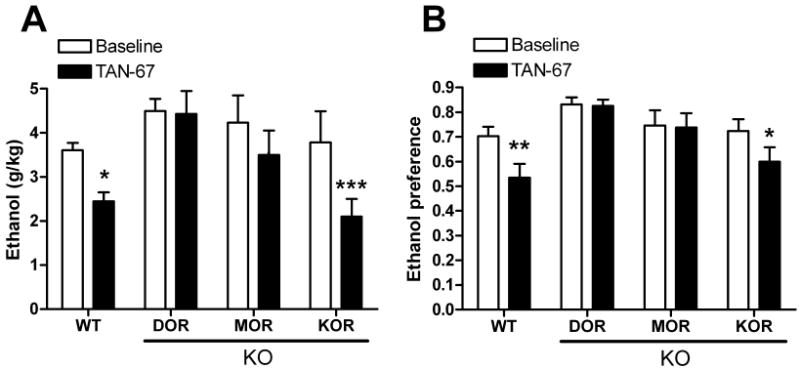
(A) TAN-67 (25 mg/kg) induced changes in ethanol consumption (A) or preference in wild-type, DOR (B) KO, MOR KO (C) and KOR KO (D) C57BL/6 mice (n=9). Thirty minutes after injection (vehicle or drug) ethanol and water consumption was measured over a 4 hour period. Percentage decrease is ((Basal-drug)/Basal)×100. Ethanol preference = ethanol consumption/(ethanol consumption + water consumption). (*p<0.05; **p<0.01; ***p<0.001; n.s., non significant).
DOR2 is a DOR homomer
We next examined the pharmacological role of the DOR, and MOR in the function of NTB. In contrast to the DOR1 agonist TAN-67, the DOR2 antagonist NTB remained effective in reducing alcohol consumption [F(2,21) = 4.47, p = 0.024] and preference [F(2,21) = 3.01, p = 0.043] in mice lacking the MOR gene suggesting that the DOR2 effect on drinking does not require the MOR (or the KOR see Supplemental Figure 6) and is most likely a DOR homomer (Figure 6).
Figure 6. NTB activity is only affected by disruption of DOR.

Naltriben (NTB, 6 mg/kg) induced changes in ethanol consumption (A) and preference (B) in DOR KO, and MOR KO C57BL/6 mice (n=9). Thirty minutes after injection ethanol and water consumption was measured over a 4 hour period. Ethanol preference = ethanol consumption/(ethanol consumption + water consumption). (*p<0.05; **p<0.01; n.s., non significant).
In vitro evidence that DOR1 is a DOR-MOR heterodimer
It is conceivable that DOR1 functionality requires MOR, but does not require the receptors to form heterodimers or even to be present in the same cell. To examine our hypothesis that the DOR1 is a DOR-MOR heterodimer with altered pharmacology compared to a DOR homomer, we heterologously expressed DOR in HEK 293 cells alone or together with the MOR in a set of cells lines carefully matched for expression as previously described (29). We found that, in cells expressing both MOR and DOR, heterodimers were present on the cell surface (Figure 7A). In these cells expressing the DOR-MOR heterodimer, we found that the DOR1 ligand TAN-67 had a significantly (p<0.05) higher affinity (pKi (M) = 10.1 ± 0.3, n=5), as assessed by displacement of [3H]DPDPE, than it did in cells expressing only DOR (pKi (M) = 9.3 ± 0.3, n=4). In contrast, we found that the DOR2 ligand NTB had a significantly lower affinity in cells expressing both MOR and DOR (pKi = 10.6 ± 0.4, n=4) than it did on cells expressing only the DOR (pKi (M)= 12.0 ± 0.2, n=3). The radioligand [3H]DPDPE did not bind to cells expressing only MOR under these conditions (data not shown).
We elaborated on these findings by examining the effect of the DOR subtype-selective antagonists on agonist-induced signaling in the cell lines expressing MOR and DOR, and cells expressing only DOR using a calcium mobilization assay (29). We observed that BNTX (100 nM) was 24 times more potent at reducing DPDPE-induced calcium mobilization in cells co-expressing DOR and MOR than in cells expressing DOR alone. In contrast, NTB (100 nM) is a more potent antagonist in cells expressing DOR alone (pEC50 (M) < 5) than in cells co-expressing DOR and MOR (pEC50 (M) = 5.3) (Figure 7B and 7C).
Non selective ligands do not provide optimal therapeutic efficacy
In support of our hypothesis that non-selective opioid ligands such as NTX and NTI have reduced efficacy compared to NTB because of their antagonism at DOR1, we tested the effectiveness of the non-selective DOR antagonist NTI on drinking in the DOR and MOR KO mice. We predicted that disruption of MOR would disrupt DOR1s, essentially converting NTI into a DOR2 antagonist. Indeed, while NTI (10 mg/kg) was ineffective at reducing drinking in WT mice and DOR (and KOR KO mice, Supplemental Figure 6), it reduced ethanol consumption [F(2,23) = 3.94, p = 0.034] in MOR KO mice. (Figure 8).
Figure 8. Disruption of the MOR turns NTI into a DOR2-selective antagonist for drinking.
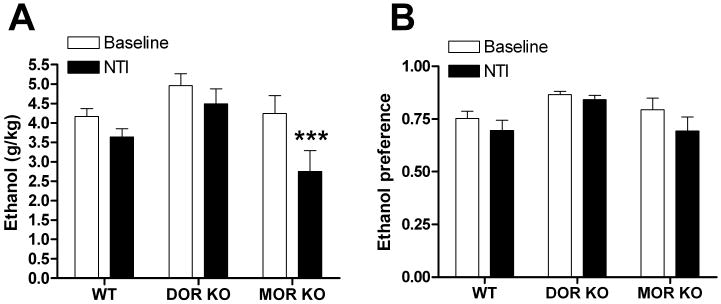
Naltrindole (NTI, 10 mg/kg) induced changes in ethanol consumption (A) and preference (B) in wild-type, DOR KO and MOR KO C57BL/6 mice (n=9). Thirty minutes after injection ethanol and water consumption was measured over a 4 hour period. Ethanol preference = ethanol consumption/(ethanol consumption + water consumption). (*p<0.05; **p<0.01).
TAN-67 is not rewarding by itself
The mesocorticolimbic dopamine pathway is well known to play a role in assigning rewarding properties to a variety of substances of abuse including ethanol (34). Because DORs are present in striatum and nucleus accumbens (35), activation of these receptors with an agonist, such as TAN-67, may induce dopamine release and signaling. To investigate whether TAN-67 reduces drinking by providing a “substitute” reward, we examined the rewarding properties of TAN-67 using a conditioned place preference paradigm. Importantly, mice conditioned with 3 sessions of 25 mg/kg TAN-67, a dose that reduced ethanol consumption, did not produce any significant [F(1,21) = 0.13, p = 0.72] place preference to TAN-67 (Figure 9).
Figure 9. Mice show no place preference to TAN-67.
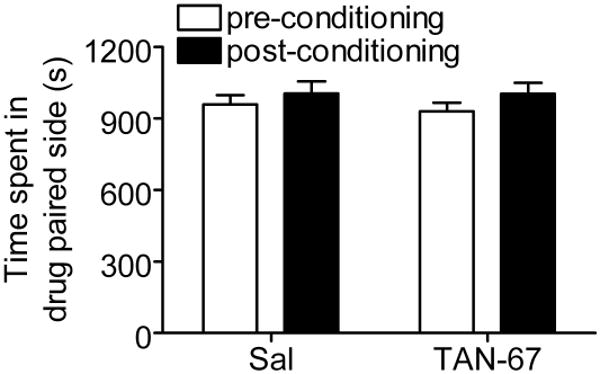
Mice (n=12) were conditioned to saline or 25 mg/kg TAN-67 for 3 days using a CPP paradigm (see materials and methods). Time spent in the drug paired site was measured on the days before and after the conditioning sessions for 30 minutes.
Discussion
Naltrexone is one of the few drugs approved by the FDA for alcoholism. However, the non-selective nature of this opioid antagonist likely contributes to its side effects and poor therapeutic efficacy. Whereas the role of the MOR in promoting alcohol intake is well-established (6), the role of the DOR has remained less clear. Several drinking studies have been conducted using non-selective DOR antagonists and DOR KO mice; however they have produced mixed and often conflicting results. We hypothesized that many of these contradictions could be resolved if the two DOR subtypes had opposing effects on ethanol consumption. These two subtypes have long been recognized in analgesia studies (12; 36). Here we examined whether the distinct pharmacologies of these two receptors, DOR1 and DOR2, could be used to influence ethanol consumption.
We provide evidence that the DOR1 and DOR2 have opposing effects on ethanol consumption in mice. We found that, in our drinking paradigm, non-selective antagonists such as NTX and NTI showed poor efficacy for reducing ethanol consumption in mice. Reports on the effectiveness of NTX and NTI vary; however, our findings are in agreement with several studies showing no effect of NTI (20; 21; 23), as well as little effect of NTX after repeated doses (37) or at high, more non-selective doses (38). We show that, similar to its effects in rats, the DOR2-selective antagonist NTB is effective at reducing ethanol consumption in mice. Conversely, we show that the DOR1 agonist TAN-67 decreases ethanol but not sucrose consumption, and does not interfere with ethanol uptake or metabolism. Nor did we find TAN-67 itself to be rewarding. In addition, we found that the pharmacological targets of the DOR1 agonist TAN-67 and the DOR2 antagonist NTB are different, and that the selective ligands for these two targets can act synergistically to decrease ethanol consumption. Furthermore, we provide evidence that the DOR1 subtype, targeted by TAN-67 in this drinking paradigm, is a heterodimer of MOR and DOR. Specifically, we show that the effects of TAN-67 are diminished not only in DOR KO mice, but also in MOR KO mice, whereas NTB effectiveness is only affected by disruption of the DOR gene and the effects of the non-selective DOR antagonist NTI are enhanced in mice with a disruption of MOR. Also we observe a pharmacological profile that more closely mimics DOR1 in HEK293 cells co-expressing DOR and MOR.
It has been established that G protein-coupled receptors, including the opioid receptors, can form homo- and heterodimers and that heterodimerization can have an impact on receptor pharmacology (26; 27). Moreover, it has been proposed that either the DOR1 or the DOR2 (39) could be an opioid receptor heterodimer. There are several lines of evidence suggesting that the DOR1 is a heterodimer of MOR and DOR. For example, disruption of the MOR eliminates the respiratory suppressive effects of the DOR1 agonists DPDPE (40), suggesting that a “complexed” DOR-MOR is responsible for the respiratory suppressive effects. However, other reports suggest that the DOR1 may be a DOP-KOP heterodimer (41; 42). Importantly, opioid receptor heterodimers have been shown to couple to different G-proteins than do the individual monomeric or homomeric receptors (27; 43; 44). This could potentially explain how DOR1 inhibits, but DOR2 enhances alcohol consumption.
Our finding that DOR1 and DOR2 have opposing roles in ethanol consumption provided an ideal model in which to study the molecular nature of the DOR subtypes, and explore whether one or the other is an opioid receptor heterodimer. To address this question, we utilized mice with a disruption of the DOR, MOR or KOR. We show that the ability of TAN-67 to decrease ethanol consumption is lost not only in mice disrupted for the DOR, but also in mice disrupted for the MOR, suggesting that TAN-67 targets a DOR-MOR heterodimer. Moreover, we found that effects of TAN-67 are blocked by the DOR1 antagonist BNTX, but are enhanced by the DOR2 antagonist NTB, suggesting that DOR1 is a target distinct from, and opposing, DOR2. We observed that DOR2 inhibition by NTB, is regulated by DOR but not MOR or KOR, suggesting DOR2 is most likely a DOR homomer, although we cannot exclude that DOR2 functionality requires a receptor other than MOR or KOR. It is possible that the requirement of MOR for DOR1 ligand effects on drinking is a consequence of convergent circuits expressing MOR and DOR rather than a result of a direct interaction between the DOR and MOR. Nevertheless, cells co-expressing the MOR and DOR show a pharmacological profile that more closely resembles DOR1 than DOR2. We could detect this change in pharmacology despite the fact that cells expressing MOR and DOR express a mixture of homomers and heterodimers (Figure 7A, lane 3 and 4). Hence, taken together, the in vivo and in vitro data suggest that the DOR1 is a DOR-MOR heterodimer whose activity opposes the actions of the DOR2 homomers on alcohol consumption.
Unlike the MOR, which is primarily expressed on the surface of cells, the DOR is stored mainly in dense core vesicles in primary afferent neurons of the dorsal horn and in brain regions including the peri-aqueductal grey (45) and nucleus raphe magnus (46). Its redistribution to the plasma membrane is facilitated by several stimuli including inflammatory pain (47), stress (48), and chronic morphine treatment (49). Whether ethanol changes the distribution of DOR is not clear. However, both in vitro, (50) and in vivo (35), ethanol does appear to stimulate increased DOR density at the cell surface. If this were the case, it is possible that the ratio of DOR1 to DOR2 is altered by ethanol drinking. Specifically, because DOR distribution is dynamically regulated, it is possible that subtypes of the DOR could be selectively expressed on the surface only under certain physiological conditions, such as stress, drug, or ethanol exposure, when the DORs are translocated out of the dense core vesicles to the surface. Further research needs to be performed to investigate this intriguing hypothesis.
Our finding that the non-selective DOR antagonist NTI has improved efficacy to decrease drinking in MOR KO mice, not only provides additional evidence that DOR1 is a DOR-MOR heterodimer, but also supports the hypothesis that non-selective ligands, such as NTI and NTX, are indeed less efficacious because they antagonize both DOR1 and DOR2. In fact, we observed that NTX, even at the relatively low dose of 1.5 mg/kg, has some effects on DOR and KOR (Supplemental Figure 7). From a therapeutic point of view, it may be more effective to use ligands that selectively target distinct subtypes of the opioid receptor family to treat alcoholism. Currently, no highly-selective, reversible, small molecule, MOR antagonists are available. This is why NTX, even though it has only modest selectivity at the MOR, is used therapeutically. However, there are several non-peptidergic ligands that are not only selective to DOR, but can distinguish between the DOR1 and DOR2 subtypes, and potently decrease ethanol consumption and preference, at least in these rodent models. Therefore, we propose that selective antagonism of DOR2s and/or selective agonism at DOR1s could provide more effective control of drinking than the currently available therapeutic NTX.
In conclusion, we have provided further evidence for the existence of two DOR subtypes in vivo. Importantly, we have shown that these subtypes act independently and in opposition to one other with respect to ethanol drinking. Moreover, we propose that the DOR1 is a DOR-MOR heterodimer, whereas DOR2 is a DOR homomer. Taken together these data suggest that more selective opioid ligands, specifically DOR1 agonists and DOR2 antagonists, could show increased efficacy in the treatment of alcoholism, compared to the non-selective antagonist, NTX, currently used therapeutically. Thus, we provide evidence that opioid receptor heterodimers represent novel in vivo targets for this indication.
Supplementary Material
Acknowledgments
We would like to thank Shruti Chander for technical assistance and Joan Holgate and Sebastien Carnicella for comments and suggestions. We thank Howard Fields, Amy Chang and Anuradha Madhavan for their valuable input on the manuscript. This work was funded by the Department of Defense grant DAMD62-10-5-071 (J.L.W.), National Institute on Drug Abuse Grants R01 DA015232 and DA019958 (J.L.W.) and funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco (J.L.W.).
Footnotes
Author contrbution: RMvR (designed research, performed research, analyzed data, wrote paper), JLW (designed research, analyzed data, wrote paper)
Financial disclosures: The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Ree JM, Gerrits MA, Vanderschuren LJ. Opioids, reward and addiction: An encounter of biology, psychology, and medicine. Pharmacol Rev. 1999;51:341–396. [PubMed] [Google Scholar]
- 2.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- 3.Froehlich JC, Li TK. Opioid involvement in alcohol drinking. Ann N Y Acad Sci. 1994;739:156–167. doi: 10.1111/j.1749-6632.1994.tb19817.x. [DOI] [PubMed] [Google Scholar]
- 4.Rohsenow DJ. What place does naltrexone have in the treatment of alcoholism? CNS Drugs. 2004;18:547–560. doi: 10.2165/00023210-200418090-00001. [DOI] [PubMed] [Google Scholar]
- 5.Anton RF, O'Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. Jama. 2006;295:2003–2017. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- 6.Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- 7.Kovacs KM, Szakall I, O'Brien D, Wang R, Vinod KY, Saito M, et al. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- 8.Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, et al. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- 9.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 10.Simonin F, Befort K, Gaveriaux-Ruff C, Matthes H, Nappey V, Lannes B, et al. The human delta-opioid receptor: genomic organization, cDNA cloning, functional expression, and distribution in human brain. Mol Pharmacol. 1994;46:1015–1021. [PubMed] [Google Scholar]
- 11.Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Opioid receptor types and subtypes: the delta receptor as a model. Annu Rev Pharmacol Toxicol. 1996;36:379–401. doi: 10.1146/annurev.pa.36.040196.002115. [DOI] [PubMed] [Google Scholar]
- 12.Mattia A, Vanderah T, Mosberg HI, Porreca F. Lack of antinociceptive cross-tolerance between [D-Pen2, D-Pen5]enkephalin and [D-Ala2]deltorphin II in mice: evidence for delta receptor subtypes. J Pharmacol Exp Ther. 1991;258:583–587. [PubMed] [Google Scholar]
- 13.Le AD, Poulos CX, Quan B, Chow S. The effects of selective blockade of delta and mu opiate receptors on ethanol consumption by C57BL/6 mice in a restricted access paradigm. Brain Res. 1993;630:330–332. doi: 10.1016/0006-8993(93)90672-a. [DOI] [PubMed] [Google Scholar]
- 14.Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK, et al. The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology (Berl) 1995;120:177–185. doi: 10.1007/BF02246191. [DOI] [PubMed] [Google Scholar]
- 15.Krishnan-Sarin S, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben selectively attenuates alcohol intake in rats bred for alcohol preference. Pharmacol Biochem Behav. 1995;52:153–159. doi: 10.1016/0091-3057(95)00080-g. [DOI] [PubMed] [Google Scholar]
- 16.June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology (Berl) 1999;147:81–89. doi: 10.1007/s002130051145. [DOI] [PubMed] [Google Scholar]
- 17.Froehlich JC, Zweifel M, Harts J, Lumeng L, Li TK. Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology (Berl) 1991;103:467–472. doi: 10.1007/BF02244246. [DOI] [PubMed] [Google Scholar]
- 18.Franck J, Lindholm S, Raaschou P. Modulation of volitional ethanol intake in the rat by central delta-opioid receptors. Alcohol Clin Exp Res. 1998;22:1185–1189. [PubMed] [Google Scholar]
- 19.Williams KL, Woods JH. Oral ethanol-reinforced responding in rhesus monkeys: effects of opioid antagonists selective for the mu-, kappa-, or delta-receptor. Alcohol Clin Exp Res. 1998;22:1634–1639. doi: 10.1111/j.1530-0277.1998.tb03960.x. [DOI] [PubMed] [Google Scholar]
- 20.Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O'Brien CP. A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol. 1998;15:281–289. doi: 10.1016/s0741-8329(97)00131-6. [DOI] [PubMed] [Google Scholar]
- 21.Middaugh LD, Kelley BM, Groseclose CH, Cuison ER., Jr Delta-opioid and 5-HT3 receptor antagonist effects on ethanol reward and discrimination in C57BL/6 mice. Pharmacol Biochem Behav. 2000;65:145–154. doi: 10.1016/s0091-3057(99)00184-7. [DOI] [PubMed] [Google Scholar]
- 22.Hyytia P. Involvement of mu-opioid receptors in alcohol drinking by alcohol-preferring AA rats. Pharmacol Biochem Behav. 1993;45:697–701. doi: 10.1016/0091-3057(93)90527-z. [DOI] [PubMed] [Google Scholar]
- 23.Honkanen A, Vilamo L, Wegelius K, Sarviharju M, Hyytia P, Korpi ER. Alcohol drinking is reduced by a mu 1- but not by a delta-opioid receptor antagonist in alcohol-preferring rats. Eur J Pharmacol. 1996;304:7–13. doi: 10.1016/0014-2999(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 24.Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, et al. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- 25.Parkhill AL, Bidlack JM. Several delta-opioid receptor ligands display no subtype selectivity to the human delta-opioid receptor. Eur J Pharmacol. 2002;451:257–264. doi: 10.1016/s0014-2999(02)02241-0. [DOI] [PubMed] [Google Scholar]
- 26.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- 28.Zapata A, Gonzales RA, Shippenberg TS. Repeated ethanol intoxication induces behavioral sensitization in the absence of a sensitized accumbens dopamine response in C57BL/6J and DBA/2J mice. Neuropsychopharmacology. 2006;31:396–405. doi: 10.1038/sj.npp.1300833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, et al. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci U S A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gharagozlou P, Demirci H, Clark JD, Lameh J. Activation profiles of opioid ligands in HEK cells expressing delta opioid receptors. BMC Neurosci. 2002;3:19. doi: 10.1186/1471-2202-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 32.Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, et al. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- 33.Yoneyama N, Crabbe JC, Ford MM, Murillo A, Finn DA. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol. 2008;42:149–160. doi: 10.1016/j.alcohol.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellegrino SM, Druse MJ. The effects of chronic ethanol consumption on the mesolimbic and nigrostriatal dopamine systems. Alcohol Clin Exp Res. 1992;16:275–280. doi: 10.1111/j.1530-0277.1992.tb01376.x. [DOI] [PubMed] [Google Scholar]
- 35.Mendez M, Morales-Mulia M, Leriche M. [3H]DPDPE binding to delta opioid receptors in the rat mesocorticolimbic and nigrostriatal pathways is transiently increased by acute ethanol administration. Brain Res. 2004;1028:180–190. doi: 10.1016/j.brainres.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Sofuoglu M, Portoghese PS, Takemori AE. 7-Benzylidenenaltrexone (BNTX): A selective [delta]1 opioid receptor antagonist in the mouse spinal cord. Life Sciences. 1993;52:769–775. doi: 10.1016/0024-3205(93)90240-4. [DOI] [PubMed] [Google Scholar]
- 37.Middaugh LD, Bandy AL. Naltrexone effects on ethanol consumption and response to ethanol conditioned cues in C57BL/6 mice. Psychopharmacology (Berl) 2000;151:321–327. doi: 10.1007/s002130000479. [DOI] [PubMed] [Google Scholar]
- 38.Phillips TJ, Wenger CD, Dorow JD. Naltrexone effects on ethanol drinking acquisition and on established ethanol consumption in C57BL/6J mice. Alcohol Clin Exp Res. 1997;21:691–702. [PubMed] [Google Scholar]
- 39.Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI. Modulation of mu-mediated antinociception in the mouse involves opioid delta-2 receptors. J Pharmacol Exp Ther. 1992;263:147–152. [PubMed] [Google Scholar]
- 40.Matthes HWD, Smadja C, Valverde O, Vonesch JL, Foutz AS, Boudinot E, et al. Activity of the delta -Opioid Receptor Is Partially Reduced, Whereas Activity of the kappa -Receptor Is Maintained in Mice Lacking the μ-Receptor. J Neurosci. 1998;18:7285–7295. doi: 10.1523/JNEUROSCI.18-18-07285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portoghese PS, Lunzer MM. Identity of the putative delta1-opioid receptor as a delta-kappa heteromer in the mouse spinal cord. Eur J Pharmacol. 2003;467:233–234. doi: 10.1016/s0014-2999(03)01599-1. [DOI] [PubMed] [Google Scholar]
- 42.Bhushan RG, Sharma SK, Xie Z, Daniels DJ, Portoghese PS. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J Med Chem. 2004;47:2969–2972. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 43.Breit A, Lagace M, Bouvier M. Hetero-oligomerization between beta2- and beta3-adrenergic receptors generates a beta-adrenergic signaling unit with distinct functional properties. J Biol Chem. 2004;279:28756–28765. doi: 10.1074/jbc.M313310200. [DOI] [PubMed] [Google Scholar]
- 44.Hasbi A, Nguyen T, Fan T, Cheng R, Rashid A, Alijaniaram M, et al. Trafficking of preassembled opioid mu-delta heterooligomer-Gz signaling complexes to the plasma membrane: coregulation by agonists. Biochemistry. 2007;46:12997–13009. doi: 10.1021/bi701436w. [DOI] [PubMed] [Google Scholar]
- 45.Hack SP, Bagley EE, Chieng BCH, Christie MJ. Induction of {delta}-Opioid Receptor Function in the Midbrain after Chronic Morphine Treatment. J Neurosci. 2005;25:3192–3198. doi: 10.1523/JNEUROSCI.4585-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma J, Zhang Y, Kalyuzhny AE, Pan ZZ. Emergence of functional delta-opioid receptors induced by long-term treatment with morphine. Mol Pharmacol. 2006;69:1137–1145. doi: 10.1124/mol.105.019109. [DOI] [PubMed] [Google Scholar]
- 47.Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain. 2003;101:199–208. doi: 10.1016/s0304-3959(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 48.Commons KG. Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol. 2003;464:197–207. doi: 10.1002/cne.10788. [DOI] [PubMed] [Google Scholar]
- 49.Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J Neurosci. 2001;21:7598–7607. doi: 10.1523/JNEUROSCI.21-19-07598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charness ME, Hu G, Edwards RH, Querimit LA. Ethanol increases delta-opioid receptor gene expression in neuronal cell lines. Mol Pharmacol. 1993;44:1119–1127. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.