

Abstract
Age-related macular degeneration (AMD) is a common degenerative disease resulting in injury to the retina, retinal pigment epithelium and choriocapillaris. Recent data from histopathology, animal models and genetic studies have implicated altered regulation of the complement system as a major factor in the incidence and progression of this disease. A variant in the gene SERPING1, which encodes C1INH, an inhibitor of the classical and lectin pathways of complement activation, was recently shown to be associated with AMD. In this study we sought to determine the localization of C1INH in human donor eyes. Immunofluorescence studies using a monoclonal antibody directed against C1INH revealed localization to photoreceptor cells, inner nuclear layer neurons, choriocapillaris, and choroidal extracellular matrix. Drusen did not exhibit labeling. Genotype at rs2511989 did not appear to affect C1INH abundance or localization, nor was it associated with significant molecular weight differences when evaluated by Western blot. In a small number of eyes (n=7 AMD and n=7 control) AMD affection status was correlated with increased abundance of choroidal C1INH. These results indicate that C1INH protein is present in the retina and choroid, where it may regulate complement activation.
INTRODUCTION
Age-related macular degeneration (AMD) is a major cause of blindness affecting millions of individuals in the Western world (Friedman, et al., 2004; VanNewkirk, et al., 2000). Although prevalence rates vary among different populations and different reports, values as high as 64% of individuals over the age of 80 have been reported (de Jong, 2006). Loss of central vision in AMD can result from either atrophic changes of the retinal pigment epithelium (RPE) or aberrant growth of the vasculature of the choriocapillaris through Bruch’s membrane and into the RPE and/or neurosensory retina.
Recently, considerable evidence has emerged indicating a major role of the complement system in AMD pathogenesis. Complement component C5 and the complement inhibitor vitronectin were found to be major components of drusen in human eyes (Hageman, et al., 1999; Mullins and Hageman, 1997), as were the terminal complement complex C5b-9 (Johnson, et al., 2000; Mullins, et al., 2000), clusterin (Johnson, et al., 2001) and other byproducts of complement activation (Johnson, et al., 2001). The finding that eyes from patients with glomerulonephritis develop early onset drusen (Duvall-Young, et al., 1989) and the elucidation of a requirement for complement activation in an animal model of choroidal neovascularization (Bora, et al., 2005; Rohrer, et al., 2009) provided further evidence for the role of complement activation in AMD pathogenesis.
The supposition that the complement complex participates in the development of AMD was strongly bolstered by the finding that a variant in the complement factor H gene (encoding a Tyr402His variant) is a major risk factor for the development of AMD (Edwards, et al., 2005; Hageman, et al., 2005; Haines, et al., 2005; Klein, et al., 2005). This finding has been subsequently replicated by a number of groups, and variations in other genes with products involved in the complement system have also been found to affect AMD risk, including C2 (Gold, et al., 2006), complement factor B (Gold, et al., 2006), and complement factor 3 (Yates, et al., 2007).
Ennis et al. recently described a single nucleotide polymorphism (rs2511989) in SERPING1 to be associated with AMD in two independent cohorts (Ennis, et al., 2008), although a second report did not replicate this association (Park, et al., 2009). The SERPING1 gene encodes C1INH, a glycoprotein that inhibits complement activation by interfering with the proteolytic activity of C1r/C1s in the classical pathway and mannose binding protein-associated serine proteases in the lectin pathway (Wagenaar-Bos and Hack, 2006). SERPING1 mRNA is present in human retina and RPE/choroid (Ennis, et al., 2008). In the current report we evaluated the expression of C1INH protein in unaffected eyes, eyes from patients with AMD, and patients with the high or low risk SERPING1 genotypes. We found consistent labeling of photoreceptor cells and variable labeling of the choriocapillaris. Eyes from donors homozygous for either phenotype were compared, and no obvious differences in localization were noted. In addition, AMD and control eyes were compared and AMD eyes showed more C1INH labeling in the choroid than controls. These results are discussed in the context of the complement system in AMD.
MATERIALS AND METHODS
Donor eyes
Human donor eyes were obtained from the Iowa Lions Eye Bank (Iowa City, IA) following informed consent from the donors’ families. Eyes were processed immediately on receipt. Macular and extramacular tissues were punched using disposable trephines, and punches were either fixed (4% paraformaldehyde in phosphate buffered saline, for 2 hours) or divided into retinal and RPE/choroidal layers which were flash frozen separately in liquid nitrogen. For biochemical studies all samples were preserved within 8 hrs of death, which is within a time frame during which protein composition of ocular tissues is well preserved (Ethen, et al., 2006). In some cases, ophthalmic records were available, and retinal diagnoses were recorded.
Genotyping
Either post-mortem blood samples or small fragments of ciliary body were used for DNA extraction. For tissue, the DNeasy Blood and Tissue Kit (Qiagen; Valencia, CA) was utilized, according to the manufacturer’s instructions. Donors were genotyped for the intronic SNP (rs2511989) in SERPING1 using the Taqman assay, as described previously for the U.S. cohort (Ennis, et al., 2008).
Histology and immunohistochemistry
Tissues were cryopreserved in sucrose solution and embedded in Optimal Cutting Temperature Compound (Ted Pella, Redding, CA) using the methods of Barthel and Raymond (Barthel and Raymond, 1990). Immunohistochemical and lectin histochemical labeling was performed as described previously (Mullins, et al., 2005; Mullins, et al., 2006). A monoclonal antibody directed against C1INH (Abcam, monoclonal antibody raised against full length C1INH protein) was used at a concentration of 2 μg/mL and detected with Alexa-488 conjugated goat anti-mouse antibody (Invitrogen; Carlsbad, CA). In order to confirm the specificity of this antibody, for some experiments antibody dilutions were preincubated with a 10 fold excess of purified C1INH protein (R&D Systems, Minneapolis, MN), as described previously for intercellular adhesion molecule-1 (ICAM1) (Mullins, et al., 2006).
Dual labeling was also performed with anti-C1INH and biotinylated Ulex europaeus agglutinin-I (UEA-I; Vector Laboratories, Burlingame CA), visualized with avidin-Texas red (Vector Laboratories) as described previously (Mullins, et al., 2005). Antibodies directed against the bipolar cell marker PKC-alpha (1μg/mL, Santa Cruz; SC-208) were also used in conjunction with C1INH antibodies, and were detected with Alexa-546 conjugated goat anti-rabbit antibodies (Invitrogen). For some experiments, adjacent tissue sections were labeled with either C1INH antibody or with monoclonal antibodies directed against the neoepitope in complement C9 that is exposed during formation of the terminal complement complex (15μg/mL; clone aE11, DAKO, Carpinteria, CA). Sections were counterstained with 100 μg/mL 4′,6-diamidino-2-phenylindole (DAPI).
For studies on the effect of AMD on C1INH localization, superotemporal-to-macular wedges of 7 AMD eyes and 7 control eyes were labeled with anti-C1INH (2μg/mL). The retina and choroid were evaluated and patterns were recorded in a masked fashion. The 7 affected eyes (mean age 78.3 years) had either atrophic AMD (n=6), characterized by RPE mottling and atrophy and/or macular drusen, or choroidal neovascularization (one case). The unaffected eyes had a mean age of 80.4 years.
Western blot analysis
In order to evaluate C1INH protein in retinal and RPE/choroidal tissues, Western blots were performed as described previously (Mullins, et al., 2006). Briefly, punches of extramacular retina and RPE/choroid were homogenized with a Kontes pestle (Kimble Chase; Vineland NJ) in ice cold protease inhibitors (Complete Mini Tablets; Roche, Indianapolis, IN) and 10–20μg of total protein were separated on either 10% or 4–15% gradient gels, transferred to polyvinylidene difluoride (PVDF) membrane (BioRad; Hercules, CA), and blocked with filtered 5% nonfat dry milk in PBS with 0.1% Tween-20. Blots were then incubated with anti-C1INH antibody (130ng/mL), washed, probed with peroxidase conjugated anti-mouse antibody (Amersham), washed, and bands were visualized with the ECL plus kit (GE Life Sciences).
RESULTS
Immunohistochemistry
In order to evaluate the specificity of commercially available C1INH antibodies, purified human C1INH protein was preincubated with diluted antibody solutions. A monoclonal antibody (Abcam, Ab54898) showed an almost complete loss of binding on tissue sections following preadsorption with C1INH (Figure 1A, B); this antibody was used for the remainder of experiments described in this report. This antibody also recognized purified C1INH protein on Western blot (data not shown). A second polyclonal antiserum (Santa Cruz Biologicals, SC46297) did not show decreased labeling on tissue sections following preadsorption and was not used for additional studies.
Figure 1.

Labeling of C1INH in the normal retina. (A) Labeling of human retina section with anti-C1INH (green fluorescence) reveals labeling of plasma C1INH in the lumens of large vessels and striking immunoreactivity against rod and cone inner segments, cone cell bodies and axons, as well as a population of inner nuclear layer cells. (B) Preadsorption of the monoclonal anti-C1INH antibody with an excess of purified C1INH abrogates this labeling pattern in an adjacent section of the same sample. Labeling of the bipolar cell marker anti-PKC (C) and anti-C1INH (D) appear as red and green fluorescence respectively (E, merged image). Blue fluorescence is due to DAPI nuclear counterstain. Scale bars are 50μm (A, B) and 25μm (C–E).
Tissue sections of human retina labeled with antibodies directed against human C1INH revealed reactivity with cone photoreceptor cells, including the inner and outer segments, cell bodies, axons and pedicles (Figure 1A). Rod inner segments showed some reactivity, although this was less notable than cone inner segments. In addition, inner nuclear layer neurons corresponding to bipolar cells appeared positive when labeled with anti-C1INH. Dual labeling with anti-PKC antibody showed colocalization of this bipolar cell antigen with C1INH (Figure 1C). Variable labeling was also noted in the ganglion cell layer.
Choroidal labeling revealed that blood vessel lumens were typically immunoreactive, presumably due to circulating C1INH adherent to the vessel walls. The walls of choroidal capillaries were also labeled in some donors (Figure 2). Dual labeling with the endothelial cell binding lectin UEA-I (Mullins, et al., 2005) (Figure 2A, B) revealed most of the vascular labeling to be within or around the choriocapillaris. The choroidal extracellular matrix including choriocapillary pillars in some donors showed reactivity.
Figure 2.
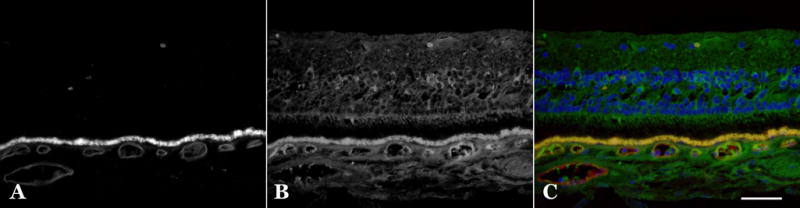
Labeling of C1INH in the normal choroid. Labeling of a human retina/choroid section with the vasculature binding lectin Ulex europaeus agglutin-I (A) and monoclonal antibody directed against C1INH (B), red and green respectively in merged panel (C). Labeling of anti-C1INH in the choroid includes vascular lumens and perivascular domains surrounding the choriocapillaris, including some intercapillary pillars. The choroidal extracellular matrix also shows labeling. Yellow fluorescence in C is due to lipofuscin autofluorescence. Scalebar = 50μm.
Based on our findings that a variation in the gene encoding C1INH is associated with AMD, and based on observations that a number of other complement proteins are drusen constituents, we predicted that drusen, basal laminar deposits and/or basal linear deposits might show immunoreactivity with this antibody. However, subRPE deposits were invariably very weak or negative for C1INH reactivity, with the exception of minor diffusion into the base of some large drusen (Figure 3A, B) which did not appear to correspond to drusen cores (Mullins and Hageman, 1999). This finding is in contrast to observations with C5b-9 terminal complement complex, which is a component of drusen. Some overlap was observed between C5b-9 and C1INH in the choriocapillaris on adjacent sections, in which the same capillaries showed immunoreactivity for both antibodies (Figure 4).
Figure 3.
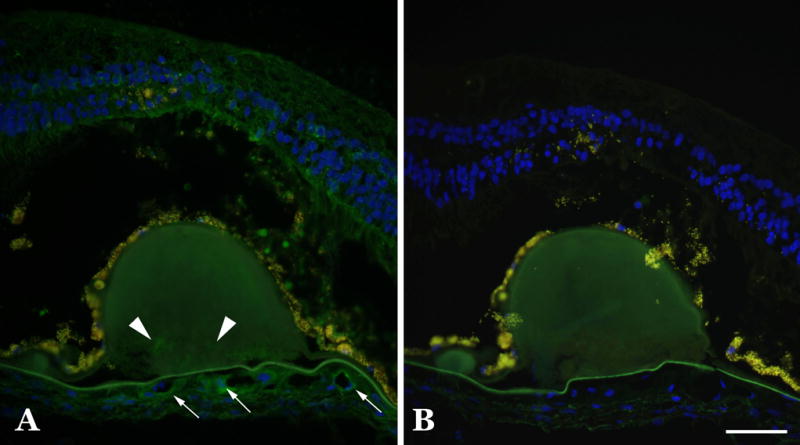
C1INH immunohistochemistry of drusen in an eye with geographic atrophy. Comparison of C1INH binding in a section with large drusen (A) compared to an adjacent section in which the primary antibody was omitted (B) shows that only minor labeling is observed within these deposits (arrowheads). Note the retinal attenuation over the large central druse and the labeling of some choriocapillaris vessels (arrows). Scale bar = 50μm.
Figure 4.
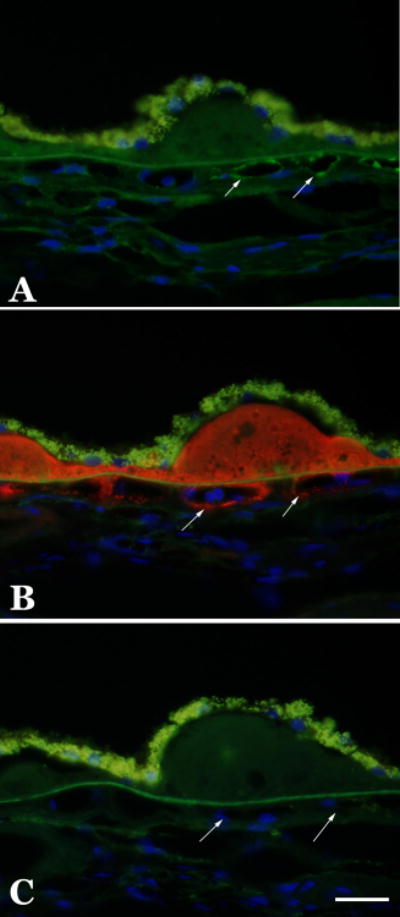
Distribution of C1INH and terminal complement complex in the human choroid. Adjacent sections were labeled with anti-C1INH antibody (A; green fluorescence), anti-terminal complement complex antibody (B; red fluorescence), or with Alexa-488-conjugated and Alexa-546-conjugated secondary antibodies only (C). Terminal complement complexes show intense labeling in drusen and throughout choriocapillary pillars and around choriocapillaris vessels (B). The C1INH antibody shows a more diffuse pattern of labeling in the choroidal extracellular matrix, as well as some individual capillaries that are also positive for terminal complement complexes (arrows). Exposures for all three panels were identical. Scalebar = 25μm.
We next sought to determine whether the genotype of an individual at the intronic SNP rs2511989 showed an obvious change in the level or distribution of C1INH protein. We compared labeling in tissue sections obtained from 6 donors that were homozygous for the low risk allele (AA) to 6 donors homozygous for the high risk allele (GG). No consistent differences were apparent in immunolabeling of the retina and choroid in homozygous AA and GG donors (data not shown).
In order to determine whether eyes diagnosed with macular degeneration showed a difference in C1INH labeling compared to control eyes, a series of 7 AMD and 7 control eyes were evaluated in a masked fashion for C1INH labeling. 5 of 7 AMD eyes and 2 of 7 control eyes showed a high degree of choroidal labeling. This was most notable in the extracellular matrix along the outer aspect of the choriocapillaris (Figure 5).
Figure 5.
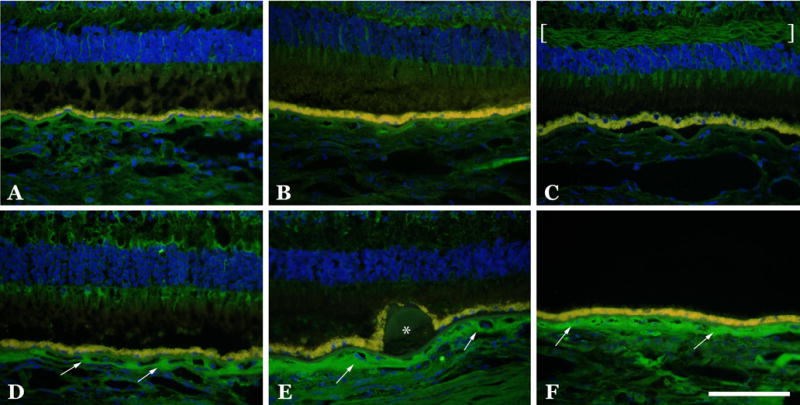
Labeling of C1INH in AMD and unaffected eyes. Representative images of eyes from unaffected (A–C) and AMD (D–F) donors are depicted. Note the increased labeling in the choroidal extracellular matrices in the eyes with AMD (arrows), as well as the layer of Henle in a control eye (C, brackets) and the unlabeled hard druse (E, asterisk). Scalebar = 100μm.
Western blot analysis
A single band with an apparent molecular weight of 106 kDa was recognized in protein samples from RPE/choroid (Figure 6), which migrated at the same molecular weight as purified C1INH protein. This signal was much more abundant in RPE/choroid compared with retina, suggesting that most of the detected protein was due to circulating C1INH protein in choroidal lumens. Comparison of samples from two donors homozygous for the A allele of rs2511989 with samples from two donors homozygous for the G allele did not show any notable differences in either molecular weight or protein abundance (Figure 6). Removal of N-linked sugars using PNGase F (as described previously (Mullins, et al., 2006)) resulted in a loss of approximately 10 kDa of apparent molecular weight, consistent with previous reports showing a significant contribution of N-linked carbohydrate chains to the molecular weight of C1INH (Liu, et al., 2004)(data not shown).
Figure 6.
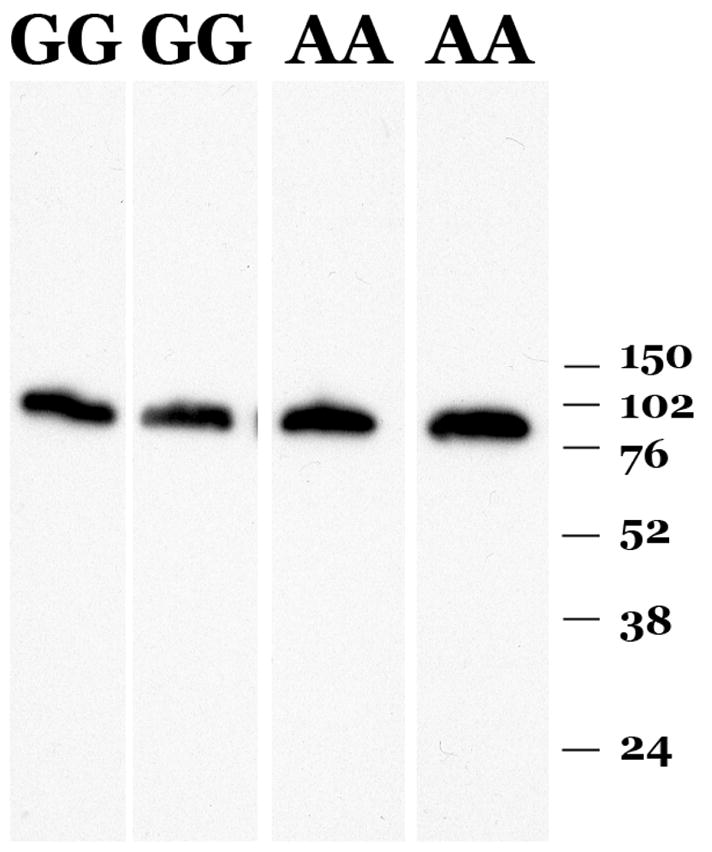
Characterization of C1INH protein. Samples of human RPE-choroid genotyped as either homozygous for the SNP rs2511989 “AA” or homozygous for the canonical allele “GG” were separated by SDS-PAGE, transferred to PVDF membrane and probed with anti-C1INH antibody. Molecular weight marker positions in kDa are indicated.
DISCUSSION
The hypothesis that the complement system participates in the pathology of AMD has been supported by a research in a number of disciplines, including animal models of neovascularization, histopathology and genetics (recently reviewed (Lotery and Trump, 2007)). To date, most of the emphasis in this field has revolved around the alternative complement pathway (see, for example, (Scholl, et al., 2008)) as variants in genes associated with the classical and lectin pathway have not been as conclusively described as AMD risk factors. One variant in the SERPING1 gene was found to be associated with AMD in one study (Ennis, et al., 2008), and other, cosegregating variants are computationally predicted to affect behavior of the minor allele mRNA (Kralovicova and Vorechovsky, 2009). This genetic association was not reproduced in a second report, however (Park, et al., 2009). Thus, the status of this gene as a risk factor for AMD is not entirely clear.
In light of both this genetic association, as well as the overall role of the complement system and its inhibitors in AMD, we evaluated the localization of the SERPING1 gene product in human donor eyes. As an inhibitor of the classical and lectin pathways of complement activation, SERPING1 expression could plausibly be protective against complement injury in AMD.
We found robust localization of C1INH protein in cone photoreceptor cells and in inner retinal neurons. Given the distribution of complement complexes in aging human eyes, which are largely confined to the choriocapillaris and intercapillary pillars (Gerl, et al., 2002; Mullins, et al., 2003) this pattern was unexpected. One possibility is that C1INH in the retina serves functions other than inhibiting complement activation. C1INH is known to inhibit other serine proteases such as kallikrein, which shows widespread expression in neural retina (Ma, et al., 1996) and which regulates retinal vascular permeability (Phipps, et al., 2009). Macular edema is sometimes observed in patients with AMD, but is generally attributed to leakage from choroidal neovascular membranes. Alternatively, C1INH may protect retinal neurons from complement activation that can be triggered during retinal injury. Although complement complexes are not normally observed in the neural retina, there is significant evidence for activation in disease states including models of light damage (Rohrer, et al., 2007), physical injury (Vazquez-Chona, et al., 2004), ischemic injury (Kuehn, et al., 2008) and in human glaucoma (Kuehn, et al., 2006), which may follow the classical pathway of activation based on local synthesis of C1q. Local expression of a classical pathway inhibitor may therefore be important in the neural retina to mitigate processes that are triggered in pathologic conditions. Establishing the temporal and spatial relationships between C1INH and the proteases it regulates will provide further insight into this question.
In a small sample of eyes with AMD, an increase in choroidal labeling was observed (Figure 4). This labeling was especially notable in the extracellular matrix along the outer aspect of the choriocapillaris, in a pattern similar to that observed for terminal complement complexes. Since C1INH does not appear to be a significant component of drusen or basal laminar deposits, it is unlikely that its expression contributes directly to the biogenesis of these deposits. On the other hand, there are additional mechanisms whereby an increase in C1INH protein in the choroid could contribute to the pathophysiology of AMD. In addition to its enzymatic inhibition of proteases in the classical and alternative pathways, C1INH has also been found to interact with endothelial cell adhesion molecules including selectins (Cai, et al., 2005) and vascular cell adhesion molecule-1 (Zhang, et al., 2007). These molecular interactions have been shown to inhibit endothelial cell-leukocyte binding (Cai and Davis, 2003). Thus, increased expression of C1INH in the choroid could impair transmigration of leukocytes.
The role of different classes of leukocytes in AMD has become controversial, with different investigators proposing either helpful or harmful roles for these cells (recently reviewed (Ferguson and Apte, 2008; Skeie and Mullins, 2008)). Modulating leukocyte-endothelial cell interactions could plausibly affect AMD risk and progression, as leukocytes have been found to be elevated in eyes with AMD (Penfold, et al., 1985; Mullins, unpublished) and to be major components of neovascular membranes in human eyes (Grossniklaus, et al., 2000; Penfold, et al., 1987). Validation of the elevation of C1INH protein in additional AMD eyes, and evaluation of the effects of C1INH on leukocyte recruitment to the choroid, will help to clarify the role of this molecule in AMD.
Acknowledgments
Supported in part by NIH grants EY-017451(RFM), EY-016822 (EMS), the Macula Vision Research Foundation (RFM and AJL), American Health Assistance Foundation (AJL) and a center grant from the Foundation Fighting Blindness. EMS is an investigator in the Howard Hughes Medical Institute. The authors gratefully acknowledge the Iowa Lions Eye Bank and the eye donors and their families.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barthel L, Raymond P. Improved method for obtaining 3-microns cryosections for immunocytochemistry. J Histochem Cytochem. 1990;38:1383–8. doi: 10.1177/38.9.2201738. [DOI] [PubMed] [Google Scholar]
- Bora P, Sohn J, Cruz J, Jha P, Nishihori H, Wang Y, Kaliappan S, Kaplan H, Bora N. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005;174:491–7. doi: 10.4049/jimmunol.174.1.491. [DOI] [PubMed] [Google Scholar]
- Cai S, Davis AE., 3rd Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion. J Immunol. 2003;171(9):4786–91. doi: 10.4049/jimmunol.171.9.4786. [DOI] [PubMed] [Google Scholar]
- Cai S, Dole VS, Bergmeier W, Scafidi J, Feng H, Wagner DD, Davis AE., 3rd A direct role for C1 inhibitor in regulation of leukocyte adhesion. J Immunol. 2005;174(10):6462–6. doi: 10.4049/jimmunol.174.10.6462. [DOI] [PubMed] [Google Scholar]
- de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355(14):1474–85. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- Duvall-Young J, Short C, Raines M, Gokal R, Lawler W. Fundus changes in mesangiocapillary glomerulonephritis type II: clinical and fluorescein angiographic findings. Br J Ophthalmol. 1989;73:900–906. doi: 10.1136/bjo.73.11.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A, Ritter R, Abel K, Manning A, Panhuysen C, Farrer L. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Ennis S, Jomary C, Mullins R, Cree A, Chen X, MacLeod A, Jones A, Collins A, Stone E, Lotery A. Association between the SERPING1 gene and age-related macular degeneration: a two-stage case–control study. The Lancet. 2008;372:1828–34. doi: 10.1016/S0140-6736(08)61348-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47(6):2280–90. doi: 10.1167/iovs.05-1395. [DOI] [PubMed] [Google Scholar]
- Ferguson TA, Apte RS. Angiogenesis in eye disease: immunity gained or immunity lost? Semin Immunopathol. 2008;30(2):111–9. doi: 10.1007/s00281-008-0113-8. [DOI] [PubMed] [Google Scholar]
- Friedman D, O’Colmain B, Munoz B, Tomany S, McCarty C, de Jong P, Nemesure B, Mitchell P, Kempen J Group. EDPR. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Gerl V, Bohl J, Pitz S, Stoffelns B, Pfeiffer N, Bhakdi S. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. 2002;43:1104–1108. [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossniklaus H, Cingle K, Yoon Y, Ketkar N, L’Hernault N, Brown S. Correlation of histologic 2-dimensional reconstruction and confocal scanning laser microscopic imaging of choroidal neovascularization in eyes with age-related maculopathy. Arch Ophthalmol. 2000;118:625–9. doi: 10.1001/archopht.118.5.625. [DOI] [PubMed] [Google Scholar]
- Hageman G, Anderson D, Johnson L, Hancox L, Taiber A, Hardisty L, Hageman J, Stockman H, Borchardt J, Gehrs K, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman G, Mullins R, Russell S, Johnson L, Anderson D. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB Journal. 1999;13(3):477–84. doi: 10.1096/fasebj.13.3.477. [DOI] [PubMed] [Google Scholar]
- Haines J, Hauser M, Schmidt S, Scott W, Olson L, Gallins P, Spencer K, Kwan S, Noureddine M, Gilbert J, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Johnson L, Leitner W, Staples M, Anderson D. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–96. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Johnson L, Ozaki S, Staples M, Erickson P, Anderson D. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–9. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- Klein R, Zeiss C, Chew E, Tsai J, Sackler R, Haynes C, Henning A, Sangiovanni J, Mane S, Mayne S, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. SERPING1 rs2511988 and age-related macular degeneration. Lancet. 2009;373(9662):461–2. doi: 10.1016/S0140-6736(09)60168-9. [DOI] [PubMed] [Google Scholar]
- Kuehn MH, Kim CY, Jiang B, Dumitrescu AV, Kwon YH. Disruption of the complement cascade delays retinal ganglion cell death following retinal ischemia-reperfusion. Exp Eye Res. 2008;87(2):89–95. doi: 10.1016/j.exer.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Kuehn MH, Kim CY, Ostojic J, Bellin M, Alward WL, Stone EM, Sakaguchi DS, Grozdanic SD, Kwon YH. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res. 2006;83(3):620–8. doi: 10.1016/j.exer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Liu D, Gu X, Scafidi J, Davis AE., 3rd N-linked glycosylation is required for c1 inhibitor-mediated protection from endotoxin shock in mice. Infect Immun. 2004;72(4):1946–55. doi: 10.1128/IAI.72.4.1946-1955.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotery A, Trump D. Progress in defining the molecular biology of age related macular degeneration. Hum Genet. 2007;122(3–4):219–36. doi: 10.1007/s00439-007-0406-3. [DOI] [PubMed] [Google Scholar]
- Ma JX, Song Q, Hatcher HC, Crouch RK, Chao L, Chao J. Expression and cellular localization of the kallikrein-kinin system in human ocular tissues. Exp Eye Res. 1996;63(1):19–26. doi: 10.1006/exer.1996.0087. [DOI] [PubMed] [Google Scholar]
- Mullins R, Anderson D, Russell S, Hageman G. Ocular drusen contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB Journal. 2000;14:835–46. [PubMed] [Google Scholar]
- Mullins R, Grassi M, Skeie J. Glycoconjugates of choroidal neovascular membranes in age-related macular degeneration. Mol Vis. 2005;11:509–17. [PubMed] [Google Scholar]
- Mullins R, Hageman G. Histochemical comparison of ocular “drusen” in monkey and human. In: LaVail M, Hollyfield J, Anderson R, editors. Degenerative Retinal Diseases. New York: Plenum Press; 1997. pp. 1–10. [Google Scholar]
- Mullins R, Hageman G. Human ocular drusen possess novel core domains with a distinct carbohydrate composition. Journal of Histochemistry and Cytochemistry. 1999;47:1533–9. doi: 10.1177/002215549904701205. [DOI] [PubMed] [Google Scholar]
- Mullins R, Kuehn M, Aptsiauri N, Hageman G. Distribution of Terminal Complement Complexes at the RPE-Choriocapillaris Interface in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2003;44 E-Abstract 4226. [Google Scholar]
- Mullins R, Skeie J, Malone E, Kuehn M. Macular and peripheral distribution of ICAM-1 in the human choriocapillaris and retina. Mol Vis. 2006;12:224–35. [PubMed] [Google Scholar]
- Park KH, Ryu E, Tosakulwong N, Wu Y, Edwards AO. Common variation in the SERPING1 gene is not associated with age-related macular degeneration in two independent groups of subjects. Mol Vis. 2009;15:200–7. [PMC free article] [PubMed] [Google Scholar]
- Penfold P, Killingsworth M, Sarks S. Senile macular degeneration: the involvement of immunocompetent cells. Graefe’s Archives for Clinical and Experimental Ophthalmology. 1985;223:69–76. doi: 10.1007/BF02150948. [DOI] [PubMed] [Google Scholar]
- Penfold P, Provis J, Billson F. Age-related macular degeneration: ultrastructural studies of the relationship of leukocytes to angiogenesis. Graefe’s Arch Clin Exp Ophthalmol. 1987;225:70–6. doi: 10.1007/BF02155808. [DOI] [PubMed] [Google Scholar]
- Phipps JA, Clermont AC, Sinha S, Chilcote TJ, Bursell SE, Feener EP. Plasma kallikrein mediates angiotensin II type 1 receptor-stimulated retinal vascular permeability. Hypertension. 2009;53(2):175–81. doi: 10.1161/HYPERTENSIONAHA.108.117663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 2007;48(11):5282–9. doi: 10.1167/iovs.07-0282. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS, Tomlinson S. A Targeted Inhibitor of the Alternative Complement Pathway Reduces Angiogenesis in a Mouse Model of Age-related Macular Degeneration. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, et al. Systemic complement activation in age-related macular degeneration. PLoS ONE. 2008;3(7):e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeie J, Mullins R. Macrophages in neovascular age-related macular degeneration: friends or foes? Eye. 2008 doi: 10.1038/eye.2008.206. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanNewkirk M, Nanjan M, Wang J, Mitchell P, Taylor H, McCarty C. The prevalence of age-related maculopathy: the visual impairment project. Ophthalmology. 2000;107:1593–600. doi: 10.1016/s0161-6420(00)00175-5. [DOI] [PubMed] [Google Scholar]
- Vazquez-Chona F, Song BK, Geisert EE., Jr Temporal changes in gene expression after injury in the rat retina. Invest Ophthalmol Vis Sci. 2004;45(8):2737–46. doi: 10.1167/iovs.03-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar-Bos IG, Hack CE. Structure and function of C1-inhibitor. Immunol Allergy Clin North Am. 2006;26(4):615–32. doi: 10.1016/j.iac.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357(6):553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- Zhang H, Qin G, Liang G, Li J, Chiu I, Barrington RA, Liu D. Suppression of complement regulatory protein C1 inhibitor in vascular endothelial activation by inhibiting vascular cell adhesion molecule-1 action. Biochem Biophys Res Commun. 2007;358(4):1120–7. doi: 10.1016/j.bbrc.2007.05.058. [DOI] [PubMed] [Google Scholar]