

Abstract
The ability of phospholipids to act as determinants of membrane protein structure and function is probably best exemplified by cardiolipin (CL), the signature phospholipid of mitochondria. Early efforts to reconstitute individual respiratory complexes and members of the mitochondrial carrier family, most notably the ADP/ATP carrier (AAC), often demonstrated the importance of CL. Over the past decade, the significance of CL in the organization of components of the electron transport chain into higher order assemblies, termed respiratory supercomplexes, has been established. Another protein required for oxidative phosphorylation, AAC, has received comparatively little attention likely stemming from the fact that AACs were thought to function in isolation as either homodimers or monomers. Recently however, AACs have been demonstrated to interact with the respiratory supercomplex, other members of the mitochondrial carrier family, and the TIM23 translocon. Interestingly, many if not all of these interactions depend on CL. As the paradigm for the mitochondrial carrier family, these discoveries with AAC suggest that other members of this large group of important proteins may be more gregarious than anticipated. Moreover, it is proposed that AAC and perhaps additional members of the mitochondrial carrier family might represent downstream targets of pathological states involving alterations in CL.
Keywords: ADP/ATP carrier, Cardiolipin, Mitochondrial carrier family, Oxidative phosphorylation, Respiratory supercomplex
1. Introduction
Whereas the importance of lipids in maintaining and establishing membrane barriers is accepted as dogma, the principles that guide how phospholipids influence the structure and function of the vast array of proteins associated with lipid bilayers are still emerging. At its most elemental, the lipid bilayer, a mixture of distinct lipid components, provides the matrix for membrane proteins. It is much more than simply a random matrix, however; for within this pool of lipids, specific protein-lipid interactions occur that have been demonstrated critical for the structure, incorporation, and/or assembly of proteins, protein complexes, or complexes of protein complexes [1-8]. The importance of specific protein-lipid interactions for the proper functioning of an organelle is perhaps best exemplified by mitochondria, the predominant (if not exclusive) subcellular location of the unique phospholipid, cardiolipin (CL).
CL is unique for at least three reasons. First, CL is almost exclusively found in association with the mitochondrial inner membrane, the membrane compartment in which it is synthesized. In fact, cardiolipin synthase, Crd1p, synthesizes CL in the context of the matrix-facing leaflet of the inner membrane [9]. Thus, to obtain its final distribution between both leaflets of the inner membrane [10-12], CL must flip to the intermembrane space apposed leaflet [13]. It has not yet been determined how this is accomplished mechanistically. Also noteworthy, while the majority of phospholipids are synthesized in a defined compartment, the endoplasmic reticulum [14], and then disseminated throughout the cell, CL, by-and-large, remains at its site of biosynthesis. This would seem to indicate that CL is critically important for this compartment. As a corollary, it also suggests that CL may be detrimental to the normal functioning of other membrane-bound organelles.
Second, as its pseudonym diphosphatidylglycerol implies, CL is a lipid dimer consisting of two phosphatidyl residues bridged by a glycerol [15]. Thus, CL has two phosphate headgroups that are associated at physiologic pH with a single negative charge [16, 17] and four attached fatty acyl chains. While none of the enzymes involved in the biosynthesis of CL display any acyl chain specificity [18, 19], the fatty acyl chain profile of CL in a given tissue or organism is not random, although it is often different in given tissues and organisms [20, 21]. Instead, steady state CL typically contains more unsaturated fatty acyl chains that exhibit a high degree of molecular symmetry [20-22]. Thus, newly synthesized CL is remodeled to obtain its final collection of attached fatty acyl chains [23, 24]. One pathway of CL remodeling involves the CL transacylase, tafazzin, the mutant gene product associated with the X-linked cardioskeletal myopathy, Barth syndrome [25-28]. The mere presence of a disease that is due to the absence of an enzyme that mediates CL remodeling suggests the physiological importance of this process. There are three significant changes in the phospholipid composition of mitochondria in Barth syndrome patients that confound this conclusion, however. Specifically, in Barth syndrome patients and models alike [20, 22, 29-36], mitochondria contain a reduced steady state level of CL, the remaining CL is asymmetrical and contains more saturated fatty acyl chains, and monolyso-CL, the intermediate in the CL remodeling pathway (which contains only three fatty acyl chains), accumulates. Any or all of these alterations could contribute to the numerous mitochondrial abnormalities observed in Barth syndrome patients.
Third, CL is a so-called structural phospholipid capable of vacillating between lamellar and inverted hexagonal structures in the absence or presence of divalent cations, respectively [37]. Nonbilayer lipids are hypothesized to participate in membrane curving and fusion events [38, 39]. Mitochondria are known to fuse and divide continuously [40-42]. Furthermore, two morphological hallmarks of mitochondria, namely cristae of the inner membrane and contact sites between the inner and outer membrane, are likely to involve nonbilayer lipid structures. The ability of CL to adopt nonbilayer hexagonal formations directly relates to its primary structure. Specifically, the phosphate headgroups of CL allow binding of divalent cations while the four attached typically unsaturated fatty acyl chains give CL a cone-shaped structure. While the physiological significance of the structural properties of lipids has not been clearly demonstrated, it is noteworthy that in yeast the combined absence in mitochondria of phosphatidylethanolamine, another cone-shaped phospholipid, and CL is synthetically lethal [43]. Interestingly, changes in the respiratory capacity of mitochondria include significant ultrastructural rearrangements of the inner mitochondrial membrane [44-48]. Moreover, actively respiring yeast contain more CL than yeast grown on fermentable carbon sources [30, 49, 50]. Again, the maintenance of CL in the mitochondrial inner membrane suggests that these non-bilayer capabilities are a) important for the normal functioning of the inner membrane, and b) potentially detrimental to the normal functioning of other membrane-bound structures.
CL is almost exclusively associated with membranes charged with the task of generating an electrochemical gradient that is used to produce ATP. Such membranes include the mitochondrial inner membrane and the bacterial plasma membrane. The mitochondrial inner membrane has an unusually high ∼3-4:1 protein:lipid ratio. In contrast, the protein:lipid ratio of the mitochondrial outer membrane is ∼1–1.6:1 [51-53]. While the contribution of CL to the establishment of this remarkably high concentration of proteins in the inner membrane has yet to be demonstrated, it is noteworthy that CL has the ability to interact with a number of different proteins, including all of the major players involved in oxidative phosphorylation [4, 6, 54-65]. Much recent attention has focused on the importance of CL for the structural organization of the respiratory complexes in higher order structures of functional importance [5, 7, 8]. A modern appreciation of the importance of CL in the structure and function of another critical component of the oxidative phosphorylation machinery, the ADP/ATP carrier (AAC), has ostensibly been forgotten. A comparison of the assembly status of oligomers of the ATP synthase, the respiratory supercomplexes, and AAC in wild type (+CL) and Δcrd1 (- CL) yeast clearly demonstrates the relative importance of CL for AAC (Fig. 1). Therefore, in this review, I will briefly discuss the importance of CL in the formation of the respiratory supercomplexes. I will then turn my attention to the critical role of CL for the normal structure and function of AAC, the charter member of the expanding mitochondrial carrier family. Used as a paradigm for this family of proteins, the demonstrated importance of CL for AAC biology suggests a general requirement of CL for the entire mitochondrial carrier family.
Figure 1. Assembly of ATP synthase oligomers, the respiratory supercomplex, and AAC2 complexes in the presence and absence of CL.
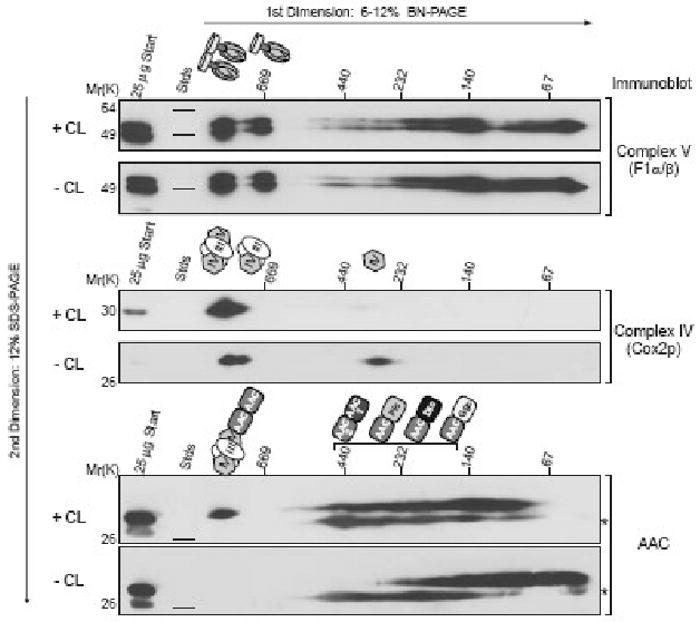
100 μg of 1.5% (wt/vol) digitonin extracts from mitochondria derived from wild type (+CL) or Δcrd1 (-CL) yeast were resolved by 2D BN/SDS–PAGE and immunoblots performed for complex V (F1α/β), complex IV (Cox2p), and AAC2. * highlights crossreaction with porin of the AAC antiserum. The migrations of the Vdimer, Vmonomer, III2IV2, III2IV, and IV supercomplexes are indicated schematically above the appropriate set of panels. The composition of the different AAC2 complexes is indicated above the AAC immunoblots. AAC1, ADP/ATP carrier isoform 1; Pic, phosphate carrier; Dic, dicarboxylate carrier; Ggc, GTP/GDP carrier. The interaction of AAC2 with the indicated mitochondrial carriers occurred in complexes ranging in size from ∼400-160 kDa. The exact size of each complex has not been determined; thus, the order of the depicted AAC2-carrier protein interactions is for illustrative purposes only. Details provided in text.
2. CL and Respiratory Supercomplexes
CL interacts with all of the major players in oxidative phosphorylation, including respiratory complexes I, III, IV, and V and the two members of the mitochondrial carrier family required for this process, AAC and the phosphate carrier (PiC; [4, 6, 54-65]). In vitro, CL is required to fully reconstitute the activity of respiratory complexes I, III, and IV [58, 65]. In contrast, yeast completely devoid of CL retained the capacity to perform oxidative phosphorylation, albeit at a reduced efficiency, under normal conditions [3, 66]. However, in the absence of CL, mitochondria failed to generate ATP under stressful conditions such as elevated temperatures [66]. Thus, while not absolutely required for oxidative phosphorylation in vivo, CL increases the dynamic range of conditions in which this process can occur. Even under optimal conditions, CL significantly enhances the efficiency of energy production.
Respiratory complexes, multisubunit complexes themselves, assemble in higher order structures referred to generically as respiratory supercomplexes [67, 68]. There are two general types of supercomplex, those involving components of the electron transport chain (complexes I, III, and IV) and those assembled using monomeric complex V (ATP synthase) as its building block. In mammalian mitochondria, the electron transport chain-containing supercomplex consists of respiratory complex I associated with a complex III dimer and from one to four copies of complex IV [68, 69]. In the yeast Saccharomyces cerevisiae which lacks complex I of the electron transport chain, the equivalent supercomplex is assembled using two complex IIIs as a central dimer scaffold with either one or two affiliated complex IVs (III2IV2 or III2IV; [68]). Electron transport chain-containing supercomplexes, herein called respiratory supercomplexes (also called respirasomes in the literature) are hypothesized to increase the efficiency of substrate channeling between individual complexes (e.g. cytochrome c between complexes III and IV). Consistent with this postulate, the respiratory complexes in yeast behave in vivo as a single functional unit [70]. Moreover, the cooperative activity of yeast complexes III and IV is lost upon solubilization of mitochondria with detergents that disrupt the respiratory supercomplex while preserving the integrity of the individual complexes [68]. Both the stability of the respiratory supercomplex and the in vivo cooperation of the electron transport chain is compromised in yeast lacking CL [1, 5, 7, 8]. Specifically, in the absence of CL there is an increased relative abundance of the small form of the respiratory supercomplex (III2IV) and free complex IV is detected (Fig.1 and [1, 5]). Importantly, these structural alterations in supercomplex stability are associated with a reduced coupling of ATP synthesis to oxygen consumption by the electron transport chain [1, 66, 71, 72], a commonly employed measure of the efficiency of oxidative phosphorylation. Thus, in yeast lacking CL, structural alterations in respiratory supercomplexes are observed that are directly associated with a functional consequence. The importance of CL for respiratory supercomplex assembly and function in mammalian mitochondria has not been reported. However, it is worth mentioning that aberrant respiratory supercomplexes were observed in fibroblasts derived from Barth syndrome patients [73]. While not a black-and-white situation for the reasons outlined in the Introduction, these observations do support a general role of CL in respiratory supercomplex stability in mammalian mitochondria.
Oligomers of the ATP synthase have been observed in yeast and mammalian mitochondria [68, 74]. In contrast to respiratory supercomplexes, ATP synthase oligomerization does not require CL, at least in yeast (Fig. 1 and [1, 5]). Oligomers of the ATP synthase are critical in the establishment and maintenance of normal cristae morphology [75-77].
3. AAC and CL
Like other members of the mitochondrial carrier family (also known as the SLC25 family), AACs are ∼300 amino acid polypeptides that encode proteins of 28-35 kDa with six transmembrane domains and a threefold pseudosymmetry [78]. Of this large family (∼50 human genes and 35 yeast genes), only AACs and PiCs are strictly required for oxidative phosphorylation. AACs (also called ANTs for adenine nucleotide transporter) mediate the 1:1 exchange of ADP into and ATP out of the mitochondrial matrix across the inner membrane. PiCs, either in symport with H+ or antiport with hydroxyl ions, transport Pi into the matrix. Thus, the combined activity of AACs and PiCs delivers both substrates (ADP and Pi) that the ATP synthase utilizes to generate ATP. Moreover, both processes are energetically costly due to their partial collapse of the electrochemical gradient established by the electron transport chain. In fact, it has been estimated that as much as one half of the energy generated by the electron transport chain is used to drive these two transport pathways [79]. In addition to this critical physiological activity, AAC and PiC have been suggested to participate in certain forms of apoptosis which may depend on the capacity of AAC to form a large Ca2+–stimulated, slightly cation-selective channel and the ability of both AAC and PiC to associate with additional mitochondrial proteins implicated in the formation of the mitochondrial permeability transition pore [80-82]. Furthermore, mutated AACs are associated with AAC1 deficiency, Senger's syndrome, and certain forms of autosomal dominant and recessive progressive external ophthalmoplegia [83-87]. A mutation in PiC has been observed in two patients with PiC deficiency [88]. Consistent with their critical role in oxidative phosphorylation, these diseases are associated with deficits in energy production.
In yeast and man, three isoforms of AAC have been identified. AAC2, the major isoform in yeast (equivalent to the heart and muscle-specific human AAC1), is one of the most abundantly expressed proteins in mitochondria and the only isoform required for respiration [89]. Initial efforts to purify and reconstitute ADP/ATP transport suggested the importance of CL for high in vitro activity [90, 91]. Consistent with this notion, six molecules of CL are tightly bound per AAC dimer [54, 92]. Interestingly, it is the headgroup structure and not the acyl chain composition of CL that promotes the high affinity interaction between CL and AAC [93, 94]. The absolute importance of CL for AAC function was for a time obscured by the six tightly bound CL molecules due to the fact that CL was dissociated only upon AAC denaturation. During a systematic analysis of yeast AAC2 mutants [95], a mutant harboring a single amino acid substitution at position 73, C73S, was identified that subsequently clarified the importance of CL for AAC function [92]. Specifically, while yeast only expressing the C73S AAC2 variant retained the ability to grow on nonfermentable carbon sources (requiring functional oxidative phosphorylation), the mutant carrier lacked any transport activity upon in vitro reconstitution unless CL was incorporated into the proteoliposomes. The lack of reconstituted transport in the absence of CL was linked to the specific loss of most of the six tightly bound CLs per AAC dimer during the purification of the C73S mutant. Thus, it was concluded that CL indeed was an “activator” of yeast AAC and possibly AACs from other organisms and/or other members of the mitochondrial carrier family [92].
As aforementioned, yeast lacking CL entirely are able to grow on nonfermentable carbon sources while Δaac2 yeast cannot. These observations alone indicate that AAC2 function does not absolutely require the presence of CL.
Although not yet thoroughly investigated, four observations suggest that the ability of CL to activate AACs is not a yeast-specific phenomenon. First, purified beef heart AAC and wild type yeast AAC2 both contain six tightly bound CL molecules per carrier dimer [54, 92]. This suggests a structural homology between yeast and bovine AACs. Second, four mutations in the heart and skeletal muscle specific isoform of AAC, AAC1, that are associated with certain types of autosomal dominant and recessive, progressive external ophthalmoplegia can be modeled in yeast AAC2 [83, 86]. Thus, the high degree of structural similarity between yeast AAC2 and human AAC1 is associated with functional similarity as well. Third, the crystal structure of bovine AAC isolated from heart muscle mitochondria includes three partially ordered CLs [62]. Fourth, reconstitution of AAC from rat brain mitochondria requires either CL or phosphatidylglycerol [96]. One notable difference in the mitochondrial phospholipid profile in Δcrd1 yeast is the accumulation of phosphatidylglycerol, the precursor of CL [3, 5, 7, 50, 97]. Thus, the ability of AAC2 to function in yeast lacking CL has been explained by a partial functional compensation by phosphatidylglycerol. Consistent with this, yeast lacking both phosphatidylglycerol and CL are unable to grow on nonfermentable carbon sources [98]. Collectively, CL is strongly implicated as being of fundamental importance for full AAC function. However, what is not known is how CL “activates” AAC activity and facilitates normal AAC physiology. The recently defined AAC2 interactome and the demonstration of the importance of CL for the AAC2 interactome have started to provide clues to these pressing questions.
4. AAC monomers or homodimers
In contrast to other components of the oxidative phosphorylation machinery, the two carrier proteins required for this process, AAC and PiC, have generally been modeled to work in isolation. This reflected the consensus that members of the mitochondrial carrier family exist and function as homodimers [99-110]. Recently, the laboratory of Kunji has challenged this dogma, providing evidence that yeast AACs assemble as monomers in mitochondrial membranes [111, 112] and that, in fact, all AAC transport activity can be fulfilled by an AAC monomer [113]. The absence of AAC2 homodimers was directly determined by the failure of either His6- or HA- epitope tagged AAC2 constructs to co-affinity purify untagged AAC2 [112]. However, utilizing a similar strategy, Deinhart and Stuart recently determined that His6-AAC2 did, in fact, co-purify endogenous, untagged AAC2 [114]. An explanation for these discrepant results is not immediately apparent.
In a very elegant study, monomeric AAC was demonstrated to be capable of performing all AAC-mediated transport [113]. The experimental design involved co-expressing two forms of AAC, one sensitive and one resistant to chemical inhibition. Results would indicate that, if AAC functions as an obligate homodimer, then the sensitive form of AAC should be able to act in a dominant-negative manner with respect to the AAC that is otherwise resistant to inhibition. Based on this rationale, it was concluded that yeast AAC2 functioned as a monomer. However, there is reason to approach this conclusion with some hesitation. In these experiments, inhibition of AAC2 function was achieved using sulfhydryl reagents. Therefore, to generate an AAC2 construct resistant to inhibition by such reagents, all four cysteine residues in AAC2 were changed to alanine, including that at position 73. As previously discussed, the C73S AAC2 mutant was critical in defining the importance of CL with respect to AAC transport [92]. The C73S mutant functioned in vivo but not in vitro unless CL was included in the assay. A molecular explanation for this dichotomy has not been provided but it is tempting to hypothesize that it might reflect an important role for CL in AAC2 oligomerization for the following four reasons. First, during the purification of C73S AAC2 most of the tightly bound CL molecules were lost, explaining the requirement of CL in the reconstitution assay [92]. This suggests that C73 is somehow involved in the tight association of CL with AAC. Second, import-assembly assays demonstrated that yeast AAC1 harboring an equivalent mutation, C63S, was imported and incorporated into the inner membrane normally but exhibited impaired assembly [101]. This implies that this conserved cysteine residue is important for AAC oligomerization. Third, while the initial crystal structure of bovine AAC contained only monomers [63], a subsequent crystal structure included a potential AAC homodimer that was mediated by two tightly bound CL molecules sandwiched between each AAC monomer on the matrix side [62]. Fourth, the potential importance and physiologic relevance of the CL-mediated greasy handshake has already been suggested. In Δcrd1 yeast, both the assembly of AAC2, as assessed by 1D blue native (BN)-PAGE, and AAC2 function are altered [3]. With respect to AAC2 assembly, 1D BN-PAGE immunoblot analyses of AAC2 following solubilization of wild type yeast mitochondria with the mild detergent digitonin revealed the presence of bands that, based on their mobility, were presumed to represent AAC2 homodimers and possibly homotetramers [3]. In the absence of CL, the largest form of AAC2 was not observed, while the most abundant smaller form, thought to reflect AAC2 homodimers, migrated as a smaller complex. While the molecular composition of the detected AAC2 adducts was not provided, CL clearly played a significant role in AAC2 assembly and function. Thus, the cysteine-less AAC2 construct may have had general assembly problems due to a weakened association with CL. The assembly status of either the C73S or cysteine-less AAC2 variants as expressed in yeast has not been documented. Obviously, the issue of whether AACs function as monomers or homodimers has not been resolved. Furthermore, if AAC does form homodimers, a potential requirement of CL for this should be determined. Finally, it is worth mentioning that even if all AAC transport can be performed as a monomer, which is important in deciphering the transport mechanism, transport may still occur physiologically in other contexts (i.e. homodimers, heterodimers, and/or other multisubunit complexes).
5. CL and the AAC2 Interactome
Conventional 1D BN-PAGE analyses of yeast AAC2 revealed AAC2 complexes that, based on their size, have been hypothesized to represent AAC2 homodimers and homotetramers [3]. The possibility that AAC2 might assemble with other proteins and/or protein complexes had not been investigated until very recently. Personal interest in this possibility was sparked upon analyzing AAC2 complexes in both wild type and Δcrd1 yeast mitochondrial extracts by 2D BN/SDS-PAGE. These analyses revealed the presence of multiple AAC2 complexes in wild type extracts, including a very large complex of >669 kDa, and the utter disorganization of AAC2 complexes when CL is absent (Fig. 1). Utilizing a newly developed dual affinity tag, AAC2 was demonstrated to interact with the respiratory supercomplex (III2IV2 or III2IV) as well as several other members of the mitochondrial carrier family, including another isoform of AAC, AAC1, the phosphate carriers, Pic1p and Pic2p, the dicarboxylate transporter, Dic1p, and the GTP/GDP transporter, Ggc1p [1]. Importantly, the interaction of AAC2 with the respiratory supercomplex was simultaneously reported by the Stuart laboratory who additionally demonstrated an association between AAC2 and the TIM23-PAM complex of the mitochondrial inner membrane [114]. The TIM23 translocon is one of two translocases in the inner membrane involved in the import of precursor proteins into mitochondria [115]. Consistent with the significant alteration of AAC2 assembly in the absence of CL, the interaction between AAC2 and the respiratory supercomplex and AAC2 and the other carrier proteins required CL [1]. An important point concerning these interactions is that they may each represent distinct AAC2 complexes with respect to their molecular composition. Therefore, when exploring the physiological significance of these interactions, it is reasonable to discuss them individually. As a CL requirement for the interaction between AAC2 and the TIM23 translocon has not been demonstrated, this interaction will not be discussed further. However, it is of interest to note that CL has been determined to be one of the minimal requirements necessary for TIM23 function [116].
5.1 The respiratory supercomplex IS super complex
Respiratory supercomplexes are hypothesized to increase the efficiency of electron shuttling between individual respiratory complexes. CL, although not required for oxidative phosphorylation, increases the efficiency of this process under optimal conditions [1, 3, 66]. The decrease in oxidative phosphorylation efficiency in the absence of CL is associated with a partial destabilization of respiratory supercomplexes [1, 5, 7, 8]. The addition of AAC2 to the respiratory supercomplex and the demonstration that this physical association requires CL provides at least two additional mechanisms by which CL facilitates efficient ATP production. The first additional mechanism focuses on perceived benefits to AAC2 function resulting from this association (Fig. 2). Another outcome of the arrangement of respiratory complexes into supercomplexes is that in addition to increasing their activity, they result in the physical union of all of the proton-transporting components of the electron transport chain. Transport of ATP out of and ADP into the matrix is known to be energetically costly due to the partial collapse of the membrane potential upon the release of ATP and its extra negative charge. Both the directionality and absolute rate of AAC transport is known to be positively influenced by a physiologic electrochemical gradient [117]. The juxtaposition of AAC2 with respiratory supercomplexes places it in a microenvironment that is anticipated to maximize AAC2 transport activity and minimize the energetic cost of this necessary process. Thus, in the absence of CL, the absolute activity of the electron transport chain is reduced due to partial destabilization of the respiratory supercomplexes, the proton pumping capacity of the electron transport chain is decentralized, and AAC2 no longer resides immediately adjacent to the electron transport chain. If this rationale is indeed correct, then it might be anticipated that the phosphate carrier, which is itself energetically costly and required for oxidative phosphorylation, is also assembled with the respiratory supercomplex.
Figure 2. The contribution of CL to energy efficiency.
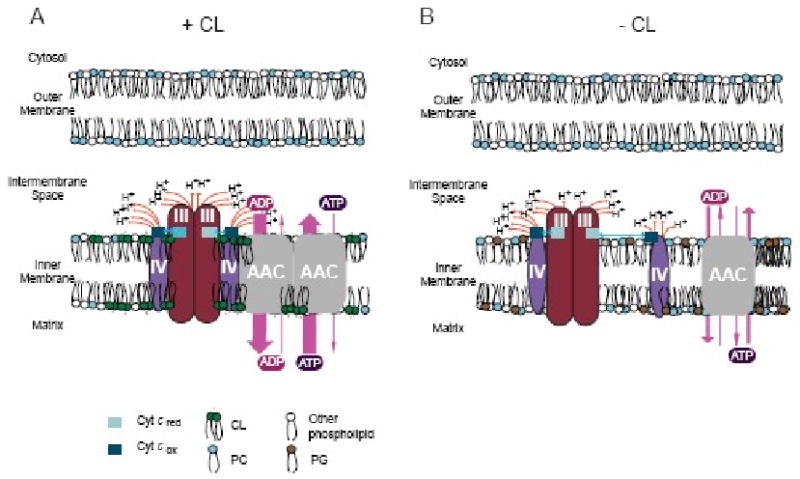
(A) CL, the “green’ phospholipid, facilitates cyt. c (blue squares) transport between complexes III (cherrywood ovals) and IV (purple ovals) by stabilizing the III2IV2-AAC2 supercomplex and stimulates AAC (gray squares) activity by placing it in an electrochemical bath provided by the proton–coupled electron transport activity of complexes III and IV. (B) In the absence of CL, the absolute activity of the electron transport chain is reduced due to partial destabilization of the respiratory supercomplexes, the proton pumping capacity of the electron transport chain is decentralized, and AAC2 no longer resides immediately adjacent to the electron transport chain. © Claypool et al., 2008. Adapted from Figure 7 originally published in The Journal of Cell Biology. doi:10.1083/jcb.200801152.
While the perceived benefits of this association for AAC2 function are admittedly speculative, the importance of the AAC2-respiratory supercomplex interaction to the proper assembly and function of the respiratory supercomplex has been demonstrated. In the absence of AAC2, the assembly of the respiratory supercomplexes is altered and complex IV activity is specifically and significantly decreased [1, 114, 118, 119]. Thus, the second new mechanism by which CL facilitates efficient ATP production is through the AAC2-mediated promotion of complex IV activity. A molecular explanation for how AAC2 promotes complex IV activity has not been provided. Based on the observation that subunits of complex IV are expressed at reduced levels in Δaac2 yeast [83, 114, 119], it has been suggested that in the absence of AAC2, complex IV is specifically destabilized. However, another Δaac2 strain maintained normal complex IV subunit expression even though complex IV activity was significantly decreased [1]. Other explanations for reduced complex IV and electron transport chain activity have been proposed [83, 120]. Pathogenic mutations or absence of AAC2 may cause structural alterations of the inner membrane and therefore affect electron transport chain organization and/or activity. Another scenario relates ATP/ADP imbalance to altered import and assembly of the electron transport chain complexes [83, 121]. While the exact mechanism by which AAC2 promotes complex IV activity has not been defined, the importance of CL for this activity is strongly implied based on the simple fact that CL is required for the interaction between AAC2 and respiratory supercomplexes [1].
5.2 A Carrier Armada
The identification of other mitochondrial carriers in the AAC2 interactome deserves additional attention [1]. First, it indicates that AAC2 participates in several distinct protein complexes, both in terms of protein composition as well as complex size. Second, the interaction between AAC2 and PiCs suggests that the transport of ADP/ATP and Pi across the inner mitochondrial membrane may be physically and spatially orchestrated, potentially representing another example of the substrate channeling phenomenon. As both ADP and Pi are required by the ATP synthase to harness the proton gradient established by the electron transport chain, such an association may increase the efficiency of the ATP synthase if the carriers were in close proximity to complex V. Interestingly, the interaction of AAC1 and the phosphate carrier was recently demonstrated in human cells, suggesting that the mixed assembly of different mitochondrial carriers might be of general physiologic importance for the mitochondrial carrier family [80]. Third, the association of AAC2 with several different mitochondrial carriers (Pic1p, Pic2p, Dic1p, Ggc1p, and AAC1) suggests that in vivo, the transport of metabolites across the inner membrane might exhibit a much higher degree of flexibility, cooperation, and coordination than previously considered. Fourth, CL is important for the interaction of AAC2 with the other carrier proteins. Based on the postulated functional benefits provided by the interaction of AAC2 with the respiratory supercomplexes, CL may also facilitate the placement of the assorted mitochondrial carriers in an environment that allows them to function most efficiently.
6. CL and the mitochondrial carrier family
As a family, the mitochondrial carriers transport a variety of metabolites across the mitochondrial inner membrane. As such, members of this family participate in a plethora of basic metabolic processes that require the cooperative activity of mitochondrial and cytosolic enzymes (extensively reviewed in [78]). Not surprisingly, defined human diseases are now known to result from mutations in several members of this family (comprehensively reviewed in [85]). All known members of the mitochondrial carrier family have the same basic structure with three ∼ 100 amino acid domains that collectively exhibit a three-fold pseudosymmetry. Given the structural and functional homology of this family, the defined importance of CL for the assembly and function of yeast AAC2 raises the issue as to whether other and/or all members of the mitochondrial carrier family depend to some extent on CL for their full range of physiological activities. This is not an unprecedented possibility as early efforts to purify and reconstitute various carrier proteins focused intensively on the potential contribution of CL [122]. In addition to AAC, the reconstituted activity of PiC and the carnitine/acylcarnitine transporter were demonstrated to require CL [57, 61, 90-92, 123-125]. Presumably based on these studies, CL became a standard addition during the solubilization and/or reconstitution steps employed in the characterization of additional members of the mitochondrial carrier family [126-137]. As a result, direct experimental evidence demonstrating a potential critical role for CL in the assembly and/or transport activity of most of the identified carrier proteins has not been generated. This omission is all the more glaring as a result of the numerous unexpected interactions of yeast AAC2 and the importance of CL for most if not all of these associations [1, 114]. If AAC is truly a paradigm for the mitochondrial carrier family, then other carrier proteins are likely to be assembling with more than just themselves and CL is likely to influence these associations. As such, it is expected that much basic biology remains to be discovered.
7. Future Directions
The recent revelation that AAC, the most abundant protein in mitochondria, which for nearly thirty years has been hypothesized to function in isolation, interacts with the respiratory supercomplex, the TIM23 translocon, and other members of the carrier family should serve as a wake-up call that there is a lot to be learned about the mitochondrial carrier family. What is the assembly status of other carrier proteins? What is the physiological significance of the association of different mitochondrial carrier proteins? Do carrier proteins that participate in a common pathway, e.g. the citrate carrier and the oxoglutarate carrier that are involved in the citrate-malate shuttle, physically as well as functionally interact? Defining the interactome for additional carrier proteins will undoubtedly provide invaluable insight into how the transport of metabolites is integrated into many basic physiologic processes.
The importance of CL as a determinant of the AAC interactome should resurrect interest into the relative importance of CL for all of the other members of the mitochondrial carrier family. With the recent cloning of the human cardiolipin synthase gene [138-141], these studies are no longer limited to yeast. It will be of great interest to determine if mammalian AAC also associates with the respiratory supercomplex and other carrier proteins and if CL is critical for these associations. If so, then deficits in AAC function specifically and mitochondrial carrier function in general should be critically evaluated in all of the assorted pathologies that have been associated with alterations in CL (reviewed in [142]). Examples include CL peroxidation which has been linked to cytochrome c release in the early stages of apoptosis [143-145]; CL oxidation, reduced CL levels, and/or altered molecular composition have been linked to the mitochondrial dysfunction associated with aging, ischemia and reperfusion, heart failure, and Barth syndrome [22, 146-151]; deficits in CL synthesis and increased CL catabolism have been connected to diabetic cardiomyopathy [152]. Consistent with the concept that AAC may represent a downstream target subsequent to alterations to CL (Fig. 3), CL peroxidation has been shown to inactivate mammalian AAC resulting in apoptosis [96]. Future studies should begin to re-address the importance of CL with respect to carrier protein structure and function and by so doing, provide insight into their participation in both normal physiological as well as numerous patho-physiological processes.
Figure 3. Are AACs and/or other carrier proteins targets of pathological situations associated with alterations to CL?
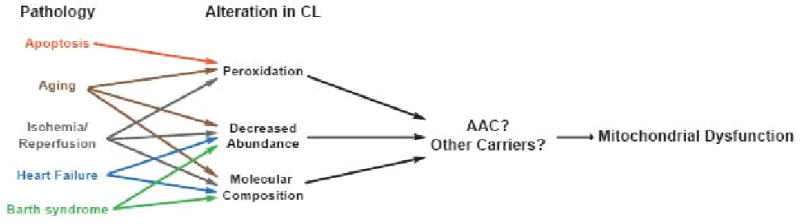
Pathologies in which alterations in CL have been implicated and the nature of the alterations in CL are indicated.
Acknowledgments
This work was supported by the National Institutes of Health (Grant R00HL089185-03).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol. 2008;182:937–950. doi: 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunte C. Specific protein-lipid interactions in membrane proteins. Biochem Soc Trans. 2005;33:938–942. doi: 10.1042/BST20050938. [DOI] [PubMed] [Google Scholar]
- 3.Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem. 2000;275:22387–22394. doi: 10.1074/jbc.M909868199. [DOI] [PubMed] [Google Scholar]
- 4.Lange C, Nett JH, Trumpower BL, Hunte C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001;20:6591–6600. doi: 10.1093/emboj/20.23.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 6.Sedlak E, Robinson NC. Phospholipase A(2) digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry. 1999;38:14966–14972. doi: 10.1021/bi9914053. [DOI] [PubMed] [Google Scholar]
- 7.Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280:29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlame M, Haldar D. Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J Biol Chem. 1993;268:74–79. [PubMed] [Google Scholar]
- 10.Cheneval D, Muller M, Toni R, Ruetz S, Carafoli E. Adriamycin as a probe for the transversal distribution of cardiolipin in the inner mitochondrial membrane. J Biol Chem. 1985;260:13003–13007. [PubMed] [Google Scholar]
- 11.Gallet PF, Petit JM, Maftah A, Zachowski A, Julien R. Asymmetrical distribution of cardiolipin in yeast inner mitochondrial membrane triggered by carbon catabolite repression. Biochem J. 1997;324(Pt 2):627–634. doi: 10.1042/bj3240627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krebs JJ, Hauser H, Carafoli E. Asymmetric distribution of phospholipids in the inner membrane of beef heart mitochondria. J Biol Chem. 1979;254:5308–5316. [PubMed] [Google Scholar]
- 13.Gallet PF, Zachowski A, Julien R, Fellmann P, Devaux PF, Maftah A. Transbilayer movement and distribution of spin-labelled phospholipids in the inner mitochondrial membrane. Biochim Biophys Acta. 1999;1418:61–70. doi: 10.1016/s0005-2736(99)00022-x. [DOI] [PubMed] [Google Scholar]
- 14.Voelker DR. New perspectives on the regulation of intermembrane glycerophospholipid traffic. J Lipid Res. 2003;44:441–449. doi: 10.1194/jlr.R200020-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Lecocq J, Ballou CE. On the Structure of Cardiolipin. Biochemistry. 1964;3:976–980. doi: 10.1021/bi00895a023. [DOI] [PubMed] [Google Scholar]
- 16.Haines TH, Dencher NA. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 2002;528:35–39. doi: 10.1016/s0014-5793(02)03292-1. [DOI] [PubMed] [Google Scholar]
- 17.Kates M, Syz JY, Gosser D, Haines TH. pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids. 1993;28:877–882. doi: 10.1007/BF02537494. [DOI] [PubMed] [Google Scholar]
- 18.Hostetler KY, Galesloot JM, Boer P, Van Den Bosch H. Further studies on the formation of cardiolipin and phosphatidylglycerol in rat liver mitochondria. Effect of divalent cations and the fatty acid composition of CDP-diglyceride. Biochim Biophys Acta. 1975;380:382–389. doi: 10.1016/0005-2760(75)90106-x. [DOI] [PubMed] [Google Scholar]
- 19.Rustow B, Schlame M, Rabe H, Reichmann G, Kunze D. Species pattern of phosphatidic acid, diacylglycerol, CDP-diacylglycerol and phosphatidylglycerol synthesized de novo in rat liver mitochondria. Biochim Biophys Acta. 1989;1002:261–263. doi: 10.1016/0005-2760(89)90296-8. [DOI] [PubMed] [Google Scholar]
- 20.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJ, DiMauro S, Blanck TJ. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 21.Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 23.Eichberg J. The reacylation of deacylated derivatives of diphosphatidylglycerol by microsomes and mitochondria from rat liver. J Biol Chem. 1974;249:3423–3429. [PubMed] [Google Scholar]
- 24.Schlame M, Rustow B. Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. Biochem J. 1990;272:589–595. doi: 10.1042/bj2720589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van 't Veer-Korthof ET, Van der Harten JJ, Sobotka-Plojhar MA. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62:327–355. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 26.Barth PG, Wanders RJ, Vreken P, Janssen EA, Lam J, Baas F. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060) J Inherit Metab Dis. 1999;22:555–567. doi: 10.1023/a:1005568609936. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Kelley RI, Blanck TJ, Schlame M. Remodeling of cardiolipin by phospholipid transacylation. J Biol Chem. 2003 doi: 10.1074/jbc.M307382200. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 29.Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174:379–390. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51:149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 31.Testet E, Laroche-Traineau J, Noubhani A, Coulon D, Bunoust O, Camougrand N, Manon S, Lessire R, Bessoule JJ. Ypr140wp, ‘the yeast tafazzin’, displays a mitochondrial lysophosphatidylcholine (lyso-PC) acyltransferase activity related to triacylglycerol and mitochondrial lipid synthesis. Biochem J. 2005;387:617–626. doi: 10.1042/BJ20041491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valianpour F, Mitsakos V, Schlemmer D, Towbin JA, Taylor JM, Ekert PG, Thorburn DR, Munnich A, Wanders RJ, Barth PG, Vaz FM. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res. 2005;46:1182–1195. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Valianpour F, Wanders RJ, Barth PG, Overmars H, van Gennip AH. Quantitative and compositional study of cardiolipin in platelets by electrospray ionization mass spectrometry: application for the identification of Barth syndrome patients. Clin Chem. 2002;48:1390–1397. [PubMed] [Google Scholar]
- 34.Vaz FM, Houtkooper RH, Valianpour F, Barth PG, Wanders RJ. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J Biol Chem. 2003;278:43089–43094. doi: 10.1074/jbc.M305956200. [DOI] [PubMed] [Google Scholar]
- 35.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279:378–382. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006;103:11584–11588. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortiz A, Killian JA, Verkleij AJ, Wilschut J. Membrane Fusion and the Lamellar-to-Inverted-Hexagonal Phase Transition in Cardiolipin Vesicle Systems Induced by Divalent Cations. Biophys J. 1999;77:2003–2014. doi: 10.1016/S0006-3495(99)77041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chernomordik LV, Kozlov MM. Membrane hemifusion: crossing a chasm in two leaps. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 39.Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite! J Cell Biol. 2006;175:201–207. doi: 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Herzig S, Martinou JC. Mitochondrial dynamics: to be in good shape to survive. Curr Mol Med. 2008;8:131–137. doi: 10.2174/156652408783769625. [DOI] [PubMed] [Google Scholar]
- 42.Westermann B. Molecular machinery of mitochondrial fusion and fission. J Biol Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- 43.Gohil VM, Thompson MN, Greenberg ML. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem. 2005 doi: 10.1074/jbc.M505478200. [DOI] [PubMed] [Google Scholar]
- 44.Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966;30:269–297. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hackenbrock CR. Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc Natl Acad Sci U S A. 1968;61:598–605. doi: 10.1073/pnas.61.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J Cell Biol. 1968;37:345–369. doi: 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 48.Mannella CA, Pfeiffer DR, Bradshaw PC, Moraru II, Slepchenko B, Loew LM, Hsieh CE, Buttle K, Marko M. Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life. 2001;52:93–100. doi: 10.1080/15216540152845885. [DOI] [PubMed] [Google Scholar]
- 49.Chen S, He Q, Greenberg ML. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol Microbiol. 2008;68:1061–1072. doi: 10.1111/j.1365-2958.2008.06216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM. The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol Biol Cell. 2008;19:5143–5155. doi: 10.1091/mbc.E08-09-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem. 1990;265:18797–18802. [PubMed] [Google Scholar]
- 52.Simbeni R, Pon L, Zinser E, Paltauf F, Daum G. Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. J Biol Chem. 1991;266:10047–10049. [PubMed] [Google Scholar]
- 53.Sperka-Gottlieb CD, Hermetter A, Paltauf F, Daum FG. Lipid topology and physical properties of the outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 1988;946:227–234. doi: 10.1016/0005-2736(88)90397-5. [DOI] [PubMed] [Google Scholar]
- 54.Beyer K, Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry. 1985;24:3821–3826. doi: 10.1021/bi00336a001. [DOI] [PubMed] [Google Scholar]
- 55.Beyer K, Nuscher B. Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry. 1996;35:15784–15790. doi: 10.1021/bi9610055. [DOI] [PubMed] [Google Scholar]
- 56.Eble KS, Coleman WB, Hantgan RR, Cunningham CC. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem. 1990;265:19434–19440. [PubMed] [Google Scholar]
- 57.Fiermonte G, Dolce V, Palmieri F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J Biol Chem. 1998;273:22782–22787. doi: 10.1074/jbc.273.35.22782. [DOI] [PubMed] [Google Scholar]
- 58.Fry M, Green D. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem. 1981;256:1874–1880. [PubMed] [Google Scholar]
- 59.Gomez B, Jr, Robinson NC. Quantitative determination of cardiolipin in mitochondrial electron transferring complexes by silicic acid high-performance liquid chromatography. Anal Biochem. 1999;267:212–216. doi: 10.1006/abio.1998.2998. [DOI] [PubMed] [Google Scholar]
- 60.Gomez B, Jr, Robinson NC. Phospholipase digestion of bound cardiolipin reversibly inactivates bovine cytochrome bc1. Biochemistry. 1999;38:9031–9038. doi: 10.1021/bi990603r. [DOI] [PubMed] [Google Scholar]
- 61.Kadenbach B, Mende P, Kolbe HV, Stipani I, Palmieri F. The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Lett. 1982;139:109–112. doi: 10.1016/0014-5793(82)80498-5. [DOI] [PubMed] [Google Scholar]
- 62.Nury H, Dahout-Gonzalez C, Trezeguet V, Lauquin G, Brandolin G, Pebay-Peyroula E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 2005;579:6031–6036. doi: 10.1016/j.febslet.2005.09.061. [DOI] [PubMed] [Google Scholar]
- 63.Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 64.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vik SB, Georgevich G, Capaldi RA. Diphosphatidylglycerol is required for optimal activity of beef heart cytochrome c oxidase. Proc Natl Acad Sci U S A. 1981;78:1456–1460. doi: 10.1073/pnas.78.3.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koshkin V, Greenberg ML. Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem J. 2000;347(Pt 3):687–691. [PMC free article] [PubMed] [Google Scholar]
- 67.Cruciat CM, Brunner S, Baumann F, Neupert W, Stuart RA. The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J Biol Chem. 2000;275:18093–18098. doi: 10.1074/jbc.M001901200. [DOI] [PubMed] [Google Scholar]
- 68.Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 70.Boumans H, Grivell LA, Berden JA. The respiratory chain in yeast behaves as a single functional unit. J Biol Chem. 1998;273:4872–4877. doi: 10.1074/jbc.273.9.4872. [DOI] [PubMed] [Google Scholar]
- 71.Koshkin V, Greenberg ML. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J. 2002;364:317–322. doi: 10.1042/bj3640317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma L, Vaz FM, Gu Z, Wanders RJ, Greenberg ML. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem. 2004;279:44394–44399. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 73.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 74.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schagger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 1998;17:7170–7178. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goyon V, Fronzes R, Salin B, di-Rago JP, Velours J, Brethes D. Yeast Cells Depleted in Atp14p Fail to Assemble Atp6p within the ATP Synthase and Exhibit Altered Mitochondrial Cristae Morphology. J Biol Chem. 2008;283:9749–9758. doi: 10.1074/jbc.M800204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Strauss M, Hofhaus G, Schroder RR, Kuhlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 79.Duszynski J, Bogucka K, Letko G, Kuster U, Kunz W, Wojtczak L. Relationship between the energy cost of ATP transport and ATP synthesis in mitochondria. Biochim Biophys Acta. 1981;637:217–223. doi: 10.1016/0005-2728(81)90160-2. [DOI] [PubMed] [Google Scholar]
- 80.Alcala S, Klee M, Fernandez J, Fleischer A, Pimentel-Muinos FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene. 2008;27:44–54. doi: 10.1038/sj.onc.1210600. [DOI] [PubMed] [Google Scholar]
- 81.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- 82.Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol. 2003;50:389–404. [PubMed] [Google Scholar]
- 83.Fontanesi F, Palmieri L, Scarcia P, Lodi T, Donnini C, Limongelli A, Tiranti V, Zeviani M, Ferrero I, Viola AM. Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum Mol Genet. 2004;13:923–934. doi: 10.1093/hmg/ddh108. [DOI] [PubMed] [Google Scholar]
- 84.Jordens EZ, Palmieri L, Huizing M, van den Heuvel LP, Sengers RC, Dorner A, Ruitenbeek W, Trijbels FJ, Valsson J, Sigfusson G, Palmieri F, Smeitink JA. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann Neurol. 2002;52:95–99. doi: 10.1002/ana.10214. [DOI] [PubMed] [Google Scholar]
- 85.Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta. 2008;1777:564–578. doi: 10.1016/j.bbabio.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 86.Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, Ferrero I, Zeviani M. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- 87.Sharer JD. The adenine nucleotide translocase type 1 (ANT1): a new factor in mitochondrial disease. IUBMB Life. 2005;57:607–614. doi: 10.1080/15216540500217735. [DOI] [PubMed] [Google Scholar]
- 88.Mayr JA, Merkel O, Kohlwein SD, Gebhardt BR, Bohles H, Fotschl U, Koch J, Jaksch M, Lochmuller H, Horvath R, Freisinger P, Sperl W. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet. 2007;80:478–484. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lawson JE, Gawaz M, Klingenberg M, Douglas MG. Structure-function studies of adenine nucleotide transport in mitochondria. I. Construction and genetic analysis of yeast mutants encoding the ADP/ATP carrier protein of mitochondria. J Biol Chem. 1990;265:14195–14201. [PubMed] [Google Scholar]
- 90.Brandolin G, Doussiere J, Gulik A, Gulik-Krzywicki T, Lauquin GJ, Vignais PV. Kinetic, binding and ultrastructural properties of the beef heart adenine nucleotide carrier protein after incorporation into phospholipid vesicles. Biochim Biophys Acta. 1980;592:592–614. doi: 10.1016/0005-2728(80)90103-6. [DOI] [PubMed] [Google Scholar]
- 91.Kramer R, Klingenberg M. Enhancement of reconstituted ADP, ATP exchange activity by phosphatidylethanolamine and by anionic phospholipids. FEBS Lett. 1980;119:257–260. doi: 10.1016/0014-5793(80)80266-3. [DOI] [PubMed] [Google Scholar]
- 92.Hoffmann B, Stockl A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem. 1994;269:1940–1944. [PubMed] [Google Scholar]
- 93.Drees M, Beyer K. Interaction of phospholipids with the detergent-solubilized ADP/ATP carrier protein as studied by spin-label electron spin resonance. Biochemistry. 1988;27:8584–8591. doi: 10.1021/bi00423a012. [DOI] [PubMed] [Google Scholar]
- 94.Schlame M, Beyer K, Hayer-Hartl M, Klingenberg M. Molecular species of cardiolipin in relation to other mitochondrial phospholipids. Is there an acyl specificity of the interaction between cardiolipin and the ADP/ATP carrier? Eur J Biochem. 1991;199:459–466. doi: 10.1111/j.1432-1033.1991.tb16144.x. [DOI] [PubMed] [Google Scholar]
- 95.Nelson DR, Lawson JE, Klingenberg M, Douglas MG. Site-directed mutagenesis of the yeast mitochondrial ADP/ATP translocator. Six arginines and one lysine are essential. J Mol Biol. 1993;230:1159–1170. doi: 10.1006/jmbi.1993.1233. [DOI] [PubMed] [Google Scholar]
- 96.Imai H, Koumura T, Nakajima R, Nomura K, Nakagawa Y. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371:799–809. doi: 10.1042/BJ20021342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chang SC, Heacock PN, Mileykovskaya E, Voelker DR, Dowhan W. Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem. 1998;273:14933–14941. doi: 10.1074/jbc.273.24.14933. [DOI] [PubMed] [Google Scholar]
- 98.Chang SC, Heacock PN, Clancey CJ, Dowhan W. The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J Biol Chem. 1998;273:9829–9836. doi: 10.1074/jbc.273.16.9829. [DOI] [PubMed] [Google Scholar]
- 99.Bisaccia F, Zara V, Capobianco L, Iacobazzi V, Mazzeo M, Palmieri F. The formation of a disulfide cross-link between the two subunits demonstrates the dimeric structure of the mitochondrial oxoglutarate carrier. Biochim Biophys Acta. 1996;1292:281–288. doi: 10.1016/0167-4838(95)00215-4. [DOI] [PubMed] [Google Scholar]
- 100.Capobianco L, Ferramosca A, Zara V. The mitochondrial tricarboxylate carrier of silver eel: dimeric structure and cytosolic exposure of both N- and C-termini. J Protein Chem. 2002;21:515–521. doi: 10.1023/a:1022473504904. [DOI] [PubMed] [Google Scholar]
- 101.Dyall SD, Agius SC, De Marcos Lousa C, Trezeguet V, Tokatlidis K. The dynamic dimerization of the yeast ADP/ATP carrier in the inner mitochondrial membrane is affected by conserved cysteine residues. J Biol Chem. 2003;278:26757–26764. doi: 10.1074/jbc.M302700200. [DOI] [PubMed] [Google Scholar]
- 102.Hackenberg H, Klingenberg M. Molecular weight and hydrodynamic parameters of the adenosine 5′-diphosphate--adenosine 5′-triphosphate carrier in Triton X-100. Biochemistry. 1980;19:548–555. doi: 10.1021/bi00544a024. [DOI] [PubMed] [Google Scholar]
- 103.Klingenberg M. Membrane protein oligomeric structure and transport function. Nature. 1981;290:449–454. doi: 10.1038/290449a0. [DOI] [PubMed] [Google Scholar]
- 104.Klingenberg M, Appel M. The uncoupling protein dimer can form a disulfide cross-link between the mobile C-terminal SH groups. Eur J Biochem. 1989;180:123–131. doi: 10.1111/j.1432-1033.1989.tb14622.x. [DOI] [PubMed] [Google Scholar]
- 105.Kotaria R, Mayor JA, Walters DE, Kaplan RS. Oligomeric state of wild-type and cysteine-less yeast mitochondrial citrate transport proteins. J Bioenerg Biomembr. 1999;31:543–549. doi: 10.1023/a:1005460810527. [DOI] [PubMed] [Google Scholar]
- 106.Lin CS, Klingenberg M. Isolation of the uncoupling protein from brown adipose tissue mitochondria. FEBS Lett. 1980;113:299–303. doi: 10.1016/0014-5793(80)80613-2. [DOI] [PubMed] [Google Scholar]
- 107.Palmisano A, Zara V, Honlinger A, Vozza A, Dekker PJ, Pfanner N, Palmieri F. Targeting and assembly of the oxoglutarate carrier: general principles for biogenesis of carrier proteins of the mitochondrial inner membrane. Biochem J. 1998;333(Pt 1):151–158. doi: 10.1042/bj3330151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Postis V, De Marcos Lousa C, Arnou B, Lauquin GJ, Trezeguet V. Subunits of the yeast mitochondrial ADP/ATP carrier: cooperation within the dimer. Biochemistry. 2005;44:14732–14740. doi: 10.1021/bi051648x. [DOI] [PubMed] [Google Scholar]
- 109.Riccio P, Aquila H, Klingenberg M. Purification of the carboxy-atractylate binding protein from mitochondria. FEBS Lett. 1975;56:133–138. doi: 10.1016/0014-5793(75)80127-x. [DOI] [PubMed] [Google Scholar]
- 110.Schroers A, Burkovski A, Wohlrab H, Kramer R. The phosphate carrier from yeast mitochondria. Dimerization is a prerequisite for function. J Biol Chem. 1998;273:14269–14276. doi: 10.1074/jbc.273.23.14269. [DOI] [PubMed] [Google Scholar]
- 111.Bamber L, Harding M, Butler PJ, Kunji ER. Yeast mitochondrial ADP/ATP carriers are monomeric in detergents. Proc Natl Acad Sci U S A. 2006;103:16224–16229. doi: 10.1073/pnas.0607640103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bamber L, Slotboom DJ, Kunji ER. Yeast mitochondrial ADP/ATP carriers are monomeric in detergents as demonstrated by differential affinity purification. J Mol Biol. 2007;371:388–395. doi: 10.1016/j.jmb.2007.05.072. [DOI] [PubMed] [Google Scholar]
- 113.Bamber L, Harding M, Monne M, Slotboom DJ, Kunji ER. The yeast mitochondrial ADP/ATP carrier functions as a monomer in mitochondrial membranes. Proc Natl Acad Sci U S A. 2007;104:10830–10834. doi: 10.1073/pnas.0703969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dienhart MK, Stuart RA. The Yeast Aac2 Protein Exists in Physical Association with the Cytochrome bc1-COX Supercomplex and the TIM23 Machinery. Mol Biol Cell. 2008;19:3934–3943. doi: 10.1091/mbc.E08-04-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Koehler CM. New developments in mitochondrial assembly. Annu Rev Cell Dev Biol. 2004;20:309–335. doi: 10.1146/annurev.cellbio.20.010403.105057. [DOI] [PubMed] [Google Scholar]
- 116.van der Laan M, Meinecke M, Dudek J, Hutu DP, Lind M, Perschil I, Guiard B, Wagner R, Pfanner N, Rehling P. Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat Cell Biol. 2007;9:1152–1159. doi: 10.1038/ncb1635. [DOI] [PubMed] [Google Scholar]
- 117.Kramer R, Klingenberg M. Modulation of the reconstituted adenine nucleotide exchange by membrane potential. Biochemistry. 1980;19:556–560. doi: 10.1021/bi00544a025. [DOI] [PubMed] [Google Scholar]
- 118.Heidkamper D, Muller V, Nelson DR, Klingenberg M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations on transport and the four ATP versus ADP exchange modes. Biochemistry. 1996;35:16144–16152. doi: 10.1021/bi960668j. [DOI] [PubMed] [Google Scholar]
- 119.Muller V, Basset G, Nelson DR, Klingenberg M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations of oxidative phosphorylation and AAC expression. Biochemistry. 1996;35:16132–16143. doi: 10.1021/bi960667r. [DOI] [PubMed] [Google Scholar]
- 120.Scherer B, Klingenberg M. Demonstration of the relationship between the adenine nucleotide carrier and the structural changes of mitochondria as induced by adenosine 5′-diphosphate. Biochemistry. 1974;13:161–170. doi: 10.1021/bi00698a025. [DOI] [PubMed] [Google Scholar]
- 121.Aprille JR. Regulation of the mitochondrial adenine nucleotide pool size in liver: mechanism and metabolic role. FASEB J. 1988;2:2547–2556. doi: 10.1096/fasebj.2.10.3290024. [DOI] [PubMed] [Google Scholar]
- 122.Kramer R, Palmieri F. Molecular aspects of isolated and reconstituted carrier proteins from animal mitochondria. Biochim Biophys Acta. 1989;974:1–23. doi: 10.1016/s0005-2728(89)80160-4. [DOI] [PubMed] [Google Scholar]
- 123.Indiveri C, Tonazzi A, Palmieri F. Identification and purification of the ornithine/citrulline carrier from rat liver mitochondria. Eur J Biochem. 1992;207:449–454. doi: 10.1111/j.1432-1033.1992.tb17070.x. [DOI] [PubMed] [Google Scholar]
- 124.Indiveri C, Tonazzi A, Prezioso G, Palmieri F. Kinetic characterization of the reconstituted carnitine carrier from rat liver mitochondria. Biochim Biophys Acta. 1991;1065:231–238. doi: 10.1016/0005-2736(91)90235-z. [DOI] [PubMed] [Google Scholar]
- 125.Noel H, Pande SV. An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur J Biochem. 1986;155:99–102. doi: 10.1111/j.1432-1033.1986.tb09463.x. [DOI] [PubMed] [Google Scholar]
- 126.Fiermonte G, Dolce V, David L, Santorelli FM, Dionisi-Vici C, Palmieri F, Walker JE. The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2003;278:32778–32783. doi: 10.1074/jbc.M302317200. [DOI] [PubMed] [Google Scholar]
- 127.Genchi G, Spagnoletta A, De Santis A, Stefanizzi L, Palmieri F. Purification and characterization of the reconstitutively active citrate carrier from maize mitochondria. Plant Physiol. 1999;120:841–848. doi: 10.1104/pp.120.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kaplan RS, Mayor JA, Johnston N, Oliveira DL. Purification and characterization of the reconstitutively active tricarboxylate transporter from rat liver mitochondria. J Biol Chem. 1990;265:13379–13385. [PubMed] [Google Scholar]
- 129.Indiveri C, Tonazzi A, Palmieri F. Identification and purification of the carnitine carrier from rat liver mitochondria. Biochim Biophys Acta. 1990;1020:81–86. doi: 10.1016/0005-2728(90)90096-m. [DOI] [PubMed] [Google Scholar]
- 130.Bisaccia F, De Palma A, Prezioso G, Palmieri F. Kinetic characterization of the reconstituted tricarboxylate carrier from rat liver mitochondria. Biochim Biophys Acta. 1990;1019:250–256. doi: 10.1016/0005-2728(90)90201-e. [DOI] [PubMed] [Google Scholar]
- 131.Nalecz MJ, Nalecz KA, Broger C, Bolli R, Wojtczak L, Azzi A. Extraction, partial purification and functional reconstitution of two mitochondrial carriers transporting keto acids: 2-oxoglutarate and pyruvate. FEBS Lett. 1986;196:331–336. doi: 10.1016/0014-5793(86)80273-3. [DOI] [PubMed] [Google Scholar]
- 132.Nalecz KA, Bolli R, Wojtczak L, Azzi A. The monocarboxylate carrier from bovine heart mitochondria: partial purification and its substrate-transporting properties in a reconstituted system. Biochim Biophys Acta. 1986;851:29–37. doi: 10.1016/0005-2728(86)90245-8. [DOI] [PubMed] [Google Scholar]
- 133.Kaplan RS, Pratt RD, Pedersen PL. Purification and characterization of the reconstitutively active phosphate transporter from rat liver mitochondria. J Biol Chem. 1986;261:12767–12773. [PubMed] [Google Scholar]
- 134.Kaplan RS, Pedersen PL. Isolation and reconstitution of the n-butylmalonate-sensitive dicarboxylate transporter from rat liver mitochondria. J Biol Chem. 1985;260:10293–10298. [PubMed] [Google Scholar]
- 135.Bisaccia F, Palmieri F. Specific elution from hydroxylapatite of the mitochondrial phosphate carrier by cardiolipin. Biochim Biophys Acta. 1984;766:386–394. doi: 10.1016/0005-2728(84)90254-8. [DOI] [PubMed] [Google Scholar]
- 136.Mende P, Huther FJ, Kadenbach B. Specific and reversible activation and inactivation of the mitochondrial phosphate carrier by cardiolipin and nonionic detergents, respectively. FEBS Lett. 1983;158:331–334. doi: 10.1016/0014-5793(83)80607-3. [DOI] [PubMed] [Google Scholar]
- 137.Mende P, Kolbe HV, Kadenbach B, Stipani I, Palmieri F. Reconstitution of the isolated phosphate-transport system of pig-heart mitochondria. Eur J Biochem. 1982;128:91–95. doi: 10.1111/j.1432-1033.1982.tb06937.x. [DOI] [PubMed] [Google Scholar]
- 138.Chen D, Zhang XY, Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J. 2006;398:169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14:597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- 140.Houtkooper RH, Akbari H, van Lenthe H, Kulik W, Wanders RJ, Frentzen M, Vaz FM. Identification and characterization of human cardiolipin synthase. FEBS Lett. 2006;580:3059–3064. doi: 10.1016/j.febslet.2006.04.054. [DOI] [PubMed] [Google Scholar]
- 141.Lu B, Xu FY, Jiang YJ, Choy PC, Hatch GM, Grunfeld C, Feingold KR. Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1) J Lipid Res. 2006;47:1140–1145. doi: 10.1194/jlr.C600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 142.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 143.Kagan KE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 144.Petrosillo G, Ruggiero FM, Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. Faseb J. 2003;17:2202–2208. doi: 10.1096/fj.03-0012com. [DOI] [PubMed] [Google Scholar]
- 145.Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001;509:435–438. doi: 10.1016/s0014-5793(01)03206-9. [DOI] [PubMed] [Google Scholar]
- 146.Lee HJ, Mayette J, Rapoport SI, Bazinet RP. Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 2006;5:2. doi: 10.1186/1476-511X-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, Gross RW. Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem. 2007;282:34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–326. doi: 10.1016/s0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- 149.Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406:136–138. doi: 10.1016/s0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- 150.Petrosillo G, Di Venosa N, Ruggiero FM, Pistolese M, D'Agostino D, Tiravanti E, Fiore T, Paradies G. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta. 2005;1710:78–86. doi: 10.1016/j.bbabio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 151.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48:1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 152.Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44:16684–16694. doi: 10.1021/bi051908a. [DOI] [PubMed] [Google Scholar]