

Abstract
DYT1 dystonia is caused by a trinucleotide deletion of GAG (ΔGAG) in DYT1, which codes for torsinA. A previous epidemiologic study suggested an association of DYT1 ΔGAG mutation with early-onset recurrent major depression. However, another study reported no significant association with depression, but instead showed an association with anxiety and dystonia. In this study, we analyzed these related behaviors in Dyt1 ΔGAG heterozygous knock-in mice. The knock-in mice showed a subtle anxiety-like behavior but did not show depression-like behaviors. The mutant mice also displayed normal sensorimotor gating function in a prepulse inhibition test. While normal hippocampus-dependent contextual fear memory and hippocampal CA1 long-term potentiation (LTP) were observed, the knock-in mice exhibited an enhancement in the formation of cued fear memories. Anatomical analysis indicated that the number of c-fos positive cells was significantly increased while the size of the central nucleus of the amygdala (CE) was significantly reduced in the knock-in mice. These results suggest that the Dyt1 ΔGAG mutation increased the activity of the CE and enhanced the acquisition of the cued fear memory.
Keywords: amygdala, anxiety, c-fos, dystonia, fear memory, knock-in mouse
1. Introduction
Dystonia is a movement disorder characterized by involuntary, repetitive, sustained muscle contractions or postures. Dystonia is defined by its symptoms and caused by a variety of factors. Acute dystonic symptoms are often observed as side effects of dopamine-receptor blockers used as antipsychotics (neuroleptics) in clinical practice (Dressler and Benecke 2005). Writer's cramp and musician's cramp, examples of focal dystonia, are caused by intensive training of the fingers (Hallett 2006; Watson 2006). Dystonia can also arise from other disorders or injuries, such as brain trauma (O'Suilleabhain and Dewey 2004). These are classified as secondary dystonia. Dystonia that arises spontaneously without any obvious cause or associated disease is primary in nature, and often has a hereditary component. Although there are 16 classifications of genetic dystonia, only about half of them have identifiable gene mutations associated with them (Breakefield et al. 2008; Camargos et al. 2008; Fuchs et al. 2009).
DYT1 primary generalized early-onset torsion dystonia is caused by mutations in DYT1 (TOR1A) that codes for torsinA. Most patients have a 3 base pair (bp) deletion, ΔGAG, in DYT1 corresponding to a loss of a glutamic acid residue in the C-terminal region of torsinA (Ozelius et al. 1997). In a rare case, an 18-bp deletion in DYT1 was also reported in a family (Leung et al. 2001; Doheny et al. 2002). Recently, another transition mutation which causes an Arg288Gln exchange was found in a dystonia patient (Zirn et al. 2008). Some biochemical and cellular studies suggest that the mutation in torsinA causes functional defects that contribute to the pathology of this disease (Hewett et al. 2000; Liu et al. 2003; Cao et al. 2005; Pham et al. 2006). Recent genetic studies suggest that it is the partial loss of torsinA function that contributes to the pathology (Goodchild et al. 2005; Dang et al. 2006a; Yokoi et al. 2008).
Depression and anxiety have been observed in a variety of movement disorder patients (Lauterbach et al. 2003; Lauterbach et al. 2004; Miller et al. 2007). In Parkinson's disease, it was suggested that anxiety and depression may manifest as the first symptoms of Parkinson's disease many years before motor symptoms appear (Lemke et al. 2004). Primary generalized dystonia has been believed to be a purely motor disease, without any associated psychiatric symptoms. However, an epidemiologic study suggested that early-onset, recurrent major depression occurs more frequently in DYT1 ΔGAG mutation carriers, regardless of the manifestation of dystonic symptoms (Heiman et al. 2004). The report raised the possibility that DYT1 ΔGAG mutation functions as a “blue” gene (Richard and McDonald 2004). However, when the data were analyzed after combining both single and recurrent major depression as a group, there was no significant difference found in the frequency between carriers and non-carriers. Another report suggested that there was no difference in the occurrence of depression between the DYT1 ΔGAG mutation carriers and non-carriers, although symptomatic DYT1 dystonia patients exhibited increased anxiety, verbal memory retroactive interference, and semantic fluency performance (Balas et al. 2006). These conflicting findings led us to investigate whether the DYT1 ΔGAG mutation by itself truly causes anxiety and depression.
We previously reported the production of Dyt1 ΔGAG knock-in heterozygous (KI) mice as a genetic animal model of DYT1 dystonia (Dang et al. 2005). KI mice showed motor and dopaminergic deficits, and brainstem protein aggregation similar to DYT1 dystonia patients. In this study, we performed standard tests for anxiety- and depression-like behaviors, sensorimotor gating, and fear memory formation on these KI mice. KI mice showed significantly enhanced cued fear memory while their contextual fear memory was normal. To determine the mechanism of this alteration, we also performed electrophysiological recordings in hippocampal slices and quantified the number of c-fos positive cells within and the size of the CE and the lateral nucleus of the amygdala (LA).
2. Materials and methods
2.1. Animals
Two groups of KI mice and their littermates were prepared for behavior tests as previously described (Dang et al. 2005). Mice were genotyped using Tcko1 and Tcko2 primer sets (Dang et al. 2005) before the behavioral experiments. A group of 21 KI (11 males and 10 females) and 20 wild-type (WT; 10 males and 10 females) littermates from 6 litters was used in anxiety and depression-like behavioral tests. Mice were housed under a 12 hr-light and 12 hr-dark cycle. Behavior tests were performed within the last 6 hours of the light period after acclimation to a sound-attenuated testing room for 1 hr. Behavior tests were performed in the following order: elevated-plus maze, open field, light-dark box, tail suspension, and forced swimming tests. Mice were allowed to rest for one week in between the tests. The second group for prepulse inhibition and fear conditioning tests consisted of 15 KI and 13 control WT males from 6 litters. All of the behavior tests and immunohistochemistry were performed by investigators blind to the genotypes.
2.2. Elevated-plus maze test
Anxiety-related behavior was tested by an elevated plus-maze (PlusMaze version 1.41; AccuScan Instruments, Inc. OH), that consisted of a black plexiglass apparatus with four arms (30 cm long × 5 cm wide) positioned in a cross from a neutral central square (5 cm × 5cm) as described previously (Cao and Li 2002). Two opposite arms were surrounded from the three sides by vertical transparent plastic walls 20 cm in height (closed), while the other arms were without walls (open). The plus-maze was located 50 cm above the floor under dim indirect lighting (approximately 100 lux on the maze platform). Each mouse (120 to 137 days old) was placed in the center of the maze facing an open arm. The activity of the mouse was videotaped for 5 min. The number of entries and the time spent in each arm were measured.
2.3. Open-field test
Locomotor activities were measured by infrared sensors of an open-field apparatus connected to a computerized Digiscan System (Accuscan Instruments, Inc. OH) as described previously (Cao and Li 2002). Each mouse (128 to 146 days old) was placed in the center of the chamber under bright illumination (approximately 1 k lux at the center by a 60 W white bulb) focused on the center of the field. Locomotor activities were automatically recorded by counting the number of breaks in the infrared beams for 15 min.
2.4. Light-dark box test
Anxiety-related locomotion was also measured in a light-dark box as described previously (File et al. 2004). A light box made of transparent plastic (27 cm × 21 cm × 21 cm; illuminated by a 60 W white bulb) and a dark box made of black plastic (27 cm × 21 cm × 21 cm) were connected by a small opening (5 × 12 cm) (Cao and Li 2002). Each mouse (137 to 154 days old) was placed in the center of the light box facing away from the dark box. Transition events between the two boxes as well as the activity and time spent in the dark box were automatically recorded for 10 min by infrared sensors of the apparatus (Accuscan Instruments, Inc. OH).
2.5. Tail suspension test
Depression-like behavior was assessed by the standard tail suspension test (TST) using mice ranging from 144 to 161 days old (Porsolt et al. 2001; Yokoi et al. 2006). Each mouse was suspended by its tail with adhesive tape attached to a rope and videotaped. The total immobility time (sec) during a 6 min interval was evaluated. Mice that climbed up the rope were excluded from the statistical analysis (3 KI males, 5 KI female, 3 WT males and 2 WT females were excluded).
2.6. Forced swim test
Depression-like behavior was also assessed by the forced swim test (FST), one week after TST (Porsolt et al. 2001). Each mouse was placed in a glass jar (diameter = 22 cm, height = 26 cm) filled with water at about 20°C to the height of 11 cm for 6 min. The test was videotaped and the total immobility time (sec) during the last 4 min was evaluated.
2.7. Prepulse inhibition
Prepulse inhibition of acoustic startle responses was measured using the Med-Associates System (Med Associates, St. Albans, VT). Mice ranging from 166 to 181 days old were first habituated to the startle chamber and plexiglass cylinder for 5 min a day for 3 days to reduce stress and unnecessary movements during the test session. On the fourth day, prepulse inhibition testing was performed. The test began with a 5 min acclimation period where the mice were left in the chamber's cylinder undisturbed. The remainder of the test session consisted of three blocks of trials. The first block consisted of six 40 msec, 120 dB sound bursts used as a startle stimulus. The second and third blocks consisted of 26 trials each, with five different trial types: startle only stimulus, no stimulus, or a 20 msec prepulse sound at 4, 8 or 16 dB above the background noise level (65 dB) presented 100 msec before the startle stimulus. The trial types were presented in pseudorandom order throughout each block with a variable inter-trial interval (ranged from 8-23 sec). The maximum startle amplitude recorded during the 50 msec sampling window was used as the dependent variable. The percent prepulse inhibition of the startle response was calculated as: (startle response to the startle stimuli alone - startle with the prepulse / startle to startle stimuli alone) × 100.
2.8. Hippocampal Slice Preparation and Electrophysiology
Preparation of hippocampal slice and electrophysiological analysis were performed as described previously (Levenson et al. 2004). The mouse brain was immersed in ice-cold cutting saline [CS (in mM): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 glucose, 0.6 ascorbate] prior to isolation of the caudal portion containing the hippocampus and entorhinal cortex. Transverse slices (400 μm) were prepared with a Vibratome (The Vibratome Company, St. Louis, MO). After isolation, cortical tissue was removed and hippocampal slices were equilibrated in a mixture of 50% CS and 50% artificial cerebrospinal fluid [ACSF (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 glucose] at room temperature. Slices were further equilibrated in 100% ACSF for 45 min at room temperature, followed by a final incubation in 100% ACSF at 32 °C for 1 h. All solutions were saturated with 95% O2/5% CO2. Electrophysiological analysis was performed in an interface chamber (Fine Science Tools, Foster City, CA). Oxygenated ACSF (95% O2/5% CO2) was perfused into the recording chamber at a rate of 1 ml/min. Electrophysiological traces were digitized and stored using Digidata (models 1200 and 1320A) and Clampex software (Axon Instruments, Union City, CA). Extracellular stimuli were administered on the border of areas CA3 and CA1 along the Schaffer-collaterals using Teflon-coated, bipolar platinum electrodes. Field excitatory postsynaptic potentials (fEPSPs) were recorded in stratum radiatum with an ACSF-filled glass recording electrode (1–3 MΩ). All subsequent experimental stimuli were set to an intensity that evoked a fEPSP that had a slope of 50% of the maximum fEPSP slope. LTP was induced by administering three trains of burst stimulation. Each train consisted of 10 sets of bursts (4 stimuli, 100 Hz) with an inter-burst interval of 200 ms. There was a 20 s interval between each stimulus train. Synaptic efficacy was monitored 30 min prior to and 120 min following induction of LTP by recording fEPSPs every 20 s (traces were average for every 2-min interval).
2.9. Contextual and cued fear conditioning
Emotional memory formation and recall was assessed by contextual and cued fear conditioning test as described (Shalin et al. 2006). Fifteen KI and 13 WT male mice from 188 to 203 days old were tested in an automated fear conditioning system (Video Freeze Ver. 1.20.0.0, Med Associates Inc.). Mice were trained by a cue-context training program on the morning of the first day in contextual chambers with wire shock grid floors. Mice were first allowed to explore the context for 3 min before presentation of the first 30 sec tone at 90 dB, which co-terminated with a 1.0 sec, 0.5 mA foot shock. Three tone-shock pairings were presented in total with an inter-trial interval of 90 sec. Mice remained in the chamber for an additional 90 sec before being returned to their home cage. The chamber was cleaned with 70% ethanol between animals. Twenty-four hrs later, mice were monitored in the context chambers (no tone or shock) for their freezing behavior for 5 min. In the afternoon on the same day, the visual, tactile and olfactory cues of the context were significantly altered. Mice were then allowed to explore this novel environment for 3 min before presentation of the tone (no shock) for 3 min. The time each mouse spent in a fixed position without movement during the 3 min tone presentation was evaluated as cued freezing. The chamber was cleaned with 75% isopropanol between animals. Freezing was monitored by video and evaluated by a Med Associates Video Freeze software. The freezing time (sec) was divided by session time (for context, 5min; for cued, 3 min) and expressed as % freezing.
2.10. Immunohistochemistry
Four KI and four WT males (37 to 40 days old) from two litters were anesthetized and perfused with ice-cold 0.1 M phosphate-buffer saline (pH 7.4; PBS) followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4; PB). The brains were soaked in the paraformaldehyde-PB at 4°C overnight and then incubated in 30% sucrose in 0.1 M PB at 4°C overnight until the brains sank. The brains were frozen using dry-ice powder and cut into 50 μm sections with a Histoslide 2000 sliding microtome (Reichert-Jung). Every fourth section was collected in 0.1 M PB. Immunohistochemisty was performed with Vectastain ABC kit for peroxidase rabbit IgG according to the manufacturer's protocol. The floating sections were treated with water for 5 min and then incubated with 0.3% H2O2 for 30 min. After washing with 10 mM PBS for 5 min, the sections were blocked with goat normal serum for 20 min and then incubated with rabbit c-fos polyclonal antibody (Santa Cruz; sc-52) at 1:50 dilution for 30 min. After washing with 10 mM PBS for 5 min, the sections were treated with biotinylated secondary antibody from the kit for 30 min and washed with 10 mM PBS for 5 min. The sections were treated with ABC reagent for 30 min and washed with 10 mM PBS for 5 min. The sections were stained with DAB peroxidase substrate kit with nickel solution (Vector Lab). After washing with water followed by 10 mM PB, the sections were mounted on slides and dried. The sections were treated with Xylene for 5 min and cover-slipped with DPX (Fluka). For every four sections collected, three continuous sections were selected for analysis of the CE and LA cell counts (Franklin and Paxinos 2008). The number of c-fos positive cells in the CE, which consists of the capsular, lateral, and medial divisions, and those in LA, which consists of the dorsolateral and ventrolateral divisions, were counted by using Nikon ECLIPSE E800M microscope with 10 × Plan Fluor objective lens and 10 × CFIUW ocular lens. The areas of the CE and LA were estimated by using ImageJ software (NIH) and a 0.01 mm objective micrometer.
2.11. Statistical analysis
Statistics were analyzed using SAS/STAT Analyst software (version 9) for anxiety tests, total immobility time for TST and FST and fear conditioning test as described (Yokoi et al. 2006). Data underwent square root transformation to obtain normal distribution to fit the model as appropriate. Genotype, sex, age and weight were considered in the models. Prepulse inhibition data was analyzed using a mixed factor ANOVA with genotype as the between subjects factor and prepulse as the within subjects factor. Fear conditioning was analyzed using Oneway ANOVA. The number of c-fos positive cells and the area (mm2) of both the right and left CE, and the right and left LA from three sections per mouse were analyzed by student's t-test. Significance was assigned at P < 0.05.
3. Results
3.1. Anxiety-like behaviors
KI and WT littermate mice were prepared as previously described (Dang et al. 2005) and were genotyped using tail DNA prior to the behavior tests (Fig. 1). Anxiety-like behavior was first evaluated using an elevated-plus maze test. KI mice exhibited significantly fewer entries into the open arms (P = 0.0191; Fig. 2A), suggesting an anxiety –like behavior. However, there was no significant difference in the number of entries into the closed arms (P = 0.4881; Fig. 2B), the percentage of open arm entries compared to the total number of arm entries (P = 0.7884; Fig. 2C), or time spent in the open arms (P = 0.4138; Fig. 2D) between KI and WT mice. Therefore the observed anxiety-like behavior appears to be subtle.
Fig. 1. PCR-based genotyping.
The top band (340bp) is the PCR product from Dyt1 ΔGAG knock-in locus and the bottom band (200bp) is from the Wild type (WT) locus. Lanes 1, 5, 6, 8 and 11: Dyt1 ΔGAG knock-in heterozygous mice. Lanes 2, 3, 4, 7, 9 and 10: WT mice.
Fig. 2. Elevated-plus maze test.

KI mice exhibited significantly less entries onto the open arms (A). However, KI mice did not exhibit any significant differences in the entries onto the closed arms (B), the percentage of open arm entries compared to total arm entries (C), or the time spent on the open arms (D). *P < 0.05. Vertical bars represent means ± standard errors (SE).
Anxiety-like behavior was also assessed by the open-field test. Because mice with high anxiety-like behavior prefer peripheral areas in the open-field apparatus, the time spent in the center vs. time spent in the entire area of the field and the ratio of distance traveled in the center vs. distance traveled in the entire area were measured. There was no significant difference between KI and WT mice in either the time spent in the central area (Fig. 3A; P = 0.9219) or the ratio of distances traveled in the center (Fig. 3B; P = 0.6964). The results suggest that KI mice do not exhibit anxiety-like behavior in the open-field test.
Fig. 3. Open-field and light-dark box tests.

In the open-field test, KI mice did not exhibit significant difference in either the time spent in the central area of the field (A) or central distance ratio (B) in the 15 min session. The data were analyzed after square root transformation to obtain a normal distribution. In the light-dark box test, KI mice did not exhibit a significant difference in time spent in the dark box (C) or total number of light-dark transitions (D). Vertical bars represent means ± SE.
Anxiety-like behavior was further assessed by a light-dark box test. Increased time and activity in the dark box or decreased number of transition between the two compartments are indicative of increased anxiety (File et al. 2004). There was no significant difference between the time KI and WT mice spent in the dark box (Fig. 3C; P = 0.1420) and the total number of transitions between light and dark box (Fig. 3D; P = 0.9633). The results suggest that KI mice do not exhibit anxiety-like behavior in the light-dark box test.
3.2. Depression-like behaviors and sensorimotor gating
Depression-like behaviors were assessed by the tail suspension test (TST) and forced swimming test (FST). The immobility times in TST were normally distributed. There was no significant difference in total immobility time between KI and WT mice (P = 0.3421; Fig. 4A). The immobility times in FST were transformed by taking the square root of each data point to obtain a normal distribution. There was no significant difference in the immobility time between KI and WT mice (P = 0.4890; Fig. 4B).
Fig. 4. Tail suspension and forced swim tests.

KI mice did not exhibit significant difference in immobility time (sec) in TST (A). KI mice did not exhibit significant difference in immobility time in FST (B). The FST data (sec) were analyzed after square root transformation to obtain a normal distribution. Vertical bars represent means ± SE.
Sensorimotor gating was assessed by a prepulse inhibition test with 3 different intensities of prepulse (4, 8, 16 dB above background). There was no significant difference between WT and KI mice in these three conditions (4dB, F(1,22) = 0.036, P = 0.852; 8dB, F(1,22) = 0.236, P = 0.632; 16dB, F(1,22) = 0.415, P = 0.526; Startle, F(1,22) = 0.000, P = 0.988; Fig. 5), suggesting that sensorimotor gating is not altered by the Dyt1 ΔGAG mutation.
Fig. 5. Prepulse inhibition.

KI mice did not exhibit a significant difference from WT mice in prepulse inhibition measured at 4, 8, and 16 dB, suggesting that their sensorimotor gating was normal. Vertical bars represent means ± SE.
3.3. Hippocampal slice electrophysiology and emotional memory
Recently, it was demonstrated that the expression of genes associated with glutamate receptor mediated synaptic plasticity and fear conditioning is altered in transgenic mice over-expressing human torsinA proteins (Grundmann et al. 2008). We first performed field recordings in hippocampal slices to determine whether synaptic plasticity is altered in KI mice. In the CA1 region of the hippocampus, glutamate receptor-mediated LTP is well documented and highly reproducible. Both WT and KI mice showed robust CA1 theta-burst-induced LTP which persisted for at least 2 h. No difference was found 2 h after LTP induction (WT, 178 ± 14%, n=22 slices from 5 mice; KI, 168 ± 16%, n=32 slices from 6 mice, P > 0.05; Fig. 6A). These results suggest that hippocampal theta-burst-induced LTP is not altered by the Dyt1 ΔGAG mutation.
Fig. 6. Hippocampal slice electrophysiology.
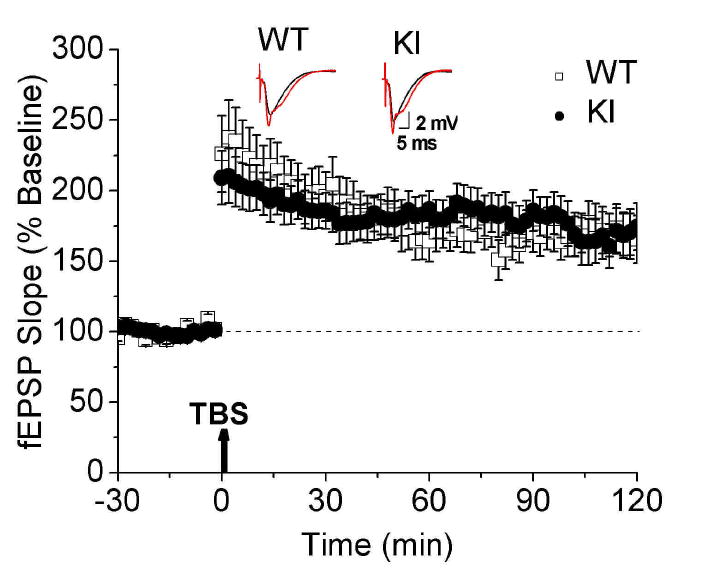
Theta-burst-induced LTP and basal synaptic transmission in the CA1 region of the hippocampus of WT and KI mice. There was no significant difference in long term potentiation between groups. The dashed lines indicate the mean basal synaptic responses and arrows show the induction of LTP. Inset shows representative traces 4 min before (black traces) and 2 h after LTP (red). Average results from 22 WT and 32 KI slices (means ± SE).
To verify our findings on CA1 LTP, we analyzed the ability of these mice to form contextual and cued fear memories. A deficiency in CA1 LTP is associated with impairment in contextual fear conditioning (Abel et al. 1997). Fear conditioning was performed by presenting three tones, each ending with a mild foot shock (Fig. 7A). The following day, fear memory was assessed as the animals' freezing response to presentation of either the context alone (hippocampus-dependent task) or cue alone (amygdala-dependent task). There was no significant difference between WT and KI mice in contextual fear conditioning (F(1,26) = 0.264, P = 0.612; Fig. 7B). However, KI mice displayed enhanced freezing in cued fear conditioning (F(1,26) = 5.08, P = 0.033; Fig. 7B), suggesting altered amygdala function in KI mice.
Fig. 7. Contextual and cued fear conditioning.

(A) Schematic diagram of training procedures on the first day. Conditioning to 30-second tones followed by electric footshocks was repeated three times with silent intervals. The black rectangles indicate the period of tone. The vertical bar is the onset and duration of the electric shock. (B) Percentage of freezing episode in contextual and cued conditioning. Although there is no significant difference in contextual conditioning, KI mice exhibited significantly increased freezing in cued conditioning, suggesting higher fear memory formation. *P < 0.05. Vertical bars represent means ± SE.
3.4. Immunoreactivity to c-fos antibody and the size of the CE and LA
To elucidate the mechanism of the increased fear memory in KI mice, we counted the number of cells that were stained with c-fos antibody in the CE and LA, and estimated the sizes of these regions as previously described (Lehner et al. 2004). We analyzed the capsular, lateral and medial divisions together as the CE between WT and KI littermates (Fig. 8A, B). The number of c-fos positive cells in the CE was significantly increased in KI mice (P = 0.0499; Fig. 8C). On the other hand, the size of the CE in KI mice was significantly smaller than that of WT mice (P = 0.0112; Fig. 8D). The density of c-fos positive cells in the CE was calculated from the above data for each mouse and normalized to WT mice. The density in KI mice was doubled (P = 0.0145; Fig. 8E). These results suggest that there were more active cells in the smaller CE in KI mice. On the other hand, there is no statistical significance in the number of c-fos positive cells (P = 0.0742; Fig. 8F), area (P = 0.1985; Fig. 8G) or the density of c-fos positive cells (P = 0.1691; Fig 8H) in the LA, suggesting the activation is specific to the CE.
Fig. 8. Immunohistochemistry of the CE with c-fos antibody.
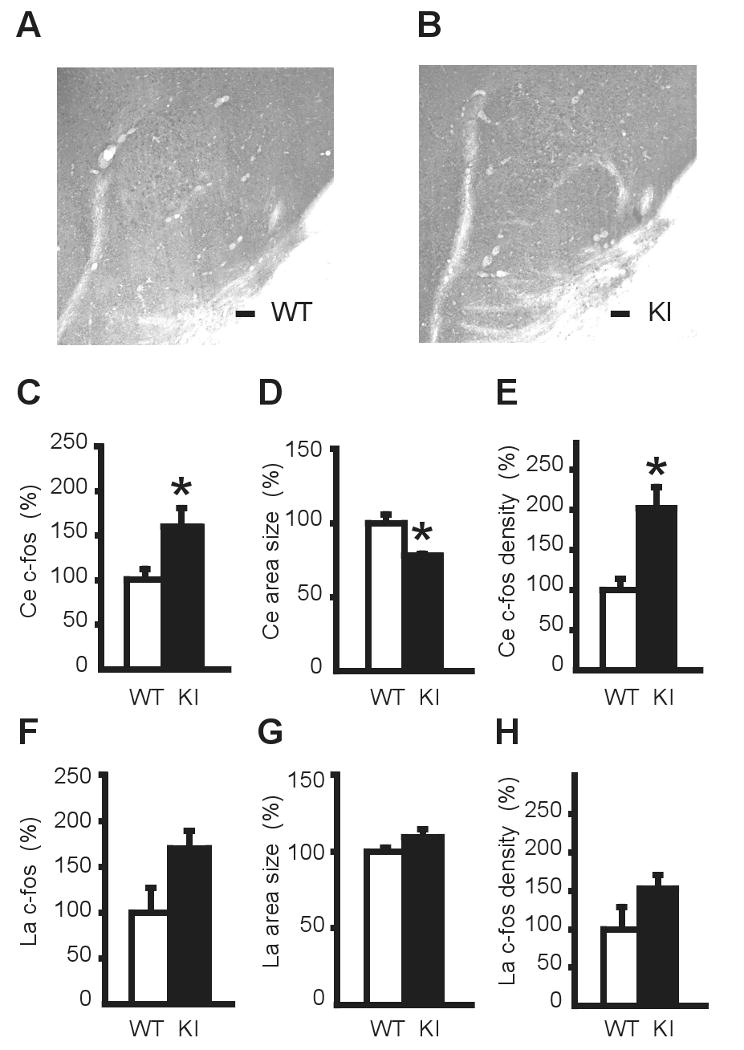
Representative microscope images of the CE in WT (A) and KI (B) mice. Scale bars: 100 μm. (C) The number of c-fos positive cells in the CE was counted. KI mice had significantly more c-fos positive cells than WT mice. The data were normalized to WT and expressed as percentage. (D) The area of the CE in KI mice was significantly smaller than that of WT mice. The area of the CE from three sections for each KI mouse was measured and normalized to that of WT mice. (E) The density of c-fos positive cells in the CE was significantly increased in KI mice. On the other hand, in the LA there is no statistical significance in the number of c-fos positive cells (F), area size (G) or the density of c-fos positive cells (H). *P < 0.05. Vertical bars represent means ± SE.
4. Discussion
In this report we analyzed the non-motor behavior of KI mouse, a genetic model of DYT1 dystonia. The KI mice showed only subtle alteration in anxiety-like behavior and did not show either depression-like behaviors or alteration in sensorimotor gating. To gain further insight into the non-motor behaviors and glutamate receptor-mediated synaptic plasticity in KI mice, we analyzed hippocampal CA1 LTP and fear memory formation. KI mice showed normal CA1 LTP and contextual fear conditioning. However, cued fear conditioning was significantly enhanced in KI mice. Anatomical analysis indicated that the number of c-fos positive cells was significantly increased while the size of the CE was significantly reduced in KI mice.
We also analyzed anxiety-like behaviors of KI mice using three different tests. The elevated-plus maze test is based on the conflict between exploration and fear of open and/or elevated places (File et al. 2004). On the other hand, anxiety-like behaviors in the open-field test are based on the conflict between exploration and fear of open space. KI mice showed significantly decreased open arm entries in the elevated-plus maze test, suggesting a possible increase of anxiety-like behavior. However, they showed normal behaviors as indicated by other parameters in the elevated-plus maze, and in both open-field and light-dark box tests, suggesting that the observed deficit was subtle.
Since there is no specific rodent test for recurrent depression, we used common tests of depression in this study, FST and TST. These well-established tests are based on two different neuronal mechanisms. FST changes monoamine metabolism in the brain while TST does not (Renard et al. 2003). There was no significant difference in total immobility time between KI mice and WT mice in either test, which showed that no gross depression-like behaviors were detected. The results support the report which suggested that there was no difference between DYT1 ΔGAG mutation carriers and non-carriers in depression (Balas et al. 2006). Moreover the results may also be consistent with another report that there was no significant difference in the frequency of depression between carriers and non-carriers when the data were analyzed after combining both single and recurrent major depression as a single group (Heiman et al. 2004).
KI mice showed enhanced cued fear conditioning response while contextual fear conditioning and prepulse inhibition were normal. Fear conditioning is a well-characterized model of mammalian associative learning (Grillon 2002; Fanselow and Poulos 2005). The amygdala is responsible for the expression of both contextual and cued fear conditioning (Phillips and LeDoux 1992). In the acquisition process of the cued fear memory, tone and shock information are carried into the lateral amygdala. On the other hand, contextual information is processed in the hippocampus and carried to the lateral amygdala and basal nucleus. The integrated information is transferred into the CE and freezing behavior is expressed as an output of fear (Sotres-Bayon et al. 2006). Therefore, the activity of the CE contributes to the expression of fear-related behaviors. Since high expression of c-fos is an indicator of high activity of these cells, the increase in c-fos expression in the CE suggest that the increased fear behavior in KI mice was based on the increased activity of the CE.
We used naïve mice for the c-fos immunohistochemistry analysis; the abnormal activation of the CE in KI mice is basal activation without any treatment to the live mouse or tissue section. Since this activation seems to be specific to the CE, a functional difference between the CE and LA may contribute to the specific activation. The CE not only functions in relaying the fear signals to the periaqueducal gray (LeDoux 2007), but also receiving peripheral pain signals via the spino(Trigemino)pontoamygdaloid pathway (Bernard and Besson 1990; Bernard et al. 1992). Therefore the CE seems to be a place that integrates both efferent fear signals and afferent pain signals. Since dystonia patients often feel pain derived from sustained muscle contraction, there is a possibility that KI mice also feel pain from muscles and the abnormal activation of the CE is caused by pain signals from the peripheral nerves in dystonic muscles. The c-fos expression as shown in this study has also been used as a neural marker of pain (Bon et al. 1998; Harris 1998). Detailed analysis of activities in the pain signal pathways in KI mice may uncover the mechanism of abnormal activation of the CE and increased fear memory. Since chronic pains also contribute to a high incidence of anxiety disorders, the observed subtle anxiety-like behavior may also be caused by chronic pains (Manchikanti et al. 2002).
The abnormal activation of the CE may be caused by the chronic activation of upstream pathways for cued fear memory formation. In the cued conditioning test, the auditory thalamus and auditory cortex process the conditioned stimulus of the tone while the somatosensory thalamus and somatosensory cortex process the unconditioned stimulus of the electric footshock (Phelps and LeDoux 2005). These signals are integrated into the lateral nucleus of the amygdala and then project to the CE. If these pathways are also chronically activated by the mutation, only cued fear memory should be enhanced. Dystonia is a hyperkinetic disorder and multiple brain regions are abnormally activated in humans (Vitek 2002). A recent report has suggested that DYT1 dystonia is a neurodevelopmental disorder involving cortico-striatal-pallido-thalamocortical and related cerebellar-thalamo-cortical pathways (Carbon and Eidelberg 2009). A possible mechanism of this enhanced cued fear memory is that abnormally enhanced signals of these pathways may overflow to the pathways related to cued fear memory formation leading to abnormal activation of the CE. Identification of a circuit that contributes to the hyperactivation of the CE remains to be elucidated.
Alternatively, cholinergic neurons in the striatum, a region of motor coordination control and motor learning (Dang et al. 2006b), may be abnormally activated in dystonia (Pisani et al. 2006). Anticholinergics are used in clinical practice for various dystonias (Jankovic 2006). Trihexyphenidyl, an antagonist to muscarinic acetylcholine receptor, is one of the effective drugs for torsion dystonia (Burke et al. 1986). Acetylcholine also modulates learning and memory in the amygdala and hippocampus (van der Zee and Luiten 1999; McIntyre et al. 2002; Gold 2003; Gold 2004). Activation of muscarinic cholinergic M1 and M2 receptors affect consolidation of fear memory (Power et al. 2003). Moreover, a recent report suggested that chronic nicotine impairs cued fear extinction (Tian et al. 2008) implying nicotinic cholinergic receptors increases fear memory. Interestingly, high memory performance was reported in DYT1 dystonia patients. DYT1 dystonia patients exhibit higher semantic fluency performance (Balas et al. 2006). Cholinergic neurons are known to contribute also to verbal memory formation and recall (Freo et al. 2002). Although previous reports also suggested a relationship between torsion dystonia and high intelligence quotient (IQ) (Eldridge et al. 1969; Eldridge et al. 1970; Eldridge et al. 1971), it is not clear whether the DYT1 mutation affects IQ. Measuring IQ of DYT1 mutation carriers may elucidate other novel neuro-cognitive aspects of this genetic disease. Finally, acetylcholine also functions in the neocortex and it is possible that acetylcholine modulates fear memory extinction through projections from prefrontal cortex to the amygdala. We have shown in a previous study the contribution of cerebral cortex in the pathogenesis of DYT1 dystonia (Yokoi et al. 2008).
The reason for the reduced size of the CE is still unclear. Since we analyzed the naïve mice, we propose that the reduction may have occurred in the developmental process to compensate for the enhanced activity of this region. The hyperactive neurons may release more neurotransmitters at excitotoxic levels and affect the development of this region. Excitotoxic effects of neurotransmitters with increased c-fos expression have been reported in several different reports (Gorman et al. 1995; Griffiths et al. 1997). Whether this is the case in DYT1 ΔGAG remains to be determined.
Acknowledgments
We thank Lisa Foster, Andrea McCullough and their staff for animal care, Mark Kilgore, Miki Jinno, Veena Ganesh, Dee Parsons, Chad C. Cheetham, Drs. Xinru Joy Li and Buffie Clodfelder-Miller for their technical assistance and advice. This work was supported by National Institutes of Health grants [NS47692, NS54246, NS57098, and NS47466] and the start up funds from the Lucille P. Markey Charitable Trust (UIUC) and Department of Neurology (UAB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Balas M, Peretz C, Badarny S, Scott RB, Giladi N. Neuropsychological profile of DYT1 dystonia. Mov Disord. 2006;21:2073–2077. doi: 10.1002/mds.21070. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1990;63:473–490. doi: 10.1152/jn.1990.63.3.473. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Huang GF, Besson JM. Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1992;68:551–569. doi: 10.1152/jn.1992.68.2.551. [DOI] [PubMed] [Google Scholar]
- Bon K, Lanteri-Minet M, Michiels JF, Menetrey D. Cyclophosphamide cystitis as a model of visceral pain in rats: a c-fos and Krox-24 study at telencephalic levels, with a note on pituitary adenylate cyclase activating polypeptide (PACAP) Exp Brain Res. 1998;122:165–174. doi: 10.1007/s002210050504. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Burke RE, Fahn S, Marsden CD. Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology. 1986;36:160–164. doi: 10.1212/wnl.36.2.160. [DOI] [PubMed] [Google Scholar]
- Camargos S, Scholz S, Simon-Sanchez J, Paisan-Ruiz C, Lewis P, Hernandez D, Ding J, Gibbs JR, Cookson MR, Bras J, Guerreiro R, Oliveira CR, Lees A, Hardy J, Cardoso F, Singleton AB. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7:207–215. doi: 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- Cao BJ, Li Y. Reduced anxiety- and depression-like behaviors in Emx1 homozygous mutant mice. Brain Res. 2002;937:32–40. doi: 10.1016/s0006-8993(02)02461-7. [DOI] [PubMed] [Google Scholar]
- Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Eidelberg D. Abnormal structure/function relationships in hereditary dystonia. Neuroscience. 2009 doi: 10.1016/j.neuroscience.2008.12.041. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y. Generation and Characterization of Dyt1 deltaGAG Knock-in Mouse as a Model for Early-Onset Dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Pence MA, Li Y. Motor deficits and hyperactivity in Dyt1 knockdown mice. Neurosci Res. 2006a;56:470–474. doi: 10.1016/j.neures.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006b;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doheny D, Danisi F, Smith C, Morrison C, Velickovic M, De Leon D, Bressman SB, Leung J, Ozelius L, Klein C, Breakefield XO, Brin MF, Silverman JM. Clinical findings of a myoclonus-dystonia family with two distinct mutations. Neurology. 2002;59:1244–1246. doi: 10.1212/wnl.59.8.1244. [DOI] [PubMed] [Google Scholar]
- Dressler D, Benecke R. Diagnosis and management of acute movement disorders. J Neurol. 2005;252:1299–1306. doi: 10.1007/s00415-005-0006-x. [DOI] [PubMed] [Google Scholar]
- Eldridge R, Edgar A, Cooper IS. Genetics, geography and intelligence in the torsion dystonias. Birth Defects Orig Artic Ser. 1971;7:167–177. [PubMed] [Google Scholar]
- Eldridge R, Harlan A, Cooper IS, Riklan M. The hereditary torsion dystonias (dystonia musculorum deformans): Geographical distribution and IQ in dominant and recessive forms. Trans Am Neurol Assoc. 1969;94:248–250. [PubMed] [Google Scholar]
- Eldridge R, Harlan A, Cooper IS, Riklan M. Superior intelligence in recessively inherited torsion dystonia. Lancet. 1970;1:65–67. doi: 10.1016/s0140-6736(70)91848-9. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- File SE, Lippa AS, Beer B, Lippa MT. Animal Tests of Anxiety. In: Crawley J, editor. Current Protocols in Neuroscience. John Wiley & Sons, Inc.; 2004. pp. 8.3.1–8.3.22. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in stereotaxic coordinates. Elsevier Inc.; San Diego: 2008. [Google Scholar]
- Freo U, Pizzolato G, Dam M, Ori C, Battistin L. A short review of cognitive and functional neuroimaging studies of cholinergic drugs: implications for therapeutic potentials. J Neural Transm. 2002;109:857–870. doi: 10.1007/s007020200070. [DOI] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- Gold PE. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem. 2003;80:194–210. doi: 10.1016/j.nlm.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Gold PE. Coordination of multiple memory systems. Neurobiol Learn Mem. 2004;82:230–242. doi: 10.1016/j.nlm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Gorman AM, Scott MP, Rumsby PC, Meredith C, Griffiths R. Excitatory amino acid-induced cytotoxicity in primary cultures of mouse cerebellar granule cells correlates with elevated, sustained c-fos proto-oncogene expression. Neurosci Lett. 1995;191:116–120. doi: 10.1016/0304-3940(95)11554-2. [DOI] [PubMed] [Google Scholar]
- Griffiths R, Malcolm C, Ritchie L, Frandsen A, Schousboe A, Scott M, Rumsby P, Meredith C. Association of c-fos mRNA expression and excitotoxicity in primary cultures of mouse neocortical and cerebellar neurons. J Neurosci Res. 1997;48:533–542. doi: 10.1002/(sici)1097-4547(19970615)48:6<533::aid-jnr6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Grillon C. Startle reactivity and anxiety disorders: aversive conditioning, context, and neurobiology. Biol Psychiatry. 2002;52:958–975. doi: 10.1016/s0006-3223(02)01665-7. [DOI] [PubMed] [Google Scholar]
- Grundmann K, Hubener J, Habig K, Reischmann B, Poths S, Hauser TK, Magg J, Riess O, Bonin M, Nguyen HP. Gene expression changes in a transgenic mouse model overexpressing human wildtype and mutant torsinA. Proteomics Clin Appl. 2008;2:720–736. doi: 10.1002/prca.200780053. [DOI] [PubMed] [Google Scholar]
- Hallett M. Pathophysiology of writer's cramp. Hum Mov Sci. 2006;25:454–463. doi: 10.1016/j.humov.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Harris JA. Using c-fos as a neural marker of pain. Brain Res Bull. 1998;45:1–8. doi: 10.1016/s0361-9230(97)00277-3. [DOI] [PubMed] [Google Scholar]
- Heiman GA, Ottman R, Saunders-Pullman RJ, Ozelius LJ, Risch NJ, Bressman SB. Increased risk for recurrent major depression in DYT1 dystonia mutation carriers. Neurology. 2004;63:631–637. doi: 10.1212/01.wnl.0000137113.39225.fa. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000;9:1403–1413. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864–872. doi: 10.1016/S1474-4422(06)70574-9. [DOI] [PubMed] [Google Scholar]
- Lauterbach EC, Freeman A, Vogel RL. Correlates of generalized anxiety and panic attacks in dystonia and Parkinson disease. Cogn Behav Neurol. 2003;16:225–233. doi: 10.1097/00146965-200312000-00004. [DOI] [PubMed] [Google Scholar]
- Lauterbach EC, Freeman A, Vogel RL. Differential DSM-III psychiatric disorder prevalence profiles in dystonia and Parkinson's disease. J Neuropsychiatry Clin Neurosci. 2004;16:29–36. doi: 10.1176/jnp.16.1.29. [DOI] [PubMed] [Google Scholar]
- LeDoux J. The amygdala. Curr Biol. 2007;17:R868–874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lehner M, Taracha E, Skorzewska A, Wislowska A, Zienowicz M, Maciejak P, Szyndler J, Bidzinski A, Plaznik A. Sensitivity to pain and c-Fos expression in brain structures in rats. Neurosci Lett. 2004;370:74–79. doi: 10.1016/j.neulet.2004.07.089. [DOI] [PubMed] [Google Scholar]
- Lemke MR, Fuchs G, Gemende I, Herting B, Oehlwein C, Reichmann H, Rieke J, Volkmann J. Depression and Parkinson's disease. J Neurol. 2004;251 6:VI, 24–27. doi: 10.1007/s00415-004-1606-6. [DOI] [PubMed] [Google Scholar]
- Leung JC, Klein C, Friedman J, Vieregge P, Jacobs H, Doheny D, Kamm C, DeLeon D, Pramstaller PP, Penney JB, Eisengart M, Jankovic J, Gasser T, Bressman SB, Corey DP, Kramer P, Brin MF, Ozelius LJ, Breakefield XO. Novel mutation in the TOR1A (DYT1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism. Neurogenetics. 2001;3:133–143. doi: 10.1007/s100480100111. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zolkiewska A, Zolkiewski M. Characterization of human torsinA and its dystonia-associated mutant form. Biochem J. 2003;374:117–122. doi: 10.1042/BJ20030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchikanti L, Fellows B, Singh V. Understanding psychological aspects of chronic pain in interventional pain management. Pain Physician. 2002;5:57–82. [PubMed] [Google Scholar]
- McIntyre CK, Pal SN, Marriott LK, Gold PE. Competition between memory systems: acetylcholine release in the hippocampus correlates negatively with good performance on an amygdala-dependent task. J Neurosci. 2002;22:1171–1176. doi: 10.1523/JNEUROSCI.22-03-01171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Okun MS, Fernandez HF, Jacobson CEt, Rodriguez RL, Bowers D. Depression symptoms in movement disorders: comparing Parkinson's disease, dystonia, and essential tremor. Mov Disord. 2007;22:666–672. doi: 10.1002/mds.21376. [DOI] [PubMed] [Google Scholar]
- O'Suilleabhain P, Dewey RB., Jr Movement disorders after head injury: diagnosis and management. J Head Trauma Rehabil. 2004;19:305–313. doi: 10.1097/00001199-200407000-00005. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Pham P, Frei KP, Woo W, Truong DD. Molecular defects of the dystonia-causing torsinA mutation. Neuroreport. 2006;17:1725–1728. doi: 10.1097/WNR.0b013e3280101220. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Brossard G, Hautbois C, Roux S. Rodent Models of Depression: Forced Swimming and Tail Suspension Behavioral Despair Tests in Rats and Mice. In: Crawley J, editor. Current Protocols in Neuroscience. John Wiley & Sons, Inc.; 2001. pp. 8.10A.11–18.10A.10. [DOI] [PubMed] [Google Scholar]
- Power AE, McIntyre CK, Litmanovich A, McGaugh JL. Cholinergic modulation of memory in the basolateral amygdala involves activation of both m1 and m2 receptors. Behav Pharmacol. 2003;14:207–213. doi: 10.1097/00008877-200305000-00004. [DOI] [PubMed] [Google Scholar]
- Renard CE, Dailly E, David DJ, Hascoet M, Bourin M. Monoamine metabolism changes following the mouse forced swimming test but not the tail suspension test. Fundam Clin Pharmacol. 2003;17:449–455. doi: 10.1046/j.1472-8206.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- Richard IH, McDonald WM. Can “blue” genes affect mood and movement? Neurology. 2004;63:610–611. doi: 10.1212/01.wnl.0000137165.34087.d7. [DOI] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Cain CK, LeDoux JE. Brain mechanisms of fear extinction: historical perspectives on the contribution of prefrontal cortex. Biol Psychiatry. 2006;60:329–336. doi: 10.1016/j.biopsych.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Tian S, Gao J, Han L, Fu J, Li C, Li Z. Prior chronic nicotine impairs cued fear extinction but enhances contextual fear conditioning in rats. Neuroscience. 2008;153:935–943. doi: 10.1016/j.neuroscience.2008.03.005. [DOI] [PubMed] [Google Scholar]
- van der Zee EA, Luiten PG. Muscarinic acetylcholine receptors in the hippocampus, neocortex and amygdala: a review of immunocytochemical localization in relation to learning and memory. Prog Neurobiol. 1999;58:409–471. doi: 10.1016/s0301-0082(98)00092-6. [DOI] [PubMed] [Google Scholar]
- Vitek JL. Pathophysiology of dystonia: a neuronal model. Mov Disord. 2002;17 3:S49–62. doi: 10.1002/mds.10142. [DOI] [PubMed] [Google Scholar]
- Watson AH. What can studying musicians tell us about motor control of the hand? J Anat. 2006;208:527–542. doi: 10.1111/j.1469-7580.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Li J, Li Y. Myoclonus, motor deficits, alterations in emotional responses and monoamine metabolism in epsilon-sarcoglycan deficient mice. J Biochem. 2006;140:141–146. doi: 10.1093/jb/mvj138. [DOI] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Mitsui S, Li J, Li Y. Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. J Biochem. 2008;143:39–47. doi: 10.1093/jb/mvm191. [DOI] [PubMed] [Google Scholar]
- Zirn B, Grundmann K, Huppke P, Puthenparampil J, Wolburg H, Riess O, Muller U. Novel TOR1A mutation p Arg288Gln in early-onset dystonia (DYT 1) J Neurol Neurosurg Psychiatry. 2008 doi: 10.1136/jnnp.2008.148270. doi10.1136:in press. [DOI] [PubMed] [Google Scholar]