

Abstract
The presenilin-1 gene is mutated in early-onset familial Alzheimer’s disease. The mutation Pro117Leu is implicated in a very severe form of the disease, with an onset of less than thirty years. The consequences of this mutation on neurogenesis in the hippocampus of adult transgenic mice have already been studied in situ. The survival of neural progenitor cells was impaired resulting in decreased neurogenesis in the dentate gyrus. Our intention was to verify if similar alterations could occur in vitro in progenitor cells from the murine ganglionic eminences isolated from embryos of this same transgenic mouse model. These cells were grown in culture as neurospheres and after differentiation the percentage of neurons generated as well as their morphology were analysed. The mutation results in a significant decrease in neurogenesis compared to the wild type mice and the neurons grow longer and more ramified neurites. A shift of differentiation towards gliogenesis was observed that could explain decreased neurogenesis despite increased proliferation of neural precursors in transgenic neurospheres. A diminished survival of the newly generated mutant neurons is also proposed. Our data raise the possibility that these alterations in embryonic development might contribute to increase the severity of the Alzheimer’s disease phenotype later in adulthood.
Keywords: ganglionic eminence, neural progenitor cell, neuritic outgrowth, neurogenesis, neuronal morphology, striatum
1. Introduction
Mutations in the presenilin-1 gene are the most commonly recognized cause of early onset familial Alzheimer’s disease (FAD). Presenilin-1 (PS-1) is an approximately 48 kDa transmembrane protein that is activated when cleaved into two sets of heterogeneous fragments, one corresponding to the N-terminal (about 30 kDa) and the other to the C-terminal part (about 18 kDa) [24]. PS-1 participates in a protein complex [23], the secretasome, which works as a γ-secretase, a protease cleaving the precursor protein of the β-amyloid peptide or APP. It leads, subsequently to the action of a β-secretase, to the release of β-amyloid peptides. The 42-43 amino acid form of the β-amyloid peptide is generated preferentially resulting in oligomeric and fibrillary compounds that are particularly toxic and proposed to be key players in the etiopathogenesis of Alzheimer’s disease according to the β-amyloid cascade hypothesis [11]. Presenilin-1 mutations also result in an increased sensitivity of neurons to various damages [10].
Some mutations in the PS-1 gene such as the Pro117Leu (P117L) missense mutation cause very-early onset Alzheimer’s disease, i.e. usually before the age of 30 years [14,27], suggesting that they might contribute to deleterious effects already during development. This hypothesis is strengthened by the fact that PS-1 is expressed widely during development including in neural progenitor cells of the primitive subventricular zone [18] and that targeted PS-1 null mutant mice die shortly after birth [21]. Furthermore PS-1 is physically and functionally associated with several intercellular signaling pathways involving, among others, Notch, GSK-3β and cadherins, all proteins known for their implications in the development of the nervous system [1,3,8]. However, PS-1 mutations might differ in their consequences. Indeed, PS-1 FAD mutations can rescue the embryonic lethality of PS-1 knock out mice suggesting that their effects on development might be rather subtle [7]. The effect of the FAD PS-1 P117L mutation has been studied in neuron-specific enolase (NSE) driven PS-1 transgenic mice. Levels of the 42 amino acid form of the β-amyloid peptide were significantly increased in the PS-1 P117L transgenic compared to the wild type mice [25]. Moreover in these studies the survival of neural progenitor cells was impaired in the dentate gyrus of adult mice by the FAD mutation leading to a decrease in neurogenesis, whereas overexpression of wild type PS-1 had no effect or was found to promote neurogenesis in the same region [25,26]. Interestingly PS-1 was also suggested to have survival promoting properties in neurodegenerative disorders [9].
Thus, given the widely distributed expression and the importance of PS-1 in neurogenesis, we wondered whether the P117L mutation might also affect or not prenatal development of the forebrain and not just the dentate gyrus. As a first approach, we decided to study the differentiation of neural progenitor cells (NPCs) isolated from the E14 murine striata. Here we show that the FAD PS-1 P117L mutation impairs neurogenesis of NPCs in vitro but promotes neuritic outgrowth in newly generated neurons. Thus, this study supports a broader role for the PS-1 P117L mutation beyond its effects limited to the metabolism of the APP in adulthood and might contribute to explaining the severity of the disease phenotype.
2. Materials and methods
2.1 Culture and differentiation of neural stem cells
B6D2 wild type mice and PS-1 P117L transgenic mice [26] were used to isolate striatal neural progenitor cells (NPCs) from embryos (E14-15). Animal manipulations were carried out in accordance with Federal Swiss Veterinary regulations and with institutional approval. The NPCs were cultured following the protocol of Reynolds and Weiss (1992, 1996) [19,20]. After animal decapitation, striata, subventricular zone included, were dissected out under sterile conditions. Cells were mechanically dissociated and then placed in basic stem cell medium: 1 to 1 mixture of Dulbecco’s modified Eagle’s medium (DMEM) and F-12 nutrient (Sigma, no D8`062, Buchs, Switzerland) including 0.6% glucose, 2 mM glutamine, 3 mM sodium bicarbonate, 25 μg/ml insulin (Sigma), 100 μg/ml apotransferrin (Sigma), 20 nM progesterone (Sigma), 60 μM putrescine (Sigma) and 30 nM sodium selenite (Sigma). To this medium, Epidermal Growth Factor (EGF; Sigma), basic Fibroblast Growth Factor (bFGF; Invitrogen, Carlsbad, USA) and heparin (Sigma) were added at 20 ng/ml, 10 ng/ml and 2.5 μg/ml final concentrations, respectively. After 4 to 6 days in culture, floating clusters of cells or neurospheres formed. The neurospheres were either used to study differentiation or NPCs proliferation. In the last case neurospheres were mechanically dissociated and cells were grown at clonal dilution [20] in 4 ml growth medium; after 7 days of proliferation the number of newly generated neurospheres and their average diameter were analysed in 100 μl neurosphere suspensions.
For cell differentiation, the procedure applied was derived from Wu et al. (2002) [29] and consisted in a first step of “preconditioning” in which dissociated cells were allowed to proliferate for 6 days as adherent cells on poly-L-ornithine (15 μg/ml) and laminin (2μg/ml; Sigma) treated glass coverslips in basic stem cell medium containing bFGF (20 ng/ml) and laminin (1μg/ml), followed by a differentiation step in basic stem cell medium containing no growth factors but B27 supplement (2% final concentration; Invitrogen) for 8 days. According to Wu et al., (2002) [29], this procedure should yield a higher proportion of newly generated neurons. Differentiated cells were immunocytochemically analysed.
2.2 Genotyping, sequencing and RT-PCR
Tail-extracted blood samples or NPCs from B6D2 wild type and PS-1 P117L hemizygous transgenic mice were genotyped by polymerase chain reaction (PCR) according to the protocol of Wen et al. (2002) [26]. The presence of mutation PS-1 P117L was verified by sequencing (Automated DNA sequencing, Applied Biosystems EBI 3100 DNA sequencer, Rotkreuz, Switzerland) a DNA fragment amplified with primers corresponding to the neuron-specific enolase (NSE) intron 1 (5′-GCTCCACCCTTCTAAGCCTC-3′; [26]) and to a PS-1 DNA sequence 5′-GGCATGGATGACCTTATAGCACCT-3′.
Transcription of the NSE driven transgene was monitored by amplifying synthesized cDNAs (First-strand cDNA synthesis; Amersham/GE Healthcare, Otelfingen, Switzerland) from DNAse I treated RNA extracted from 106 proliferating NPCs (TRIzol reagent, Invitrogen) using human PS-1 primers 5′-ATGACAGAGTTACCTGCACCG-3′ and 5′-ACAGAACCACCAGGAGGATA-3′ (L76517).
2.3 Immunoprecipitation
Five millions dissociated progenitor cells were collected from cultured wild type or PS-1 P117L neurospheres by centrifugation (5′, 1′ 000 rpm, 4°C), lysed for 30 min on ice in 500 μl NP40 buffer (20 mM Tris, 150 mM NaCl, 0.5% Na deoxycholate and 0.5% NP40, pH8.0) in the presence of 1 μl of a protease inhibitor cocktail (10 μl/ml, Sigma). They were sonicated (1 × 10 sec; Bandelin Sonopuls HD200, Bandelin, Germany) and centrifuged 10 min at 10′000 rpm to remove unsolubilized material. Ten μl of rabbit polyclonal anti-PS-1 IgG (Santa Cruz, H70; Santa Cruz, USA) that recognizes human as well as murine PS-1 was added to each lysate and incubated overnight at 4°C under mild agitation. Twenty μl protein A beads suspension (magnetic Bio-Adembeads protein A, Ademtech, Pessac, France), washed 3 times in NP40 buffer and separated each time from the buffer by magnetic separation, was added to the lysate of each sample and incubated 1 hour at room temperature under mild mixing. After magnetic separation, the pellets were washed 4 times with 500 μl NP40 buffer using magnetic separation. Finally, beads from each sample were resuspended in 20 μl of SDS polyacrylamide gel electrophoresis (PAGE) buffer 1X (50 mM Tris-HCl, pH 6.8, 10% glycerol, 2.5% β-mercaptoethanol, 2% SDS, 0.01% bromophenol blue) and boiled 1 min. The supernatants were isolated after a last magnetic separation and 15 μl per sample was loaded onto the SDS-PAGE gel (10% acrylamide; Miniprotean II dual slab cell, Bio-Rad, Reinach, Switzerland). After electrophoresis proteins were electroblotted (overnight, 35 V; Mini Trans-Blot electrophoretic transfer cell, Bio-Rad) onto a nitrocellulose membrane (Schleicher and Schuell/GE Healthcare).
After a brief rinse in distilled H2O the membrane was washed (3 × 10 min) in TB buffer (Tris-HCl 0.02M, pH 8.0, 130 mM NaCl, 0.05% Tween 20), non-specific binding sites were saturated by 1 h incubation in a 2% blocking solution (ECL advance kit, Amersham/GE Healthcare) in TB. The membrane was then incubated overnight at 4°C with a mouse monoclonal anti-human PS-1 primary antibody NT.1 (diluted 1:3′000 in blocking solution, Paul Mathews, New York University School of Medicine, Nathan Kline Institute, Orangeburg, NY 10962).
The membrane was then washed as before and incubated 1 h with a horseradish peroxidase (HRP)-conjugated anti-mouse antibody (GE Healthcare) diluted 1:100′000 in blocking solution. After washing (3 × 10 min in TB), the membrane was briefly rinsed in distilled H2O and bands detected by chemiluminescence (ECL advance Western blotting detection, Amersham/GE Healthcare) on film (Hyperfilm ECL, Amersham/GE Healthcare).
After washing (3 × 10 min in TB) the same blot was reprobed with HRP-labelled anti-rabbit IgG (1:100′000; GE Healthcare) to detect the immunoglobulin used in the immunoprecipitation. After washing, the membrane was stripped in 0.2 M glycine-HCl, pH 2.5 with 0.1% Tween 20 for 2 × 2 hours at 60°C, neutralized in TB for 2 × 15 min and incubated 1 hour in 2% blocking solution, then 2 hours at room temperature with rabbit polyclonal anti-PS-1 antibody (Santa Cruz, H70; dilution 1:1′000); after washing (3 × 10 min in TB) the membrane was incubated 1 h with the HRP-labelled anti-rabbit antibody to detect the total amount of transgenic (human) and murine PS-1 immunoprecipitated.
For the densitometric analysis, films were scanned with a GS-700 imaging densitometer (Bio-Rad) and the band intensities were measured as optical dentity unit x area (mm2) using the molecular analyst software program (Bio-Rad) and compared as percentages.
2.4 Immunocytochemistry
Indirect immunocytochemistry was carried out on preconditioned and differentiated NPCs. Cells were fixed for 30 min at room temperature in 4% paraformaldehyde (PAF) in PBS. After washing in PBS (3 × 5 min), cells were permeabilized for 5 min in PBS containing 0.25% Triton X-100. Following a second washing step in PBS (10 min), coverslips were incubated for 30 min in PBS containing 10% horse serum (PBS/HS) and then overnight at 4°C with primary antibodies diluted in PBS/HS. After washing (3 × 10 min) in PBS, coverslips were incubated with appropriate secondary antibodies diluted in PBS/HS for 1 h at room temperature and finally washed (3 × 10 min) in PBS. At the end of the procedure, coverslips were incubated in 0.2 μg/ml of bisbenzimide (Hoechst 33258; Calbiochem/Merck, Darmstadt, Germany) for 15 min and then washed three times 5 min in PBS. Hoechst reagent stains cell nuclei and allows counting the number of dead and living cells attached on coverslips. Coverslips were finally rinsed 3 times 10 min in PBS, then in distilled water, mounted on glass slides coverslipped with Fluorosave (Calbiochem).
The mouse monoclonal anti class-III beta tubulin antibody (Sigma), diluted 1/1′000, was used as a marker for newly generated neurons and the rabbit polyclonal anti-glial fibrillary acidic protein antibody (GFAP, Sigma), diluted 1:500, as a marker for astrocytes.
Secondary antibodies, diluted 1:1′000, were either anti-mouse IgG coupled to cyanide 3 (Jackson ImmunoResearch Laboratories, West Grove, USA) or coupled to AlexaFluor 488 (Molecular probes/Invitrogen), anti-rabbit IgG coupled to cyanide 3 (Chemicon) or to AlexaFluor 488 (Molecular probes/Invitrogen). Pictures of immunostained cells were taken using a Zeiss Axioskop 2 plus microscope (Carl Zeiss, Feldbach, Switzerland) equipped with epifluorescence and were digitalized with an Axiocam camera.
2.5 Cell counting and morphometric analysis
For determination of the proportion of neurons generated by wild type or transgenic NPCs, 3 separate experiments for either wild type or transgenic cells were considered. For each experiment, measures were performed independently by two collaborators. Using a 20x objective, 36 and 51 pictures corresponding to various fields were taken for wild type and PS-1 P117L cells, respectively. A total of 300 neurons were analysed. On each picture βIII tubulin positive neurons were counted and expressed as percent of total cells number (Hoechst positive nuclei).
The proportions of astrocytes and cell death generated in wild type and transgenic cell culture were measured in the same three experiments carried out to determine the proportion of neurons. About 1800 astocytes were counted in 21 or 18 pictures corresponding to various fields for wild type or PS-1 P117L cells, respectively. For each picture GFAP positive astrocytes were expressed as percent of the total number of cells (Hoechst positive nuclei). To estimate cell death pyknotic cell nuclei were counted as percentage of the total number of cells visualized by Hoechst staining in a total of 40 pictures for the three separate cell culture experiments. About 3000 cells were counted for each cell culture type, i.e. wild type or the PS-1 P117L cells.
For determination of total neuritic length (in μm) we used the NIH Image J 1.33 u program. For each neuron analysed total neuritic length corresponds to the sum of the length of all primary and secondary neurites. The number of primary and secondary neurites per neuron was also counted. Only neurites the length of which was equal or longer than twice the cell body diameter were considered according to a standard procedure [28].
For each type of analysis, mean values were determined and statistical differences were assessed using a One-Way ANOVA test with significance p < 0.05.
3. Results
3.1 Both wild type and transgenic presenilin-1 are expressed in cultured neural progenitor cells from ganglionic eminences
All studies described below were obtained with hemizygous transgenic animals compared to non-transgenic littermates used as controls. The human presenilin-1 (PS-1) P117L transgenic mice were genotyped by PCR amplification (Fig. 1A). The transgene PS-1 P117L was amplified and sequenced to corroborate the presence of the mutation. PS-1 expression was also determined in cultured neurospheres originating from the striata of E14 wild type and transgenic mice. This was done either by reverse transcription and PCR amplification (Fig. 1B) or by immunoprecipitation using a rabbit polyclonal antibody that recognizes human as well as murine PS-1 N-terminus (PS-1 NT) as precipitating antibody (Fig. 1C). In a first immunoblotting of the precipitated material, a band around 30 kDa was specifically labelled with the anti-human PS-1 antibody (NT.1; Fig. 1C, lanes 1 and 2); this band which corresponds to the N-terminal fragment of the activated PS-1 P117L can be detected in the transgenic (lane 2 from left) but not in the wild type NPCs (lane 1), indicating neuron-specific enolase (NSE) driven expression of the human PS-1 P117L transgenic protein. After incubation of the blot with an anti-rabbit IgG antiserum, large and equal amounts of the precipitating antibody (rabbit anti-PS-1) were detected both in wild type and transgenic samples as intensively labelled bands migrating at 50 kDa (IgG heavy chain; Fig. 1C, lanes 3 and 4). Finally, the blot was reprobed using the precipitating rabbit antibody (Fig. 1C, lanes 5 and 6). In both wild type and transgenic samples, a large band corresponding to the N-terminal portion of PS-1 (Fig. 1C, 30 kDa) was labelled. The intensity of the PS-1 P117L NT signal (lane 2) was about 35% of the total NT signal (murine as well as transgenic PS-1; lane 6), meaning that there is no strong increase of overall PS-1 NT signal due to PS-1 P117L transgenesis.
Fig. 1.
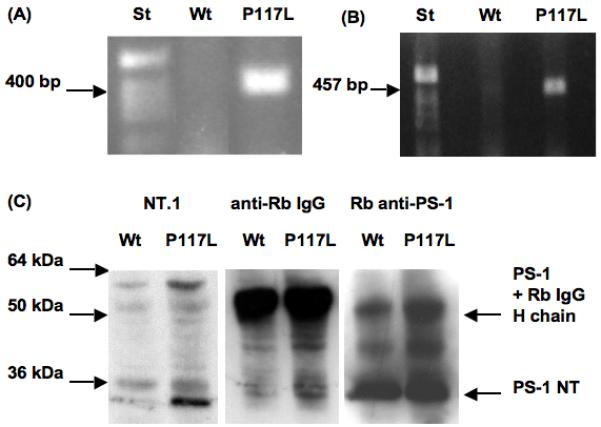
Genotype of presenilin-1 (PS-1) P117L transgenic E14 mice (A) and expression of the PS-1 P117L mutation in E14 striatal progenitors (B, C). PCR amplification of a 400 bp-human PS-1 P117L DNA fragment from transgenic but not from wild type (Wt) mouse (A), and transcription, shown by RT-PCR, of the PS-1 P117L transgene in neural stem cells (B; 457 bp). (A) and (B): 1% ethidium bromide stained agarose MP gel (Roche, Rotkreuz, Switzerland). St = 1 kb standard DNA ladder (Invitrogen). (C) Western blots of immunoprecipitated PS-1 (human and mouse) after immunolabelling with the human specific anti-PS-1 antibody NT.1 (PS-1 N-terminus or NT; <30 kDa; lanes 1 and 2 from left). Presence of equal amounts of IgG heavy chains of the immunoprecipitating rabbit (Rb) antibody was revealed using HRP-conjugated antibody to rabbit IgG (band at 50 kDa; lanes 3 and 4). The rabbit anti-PS-1 antibody can detect both, human transgene and murine wild type PS-1 NT fragments (< 30 kDa; lanes 5 and 6).
3.2 PS1 P117L mutation affects proliferation and differentiation of neural progenitor cells
In order to appreciate whether the presence of mutated PS-1 in transgenic mice has an influence upon the proportion and proliferation rate of neural progenitors, we estimated the number of newly generated neurospheres from equal amounts of wild type and PS-1 P117L NPCs as well as the average diameter of these neurospheres. Although the numbers of neurospheres generated by each population of NPCs were not differing significantly, the mean diameter of PS1 P117L neurospheres was however significantly (p < 10-6) larger (120 +/- 4.6 μm) than that of wild type neurospheres (77 +/- 3.6 μm).
In differentiation experiments, equal amounts of NPCs isolated from E14 wild type or human PS-1 P117L transgenic mice, were differentiated on glass coverslips and were then immunolabelled with antibody markers for astrocytic (anti-GFAP) and neuronal cells (anti-βIII-tubulin). Representative pictures are shown in figure 2. Both wild type and transgenic cells have generated large amounts of astrocytes, whereas neurons represented only about 3.5+/-0.5% of the total number of differentiated cells in the wild type cell culture (Fig. 3A). The proportion of PS1 P117L transgenic neurons was lower (about 1+/-0.1%) but they appeared to be more differentiated than the wild type ones. The differences in neurogenesis (Fig. 3A), in neuritic outgrowth, i.e. total neuritic length (Fig. 3B) and secondary neurites per neurons (Fig. 3D) were statistically different (p < 0.05), whereas this was not the case for the number of primary neurites per neurons (Fig. 3C). Tertiary neurites were found on very few neurons and were very short and therefore were not taken into consideration. The proportion of GFAP positive cells represented 60.5+/-10% of the total number of cells in the wild type cell culture. It increased significantly (p = 3 × 10-4) to 72.9+/-10.5% in PS-1 P117L cell culture (Fig. 3E). Estimated cell death was shown to increase significantly (p = 5 × 10-4) from 27.8+/-9.9% in the wild type cell culture to 44.1+/-14.5% in the PS-1 P117L cell culture (Fig. 3F).
Fig. 2.
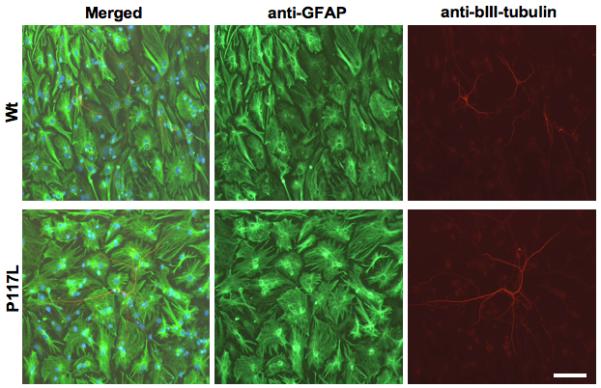
Comparison by immunocytochemistry of differentiated neural stem cells originating from B6D2 wild type (Wt) and presenilin-1 P117L transgenic mice. Glial cells are recognized by anti-GFAP (Glial fibrillary acidic protein) and neurons by anti-βIII-tubulin immunolabelling. Differentiation was carried out in basic stem cell medium containing B27 supplement for 8 days after 6 days in “preconditioning” medium. Transgenic neurons appear to be less numerous but more differentiated than the wild type ones. Scale bar = 80 μm.
Fig. 3.
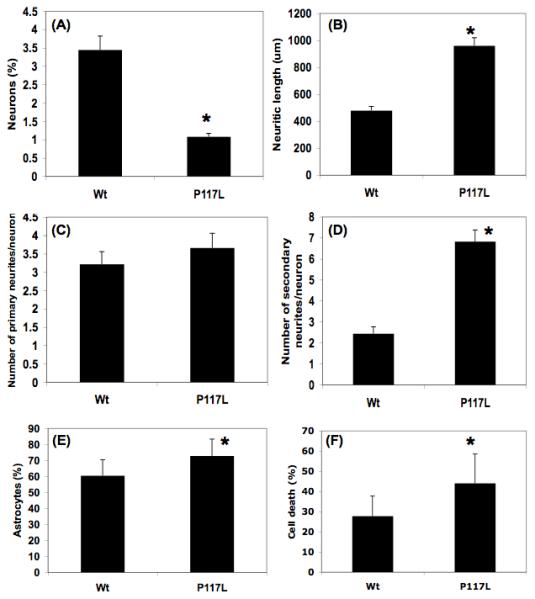
Quantitative comparison of neurogenesis (A, in %), neuritic length (B, in μm), number of primary and secondary neurites per neuron (C and D, respectively), astrocyte differentiation (E, in %) and cell death (F, in %) in neural progenitor cell cultures originating from B6D2 wild type (Wt) and presenilin-1 P117L transgenic mice. Differences are statistically significant, as indicated by a star, for neurogenesis (A: p = 3 × 10-4), neuritic length (B: p = 3.6 × 10-4), secondary neurites per neuron (D: p = 1.8 × 10-4), astrocyte differentiation (E: p = 3 × 10-4) and cell death (F: p = 5 × 10-4). Cells were analysed on photographs as the ones shown in Fig. 2, i.e. after 6 days “preconditioning” followed by 8 days differentiation and immunocytochemical staining.
4. Discussion
The aim of the present study was to investigate the effects on neurogenesis of exogeneous expression of the PS-1 P117L mutation in NPCs isolated from E14 mouse striata. For this, human PS-1 P117L transgenic mice were used in which transgene expression was under the control of NSE promotor [26]. According to these authors, the human transgene derived protein was expressed at about two to three times the level of endogeneous PS-1 as shown by Western blotting, and its expression was widespread in adult mouse brain and essentially neuron specific. In the present work we could show using RT-PCR and immunoprecipitation that the PS-1 P117L transcript and also the protein (N-terminal part) were present in NPCs isolated from mouse embryos, although at lower levels. The presence of PS-1 P117L in growing NPCs appeared to increase the size of neurospheres, an effect that could suggest that the PS-1 P117L NPCs were proliferating more rapidly and/or were more encline to form fused neurospheres [22]. The expression of the transgene in NPCs before differentiation may therefore have affected their fate. Indeed, transgenic cells did generate a lower proportion of neurons than their wild type counterpart. Thus, the PS1 P117L mutation impairs neurogenesis of NPCs. Using in situ studies, Wen et al. (2002, 2004) [25,26] obtained similar results with the progenitors of adult hippocampus of these PS-1 P117L transgenic mice. A decreased neurogenesis could be explained either by a differentiation shift from neurons to glial cells and/or by a reduced survival capacity of the newly generated neurons. A statistically significant increased proportion of astrocytes of about 12% was indeed measured in PS-1 P117L compared to wild type cell culture, an observation that can be related to the diminished proportion in neurogenesis. Decreased neurogenesis might also be due to enhanced apoptosis in PS-1 P117L cell culture as indicated by a statistically significant increased cell death of about 16% in the PS-1 P117L compared to the wild type cell culture. Numerous studies have already demonstrated that mutated PS-1 neurons were more prone to cell death e.g. in presence of proapoptotic stimuli [12].
Neurons generated by PS-1 P117L NPCs were reproducibly found to be more differentiated than those from wild type cells: their neurites were longer and exhibited a statistically significant enhanced number of secondary neurites, whereas there was a non significant increased number of primary neurites per neuron. These effects were found to be independent of culture condition. Indeed, differentiation after a “preconditioning” step as in this work or direct differentiation in a 1% fetal calf serum containing medium as most commonly used for NPC cell culture resulted in very similar results (data not shown). These observations are comparable to data obtained in the caudate nucleus of homo sapiens, a brain region originating with the globus pallidus from the ganglionic eminence. Neurons of the caudate nucleus of early onset Alzheimer disease cases compared to control patients showed increased total dendritic length and arborisation but no significant increased numbers of primary dendrites. These changes were related to a reduced neuronal density, whereas no alterations were observed in the globus pallidus that appears to be marginally vulnerable [2]. As PS-1 mutations are known to be deleterious, the most obvious hypothesis is the one of a premature differentiation or of a higher structural plasticity that would lead to dystrophic neurites interfering later in development - when synapses are massively selected - with proper neural network formation. Indeed, neuronal populations with a high degree of structural plasticity, as is the case in the caudate nucleus and not in the globus pallidus, appear to be particularly vulnerable to degenerative changes, such as neurofibrillary tangles [2]. However, we cannot exclude that the neuritic outgrowth might be compensatory, at least partly, and that it would attenuate deleterious consequences. Thus, the functional significance of reduction of neurogenesis and increased neuritic outgrowth in the developing medial and lateral ganglionic eminences in PS-1 P117L transgenic mice has still to be investigated. However, we can hypothesize that those small changes in these regions at embryonic stage E14 might weaken the organism later in adulthood. Indeed these structures give rise to neurons populating not only the striatum (including the caudate nucleus), but also the neocortex and the hippocampus [5,17]. The primate subventricular zone gives rise among others, to neurons destined to amygdala and pyriform cortex [4], two regions important for emotions and memory and atrophied in Alzheimer disease [6]. At least, as extrapyramidal stiffness, known to be related to dysfunction in the dopamine-striatum system, was described in patients with the PS-1 P117L mutation [14], we can wonder whether this phenotype might be related to early impairments in ganglionic eminence neurogenesis.
At the present time we can only speculate about the molecular mechanisms involved in the effects of the PS-1 P117L mutation. Several metabolic candidate pathways are possible or might even be involved at the same time (e.g. Notch, GSK-3β, cadherins) [1,3,8], not to speak of the overproduction of Aβ peptides [15,25]. In the subventricular zone of adult mice neurogenesis was disrupted in familial Alzheimer mutant APP transgenic mice or after Aβ injection into the subventricular zone [13]. PS-1 mutations have been shown to affect gene expression, e.g. via inhibition of N-cadherin cleavage [16], suggesting that the PS1-P117L mutation might result in decreased neurogenesis via a similar mechanism.
Overall, we can conclude that the autosomal dominant PS-1 P117L mutation leads to non lethal effects in the murine embryonic development, which might however contribute to increased cytological fragility in adulthood and thus, in human, to forms of Alzheimer’s disease with very early onset.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Alexson TO, Hitoshi S, Coles BL, Bernstein A, van der Kooy D. Notch signaling is required to maintain all neural stem cell populations - irrespective of spatial or temporal niche. Dev. Neurosci. 2006;28:34–48. doi: 10.1159/000090751. [DOI] [PubMed] [Google Scholar]
- [2].Arendt T, Brückner MK, Bigl V, Marcova L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. III. The basal forebrain compared with other subcortical areas. J. Comp. Neurol. 1995;351:223–246. doi: 10.1002/cne.903510204. [DOI] [PubMed] [Google Scholar]
- [3].Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich VL, Jr., Robakis NK. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2381–6. doi: 10.1073/pnas.041603398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11464–9. doi: 10.1073/pnas.172403999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brazel CY, Romanko MJ, Rothstein RP, Levison SW. Roles of the mammalian subventricular zone in brain development. Prog. Neurobiol. 2003;69:49–69. doi: 10.1016/s0301-0082(03)00002-9. [DOI] [PubMed] [Google Scholar]
- [6].Callen DJ, Black SE, Gao F, Caldwell CB, Szalai JP. Beyond the hippocampus: MRI volumetry confirms widespread limbic atrophy in AD. Neurology. 2001;57:1669–74. doi: 10.1212/wnl.57.9.1669. [DOI] [PubMed] [Google Scholar]
- [7].Davis JA, Naruse S, Chen H, Eckman C, Younkin S, Price DL, Borchelt DR, Sisodia SS, Wong PC. An Alzheimer’s disease-linked PS1 variant rescues the developmental abnormalities of PS1-deficient embryos. Neuron. 1998;20:603–9. doi: 10.1016/s0896-6273(00)80998-8. [DOI] [PubMed] [Google Scholar]
- [8].Figueroa DJ, Morris JA, Ma L, Kandpal G, Chen E, Li YM, Austin CP. Presenilin-dependent gamma-secretase activity modulates neurite outgrowth. Neurobiol. Dis. 2002;9:49–60. doi: 10.1006/nbdi.2001.0447. [DOI] [PubMed] [Google Scholar]
- [9].Giannakopoulos P, Kövari E, Savioz A, de Bilbao F, Dubois-Dauphin M, Hof PR, Bouras C. Differential distribution of presenilin-1, Bax, and Bcl-X(L) in Alzheimer’s disease and frontotemporal dementia. Acta Neuropathol. 1999;98:141–9. doi: 10.1007/s004010051062. [DOI] [PubMed] [Google Scholar]
- [10].Grilli M, Diodato E, Lozza G, Brusa R, Casarini M, Uberti D, Rozmahel R, Westaway D, St George-Hyslop P, Memo M, Ongini E. Presenilin-1 regulates the neuronal threshold to excitotoxicity both physiologically and pathologically. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12822–7. doi: 10.1073/pnas.97.23.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- [12].Hashimoto Y, Tsukamoto E, Niikura T, Yamagishi Y, Ishizaka M, Aiso S, Takashima A, Nishimoto I. Amino- and carboxyl-terminal mutants of presenilin-1 cause neuronal cell death through distinct toxic mechanisms: study of 27 different presenilin 1 mutants. J. Neurosci. Res. 2004;75:417–28. doi: 10.1002/jnr.10861. [DOI] [PubMed] [Google Scholar]
- [13].Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 2002;1:125–35. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- [14].Kulczycki J, Bertrand E, Lojkowska W, Dowjat W, Wisniewski T, Lyczywek-Zwierz M. Familial Alzheimer’s disease connected with mutation in presenilin gene 1 (P117L) Neurol. Neurochir. Pol. 2001;35:213–24. [PubMed] [Google Scholar]
- [15].López-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 2004;24:5439–44. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–45. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- [17].Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J. Neurosci. 2000;20:6063–76. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moreno-Flores MT, Medina M, Wandosell F. Expression of presenilin 1 in nervous system during rat development. J. Comp. Neurol. 1999;410:556–70. [PubMed] [Google Scholar]
- [19].Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992;12:4565–74. doi: 10.1523/JNEUROSCI.12-11-04565.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- [21].Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–39. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- [22].Singec I, Knoth R, Meyer RP, Maciaczyk J, Volk B, Nikkhah G, Frotscher M, Snyder E. Defining the actual sensitivity and specificty of the neurosphere assay in stem cell biology. Nature Methods. 2006;3:801–806. doi: 10.1038/nmeth926. [DOI] [PubMed] [Google Scholar]
- [23].Wakabayashi T, De Strooper B. Presenilins: members of the gamma-secretase quartets, but part-time soloists too. Physiology (Bethesda) 2008;23:194–204. doi: 10.1152/physiol.00009.2008. [DOI] [PubMed] [Google Scholar]
- [24].Ward RV, Davis JB, Gray CW, Barton AJ, Bresciani LG, Caivano M, Murphy VF, Duff K, Hutton M, Hardy J, Roberts GW, Karran EH. Presenilin-1 is processed into two major cleavage products in neuronal cell lines. Neurodegeneration. 1996;5:293–8. doi: 10.1006/neur.1996.0040. [DOI] [PubMed] [Google Scholar]
- [25].Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp. Neurol. 2004;188:224–37. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- [26].Wen PH, Shao X, Shao Z, Hof PR, Wisniewski T, Kelley K, Friedrich VL, Jr., Ho L, Pasinetti GM, Shioi J, Robakis NK, Elder GA. Overexpression of wild type but not an FAD mutant presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol. Dis. 2002;10:8–19. doi: 10.1006/nbdi.2002.0490. [DOI] [PubMed] [Google Scholar]
- [27].Wisniewski T, Dowjat WK, Buxbaum JD, Khorkova O, Efthimiopoulos S, Kulczycki J, Lojkowska W, Wegiel J, Wisniewski HM, Frangione B. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport. 1998;9:217–21. doi: 10.1097/00001756-199801260-00008. [DOI] [PubMed] [Google Scholar]
- [28].Wu G, Fang Y, Lu Z-H, Ledeen RW. Induction of axon-like and dendrite-like processes in neuroblastoma cells. J. Neurocytol. 1998;27:1–14. doi: 10.1023/a:1006910001869. [DOI] [PubMed] [Google Scholar]
- [29].Wu P, Tarasenko YI, Gu Y, Huang LY, Coggeshall RE, Yu Y. Region-specific generation of cholinergic neurons from fetal human neural stem cells grafted in adult rat. Nat. Neurosci. 2002;5:1271–8. doi: 10.1038/nn974. [DOI] [PubMed] [Google Scholar]