

Abstract
Cholinergic stimulation of dopamine neurons in the ventral tegmental area (VTA) underlies activation of the brain reward circuitry. Activation of this circuit is proposed to preferentially suppress the affective reaction to noxious stimulation. Vocalization afterdischarges (VADs) are a validated model of the affective response of rats to noxious tailshock. The antinociceptive action of the acetylcholine agonist carbachol microinjected into the VTA on VAD threshold was compared to its effect on the thresholds of other tailshock-elicited responses (VDS = vocalizations during shock, and SMR = spinal motor reflexes). Whereas VADs are organized within the forebrain, VDSs and SMRs are organized at medullary and spinal levels of the neuraxis, respectively. Carbachol (1 μg, 2 μg, and 4 μg) injected into VTA produced dose-dependent increases in VAD and VDS thresholds, although increases in VAD threshold were significantly greater than increases in VDS threshold. Administration of carbachol into VTA failed to elevate SMR threshold. Elevations in vocalization thresholds produced by intra-VTA carbachol were reversed in a dose-dependent manner by local administration of the muscarinic receptor antagonist atropine sulfate (30 μg and 60 μg). These results provide the first demonstration of the involvement of the VTA in muscarinic-induced suppression of pain affect.
Perspective
Cholinergic activation of the brain reward circuit produced a preferential suppression of rats’ affective reaction to noxious stimulation. The neurobiology that relates reinforcement to suppression of pain affect may provide insights into new treatments for pain and its associated affective disorders.
Introduction
The possibility that the neural substrates of reward and antinociception overlap has been considered for over six decades43. Evidence supporting this hypothesis derives from observations that strong analgesics (e.g. morphine, amphetamine) have high abuse potential and are self-administered in both animals and humans21,22. The reinforcing properties of these drugs are believed to contribute to their addictive liability and antinociceptive action by reducing the level of distress that normally accompanies noxious stimulation. This phenomenon is referred to as “affective analgesia” and reflects the preferential suppression of the emotional reaction to pain21.
The affective analgesia hypothesis proposes that neural substrates underlying reinforcement contribute to suppression of the affective reaction to pain. Activation of dopamine neurons in the ventral tegmental area (VTA) that project to nucleus accumbens (nAb) underlies the reinforcement produced by morphine, amphetamine, other drugs of abuse, natural reinforcers (food, water, sexual interaction), and lateral hypothalamic stimulation29,56. Activation of this mesoaccumbal dopamine system also contributes to the antinociceptive action of morphine and amphetamine2.
Mesoaccumbal dopaminegic neurons in the VTA are activated via cholinergic projections from the laterodorsal tegmental (LDTg) and pedunculopontine tegmental (PPTg) nuclei44. Injecting muscarinic or nicotinic antagonists into VTA attenuates the accumbal efflux of dopamine that accompanies electrical stimulation of LDTg19. Microinjecting nicotinic and muscarinic agonists into the VTA excite dopaminergic neurons in the VTA via activation of local cholinergic receptors15,32, and increase the release of dopamine in nucleus accumbens25,41.
Cholinergic activation of mesoaccumbal dopamine neurons is critical for reinforcement. Self-administration of lateral hypothalamic brain stimulation results in the efflux of acetylcholine in VTA48, and is attenuated by administration of muscarinic antagonists into VTA57. Infusion into the VTA of the antisense oligonucleotides targeting muscarinic M5 mRNA reduced M5 receptor density and inhibited lateral hypothalamic self-stimulation in rats58. Mutant mice with deletion of the M5 receptor exhibited reduced conditioned place preference learning with systemic morphine injections4. Alternately, the rewarding effects of lateral hypothalamic stimulation are enhanced by infusion of acetylcholine into the VTA49. Intra-VTA administration of the non-specific cholinergic agonist carbachol supports development of conditioned place preference learning, and rats learn to self-administer carbachol into the VTA30,59. These reinforcing effects of carbachol were attenuated more effectively by pre-treating rats with muscarinic versus nicotinic receptor antagonists.
The present study evaluated whether cholinergic activation of dopamine neurons in VTA suppresses the affective reaction of rats to noxious stimulation. Research in this laboratory validated a rodent model of pain affect (see Discussion). Vocalization afterdischarges (VADs) occur immediately following application of noxious tailshock, are organized within the forebrain, and have distinct spectrographic characteristics compared to vocalizations that occur during shock (VDSs)6,9,14,16. The effects of carbachol injected into the VTA on VAD threshold was compared to its effects on tailshock elicited behaviors organized at spinal (SMR = hindlimb movements and tail flexion) and medullary (VDS) levels of the neuraxis10,16. It was predicted that VAD threshold would be preferentially elevated by VTA-administered carbachol. Muscarinic receptor mediation of carbachol-induced threshold increases was assessed by pre-treating the VTA with the nonspecific muscarinic receptor antagonist atropine.
Materials and Methods
Animals
Male Long-Evans rats (Charles River, Raleigh, NC) ranging from 90 to 150 days old were used. Rats were housed as pairs in plastic cages in a climate controlled vivarium (lights on 6 A.M. to 6 P.M.), and given ad libitum access to food and water. Testing occurred during the light portion of the cycle. Rats were handled one to two times per day for at least 1 week before testing to minimize possible effects of stress from human contact. All procedures were approved by the Animal Investigation Committee of Wayne State University and followed international guideline.
Surgery & Histology
Surgeries were performed under aseptic conditions. Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) following pretreatment with atropine sulfate (1 mg/kg, i.p.), and positioned in a Kopf small animal stereotaxic frame. Guide cannule (22-gauge stainless steel hypodermic tubing) were implanted unilaterally at a 10 degree angle (lateral to medial) 2 mm above the VTA according to coordinates extrapolated from the rat brain atlas of Paxinos and Watson46. The coordinates (in mm) relative to the bregma suture and the top of the level skull were AP = − 5.7, L= 2.7, DV= − 7.2. Guides were affixed to the skull with 3 stainless steel bone screws and cranioplastic cement. Each guide cannula was fitted with a 28-gauge dummy cannula that extended the length of the guide to keep it clear and free of debris. Rats were given 7–10 days to recover before the initiation of testing.
Following testing, rats were sacrificed by carbon dioxide asphyxiation. Injection sites were marked by safrin-O dye (0.5 μl) and brains were extracted and placed in a 20% (w/v) sucrose formalin solution for 48–72 h. Brains were sectioned at 50 μm on a freezing microtome, and injection sites were localized with the aid of the Paxinos and Watson56 brain atlas by an experimenter unaware of the behavioral outcomes.
Apparatus
Testing was controlled by custom computer programs via a multifunction interface board (DT-2801, Data Translation, Marlboro, MA) installed in a PC. Rats were placed into custom made Velcro body suits and restrained on a Plexiglas pedestal using Velcro strapping that passed through loops located on the underside of the suits (see photograph in Borszcz6). This design maintained the rat in a crouching posture throughout testing, enabled rats to breathe and vocalize normally, and permitted unobstructed access to the head for intracerebral injections. Testing was conducted within a sound attenuating, lighted, and ventilated chamber equipped with a small window that enabled visual monitoring of rats during testing.
Tailshock (20 ms pulses at 25 Hz for 1,000 ms) was delivered by a computer controlled constant current shocker (STIMTEK, Arlington, MA) through electrodes (0-gauge stainless steel insect pins) placed intracutaneously (.5mm below the skin surface) on opposite sides of the tail, 7.0 cm (cathode) and 8.5 cm (anode) from the base. The utility of this form of tailshock as a noxious stimulus has been extensively discussed5,7,11. The intensity, duration, and timing of tailshocks were controlled by the computer. Current intensity was monitored by the computer via an analog-to-digital converter of the multifunction board that digitized (500 Hz sampling rate) an output voltage of the shocker that was proportional to the current delivered.
SMRs were measured with a semi-isotonic displacement transducer (Lafayette Instruments Model 76614, Lafayette, IN) attached to the rat’s tail with cotton thread. The output voltage of the transducer was amplified (×50) and then digitized (500 Hz sampling rate) by a second analog-to-digital converter of the multifunction board/computer. SMR was defined as movement of the transducer arm by at least 1.0 mm following shock onset. Once SMR criterion was exceeded the output voltage of the transducer was monitored for 2000 ms. The computer recorded the latency (ms), peak amplitude (mm), and magnitude (cm x ms) of tail movement on each trial. Displacement up to 100 mm could be detected.
Vocalizations were recorded by a pressure-zone microphone (Realistic model 33–1090, Tandy, Ft. Worth, TX) located on the wall of the testing chamber 15 cm from the rat’s head. The microphone was connected to an audio amplifier (Technics model SA-160, Tandy, Ft. Worth, TX) and a 10-band frequency equalizer adjusted to selectively amplify frequencies above 1500 Hz. The filtering of low frequencies prevented extraneous noise (i.e. rats’ respiration and movement artifacts) from contaminating vocalization records. The output of the amplifier was integrated by a Coulbourn Instruments (Allentown, PA) contour following integrator (2 ms time base) and digitized (500 Hz sampling rate) by a third analog-to-digital converter of the multifunction board/computer.
The audio system was calibrated by determining the relation between the peak digitized output of the analog-to-digital converter and the amplitude (SPL, B Scale) of a 3.0 kHz pure tone – the approximate fundamental frequency of pain-induced vocalizations of the rat6,9,14. The derived function was used to convert analog-to-digital inputs to decibels (dB). Sound intensities up to 113.0 dB could be measured. The most intense vocalization measured during any sampling period was 101.3 dB. The computer recorded the peak intensity (in decibels), latency (ms), and duration (ms) of vocalizations during the shock epoch (VDS) and for the 2,000 ms interval following shock termination (VAD).
Procedures
Pain testing
For two consecutive days prior to testing, rats were adapted to the testing apparatus for a period of 20 min/day to minimize the effects of restraint stress. Experimenters were blind to the group assignment of rats. Testing began 10 – 12 min following completion of intracerebral injections. Test sessions consisted of 20 trials. On 16 trials tailshocks between 0.01 and 2.50 mA were delivered, and on 4 trials no current was delivered so as to assess false alarm rates. Trials were presented in a randomized order to control for the impact of any particular tailshock intensity on subsequent responding, and to prevent rats from anticipating the intensity of successive tailshocks. Trials were presented with a minimum intertrial interval of 30 s and each test session concluded within 20 min. These procedures caused no observable damage to the tail. Following each test session, the testing apparatus was cleaned with 5% ammonia hydroxide to eliminate stress odors18.
Drug injections
Intracerebral injections were administered in a volume 0.5 μl at a constant rate over 2 min via 28-gauge injectors connected to a microinfusion pump (Harvard Model PHD 2000). Injectors extended 2 mm beyond the guide cannulae into the VTA. Injectors were left in place for 2 min after the completion of injections to aid the diffusion of drugs into tissue. All drugs were dissolved in sterile saline. All drugs were purchased from Sigma-Aldrich (St. Louis, MO).
Experiment 1: Dose-response Analyses
To quantify the dose-response relationship between carbachol administered into the VTA and SMR, VDS, and VAD thresholds, rats received unilateral microinjections of carbachol (1μg, 2 μg, and 4 μg) and saline into the VTA prior to four separate test sessions. Test sessions were separated by 3–5 days. The order of injections was counterbalanced using a quasi-Latin square design that maintained the saline injection at either the beginning or the end of the test sequence. This design permitted evaluation of the effects of repeated testing on baseline responding.
Experiment 2: Antagonism Analyses
The pharmacological specificity of carbachol was evaluated in a separate group of rats. Elevations in thresholds generated by 4 μg carbachol were challenged with the intra-VTA administration of the muscarinic receptor antagonist atropine sulfate. Atropine (30 μg or 60 μg) was administered 10 min prior to carbachol treatment. Administration of these doses of atropine into VTA was effective in reducing lateral hypothalamic self-stimulation in rats57. Animals received unilateral injections of saline + saline, saline + 4 μg carbachol, 30 μg atropine + 4 μg carbachol, and 60 μg atropine + 4 μg carbachol, 30 μg atropine + saline, and 60 μg atropine + saline. The order of injections was counterbalanced using a quasi-Latin square design that maintained the saline + saline injection at either the beginning or the end of the test sequence. Tests were separated by 3–5 days.
Data analysis
Some animals did not complete all testing sessions due to illness (n = 1), or blocked and damage cannulae (n = 5). This attrition resulted in unequal sample sizes necessitating that data be considered from independent groups and analyzed accordingly.
Following each test session, data were reorganized in ascending order according to tailshock intensity. SMR, VDS, and VAD thresholds for each rat were calculated as the minimum current intensity from a string of at least two consecutive intensities that generated the response. Response thresholds in the dose-response experiment (n = 5 –7 rats/group) were directly compared using repeated-measures multivariate analysis of variance (MANOVA). The effects of dose on individual responses were analyzed by one-way analysis of variance (ANOVA). The doses of carbachol that elevated response thresholds above baseline levels were determined by planned comparisons of thresholds following saline and carbachol treatments using Student’s t-test for independent groups.
The capacity of atropine to reduce carbachol-induced increases in response thresholds was analyzed for each response by two-factor ANOVA (n = 6 –7 rats/group). One factor was antagonist drug treatment (3 levels: 30 μg atropine, 60 μg atropine, and saline); the second factor was agonist drug treatment (2 levels: 4 μg carbachol and saline). Because atropine was predicted to reduce carbachol-induced increases in thresholds a significant ANOVA was followed by planned comparisons using Student t-tests for independent groups.
Data from rats (n = 7) were analyzed separately as anatomical controls when histological evaluation revealed that their injection site was outside the VTA.
Results
Behavioral profile
As demonstrated by Carroll and Lim16, SMR, VDS, and VAD reflect nociceptive processing at progressively higher levels of the neuraxis. Their analysis of rats that received transections of the neuraxis revealed that SMRs are organized at the spinal level (also see10), VDSs within the medulla below the pontomedullary border, and VADs within the forebrain. Consistent with our previous reports, responses organized rostrally within the CNS were rarely generated without those integrated more caudally within the CNS5,6,11,40. VAD generation, without concomitant elicitation of VDS and SMR occurred on 0.005% of all trials. VDS were elicited without SMR on 0.002% of the trials in which VDS was the most rostrally elicited response.
The effects of carbachol treatment on performance of each response at threshold were also analyzed. The capacity of monitored performance variables to detect decrements in performance that confound threshold measurement was established by previous studies5,11. Performance variables at threshold attained following saline treatment were compared to performance variables at threshold attained following treatment with each dose of carbachol. In Experiment 1, latency, amplitude, and magnitude of SMRs, and latency, amplitude, and duration of VDSs and VADs did not differ following saline and carbachol (1 μg, 2 μg, or 4 μg) administration except for VDS amplitude following 2 μg and 4 μg. Compared with performance following saline administration VDS amplitude was reduced following both 2 μg and 4 μg administration of carbachol into VTA, ts > 2.21, ps <.05. The reductions in VDS amplitude were modest, and the resulting amplitudes fell within the normal range observed in past experiments conducted in our laboratory: mean amplitudes (± S.E.M): saline = 78.3 dB ± 2.24, 2μg carbachol = 71.1 dB ± 2.28, and 4μg carbachol 69.8 dB ± 1.49). In Experiment 2, comparison of performance variables following saline + saline and saline + 4 μg carbachol treatments revealed no differences in performance at threshold for any response.
False alarm rates for each response were low (SMR = 0.05%, VDS = 0.007%, VAD = 0.01%). The low incidence of false alarms indicates that responses were not induced by drug treatments, were not occurring spontaneously, and were not conditioned responses to the context, but instead were generated by tailshock.
Experiment 1: Dose Response Analyses
The dose-dependent effects of carbachol (1 μg, 2 μg, and 4μg) administration into the VTA on SMR, VDS, and VAD thresholds are shown in Figure 1. Comparison of response thresholds following saline administration revealed no differences in baseline thresholds, F < 1. Repeated drug administration into the VTA did not alter baseline thresholds. No differences in response thresholds following saline treatments were observed in subgroups that were administered saline first or last in the testing sequence, ts < 1.0.
Figure 1.
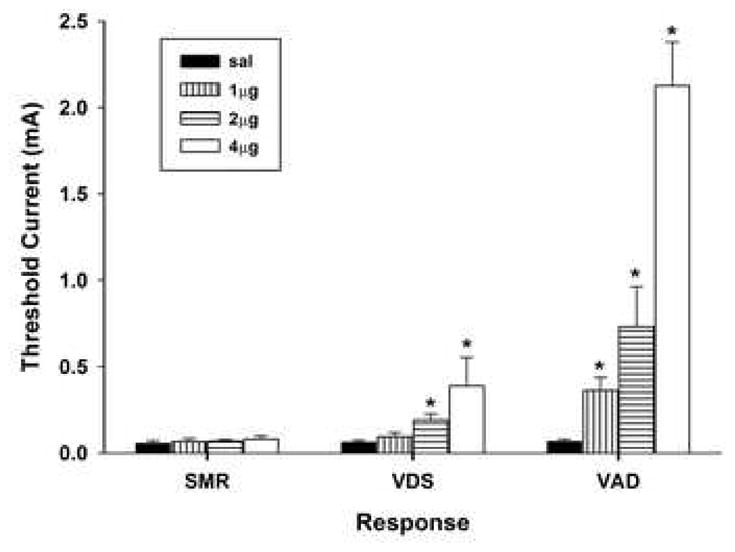
Effects of unilateral administration of carbachol (1, 2, or 4μg) into the ventral tegmental area on the mean (+S.E.M.) thresholds of spinal motor reflexes (SMRs), vocalizations during shock (VDSs), and vocalization afterdischarges (VADs). Asterisk (*) indicates significantly elevated above saline treatment.
Comparison of response thresholds across saline and carbachol treatments (repeated measures MANOVA, Wilk’s Lambda) revealed significant main effects of dose, F(3, 21) = 21.15, p <.001 and response, F(2, 20) = 44.72, p <.001, and a significant Dose x Response interaction, F(6,40) = 8.62, p <.001. This interaction reflects the finding that carbachol preferentially increased VAD threshold. Analysis of each response across saline and carbachol treatments revealed that VDS and VAD thresholds were increased in a dose-dependent manner, VDS, F(3, 24) = 3.92, p <.05, and VAD, F(3, 24) = 27.27, p <.001; whereas, SMR threshold was not elevated following carbachol administration, F < 1. Planned comparisons of VDS and VAD thresholds following saline treatment and each dose of carbachol revealed that the minimum effective doses of carbachol that elevated thresholds above baseline were 2 μg for VDS, t(11) = 3.10, p <.01, and 1 μg for VAD, t(11) = 3.59, p <.01. Direct comparison of VAD and VDS thresholds revealed that VAD threshold was significantly elevated above VDS threshold following administration of 1 μg, 2 μg, and 4 μg carbachol, all ts > 2.50, all ps <.05.
Experiment 2: Antagonism Analyses
Figure 2 depicts the effects of atropine on increases in response thresholds generated by carbachol administered into the VTA. Unilateral administration of 4 μg carbachol into the VTA produced results consistent with those from Experiment 1. VDS and VAD thresholds were significantly elevated following carbachol treatment, but there was no significant change in SMR threshold. An overall two-factor ANOVA (carbachol treatment and atropine treatment), revealed significant main effects of carbachol treatment for both VDS, F(1, 37) = 8.38, p <.01, and VAD, F(1, 37) = 36.88, p <.001 thresholds, but not SMR thresholds, F(1, 37) = 2.02, p >.15. Planned comparisons of response thresholds following administration of saline + saline and saline + carbachol treatments revealed significant increases in VDS, t(11) = 2.73, p <.05, and VAD thresholds, t(11) = 5.39, p <.001, but not SMR threshold, t(11) = 1.18, p >.25.
Figure 2.
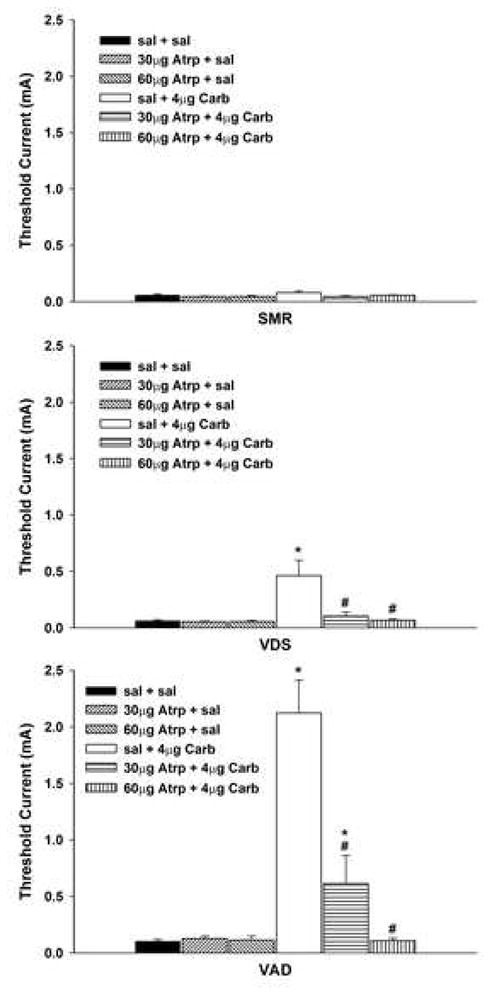
Effects of unilateral administration of atropine (Atrp: 30 μg and 60μg) into the ventral tegmental area (VTA) on increases in vocalization thresholds produced by intra-VTA injections of 4μg carbachol (Carb). Data are plotted as the mean (± SEM) threshold of spinal motor reflexes (SMRs), vocalizations during shock (VDSs), and vocalization afterdischarges (VADs). Asterisk (*) indicates thresholds significantly elevated above saline (sal) + saline treatment. Pound sign (#) indicates thresholds significantly reduced compared to sal + 4μg Carb treatment.
Administration of atropine abolished carbachol-induced threshold elevations for VDS and VAD in a dose-dependent manner. The overall two factor ANOVA also revealed significant main effects of atropine treatment for both VDS and VAD thresholds: VDS, F(2, 37) = 6.09, p <.01, and VAD, F(2, 37) = 19.53, p <.001. The Carbachol x Atropine interaction was also significant for VDS and VAD thresholds, all Fs(2,37) > 5.54, all ps <.01. This interaction reflects the finding that atropine reduced increases in VDS and VAD thresholds generated by injection of carbachol, but did not alter baseline thresholds. Planned comparisons revealed that both doses of atropine significantly reduced carbachol-induced increases in VDS threshold, all ts(11) > 2.40, all ps <.05, and returned threshold to baseline levels, all ts(10) < 1.11, all ps >.25. Carbachol-induced increase in VAD threshold was reduced after administration of 30 μg atropine, t(11) = 3.90, p <.01, but remained marginally elevated relative to baseline, t(10) = 2.08, p = .065. Administration of 60 μg atropine also reduced carbachol-induced increases in VAD threshold, t(10) = 6.40, p <.001, and returned threshold to baseline levels, t(10) < 1.
Atropine alone did not alter baseline response thresholds. One-way ANOVA comparing thresholds of each response following saline + saline, 30 μg atropine + saline, and 60 μg atropine + saline revealed no significant changes in thresholds, all Fs < 1. Repeated drug administration into the VTA did not alter baseline thresholds. No differences in response thresholds following saline + saline treatments were observed in subgroups that were administered saline first or last in the testing sequence, ts < 1.0.
Anatomical Specificity
No systematic differences were observed in the distribution of sites within the VTA that received different doses of carbachol or atropine. The effectiveness of any specific dose of carbachol to elevate response thresholds did not differ across sites. The effectiveness of atropine in antagonizing carbachol-induced increases in VAD and VDS thresholds also did not differ across injection sites. Anatomical specificity of carbachol-induced antinociception within the VTA was determined by evaluating the effects on response thresholds of 4 μg carbachol administered into sites dorsal, ventral, lateral, and medial to the VTA (Figure 3).
Figure 3.
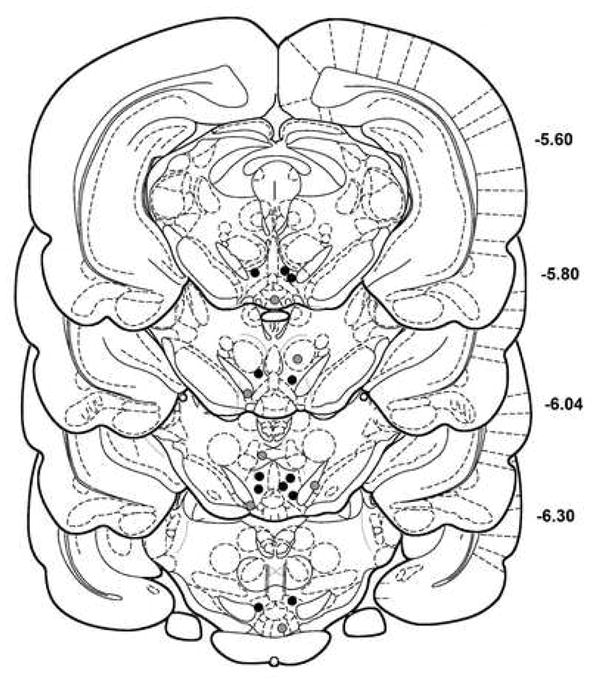
Histological reconstruction of sites that received unilateral injections of 4μg carbachol. Black circles indicate injection sites within the ventral tegmental area (VTA) that were effective in elevating response thresholds. Gray circles indicate injection sites dorsal, ventral or medial to VTA that produced no change in response thresholds. Coordinates are in millimeters posterior to bregma. Plates are derived from the rat brain atlas of Paxinos and Watson56. The right side of the midline was targeted for all injections, but for the sake of clarity of presentation injection sites are plotted on both sides of the midline.
The effects of carbachol on response thresholds when administered within the VTA versus outside the VTA are summarized in Table 1. SMR and VDS thresholds following administration of 4 μg carbachol into sites outside the VTA were not different from those observed following the administration of saline into VTA, all ts < 1. On the other hand, VAD threshold following 4 μg carbachol administration into extra-VTA sites was elevated above baseline thresholds, t(17) = 2.65, p <.05. However, the elevation in VAD threshold observed following carbachol administration into extra-VTA sites was significantly lower than the elevation of VAD thresholds observed following carbachol administered into the VTA, t(17) = 7.80, p <.001.
Table 1.
Anatomical specificity of carbachol (4 μg) injected into the ventral tegmental area (VTA) on increases in response thresholds.
| Response |
|||
|---|---|---|---|
| SMR | VDS | VAD | |
| Injection Site | M S.E.M | M S.E.M | M S.E.M |
| VTA - carb (12) a | .080 ±.011 | .432 ±.100 e | 2.124 ±.190 e |
| Other - carb (7) b | .064 ±. 013 | .069 ±.011 d | .151 ±.025 de |
| VTA - sal (12) c | .058 ±. 008 | .063 ±.007 d | .085 ±.012 d |
Values are means ± S.E.M. of threshold current (in milliamperes). Values in parentheses represent the number of injections at each site. SMR = spinal motor reflexes; VDS = vocalizations during shock, VAD = vocalizations afterdischarges; VTA = ventral tegmental area; other = injection sites dorsal, ventral, medial, or lateral to VTA (see Figure 3). carb = carbachol, sal = saline
Mean response thresholds from Experiment 1 after the administration of 4 μg carbachol plus Experiment 2 after administration of saline + 4 μg carbachol into VTA.
Mean response thresholds following misplaced injections of 4 μg carbachol (Experiment 1) plus saline + 4 μg carbachol (Experiment 2).
Mean response thresholds from Experiment 1 after the administration of saline plus Experiment 2 after administration of saline + saline into VTA.
Values with subscript ‘d’ are significantly lower than those observed following carbachol treatment in VTA (Student’s t test; p < 0.05).
Values with subscript ‘e’ are significantly higher than those observed following saline treatment (Student’s t test; p < 0.05).
Discussion
The present study provides the first demonstration of behavioral antinociception generated by administration of the acetylcholine agonist carbachol into the VTA. Administration of carbachol into VTA produced dose-dependent increases in VAD and VDS thresholds, but failed to elevate SMR threshold. Direct comparisons of response thresholds revealed that VAD threshold was preferentially elevated by the intra-VTA injection of carbachol. The minimum effective dose of carbachol that elevated VAD threshold was lower than the dose that raised VDS threshold. Also, baseline thresholds of VAD and VDS did not differ but VAD threshold was elevated to a greater extent following administration of each dose of carbachol. The preferential increase in VAD threshold cannot be attributed to drug-induced motor deficits as increases in VAD thresholds were not accompanied by performance decrements. It is unlikely that carbachol-induced increases in VDS threshold results from the observed reduction in amplitude at threshold. The reduction in amplitude was small and it was only observed in Experiment 1. In Experiment 2, administration of 4 μg carbachol produced a comparable increase in VDS threshold without any decrement in performance.
Similar to the present results, administration of carbachol, 5-HT, 8-OH-DPAT (5-HT1A/7 agonist) or morphine into either the amygdala or nucleus parafascicularis thalami produced selective increases in VAD and VDS thresholds without an accompanying increase in SMR threshold8,26,27,40. The failure to observe increases in SMR threshold does not reflect the resistance of this response to antinociceptive treatments. In previous studies, administration of morphine into the rostral ventromedial medulla or ventrolateral periaqueductal gray produced significant increases in VAD, VDS, and SMR thresholds8,12, and the intrathecal administration of morphine, serotonin, or norepinephrine was equally effective in raising VAD, VDS, and SMR thresholds13. The capacity of these central treatments to elevate SMR threshold also demonstrates that SMRs are not generated by direct stimulation of the tail musculature. These findings indicate that the capacity to elevate SMR threshold depends on the site within the CNS at which antinociceptive treatments are administered.
The selective increase in vocalization thresholds observed in the present study presumably reflects carbachol-induced activation of dopamine neurons within the VTA that project to nAb. This interpretation is supported by findings that morphine-induced increases in the accumbal efflux of dopamine are modulated by muscarinic receptors in VTA38. Additionally, infusion of morphine into the VTA or amphetamine into nAb (induces release from dopamine axon terminals and blocks re-uptake of dopamine) suppresses paw-licking in the formalin test, but does not alter withdrawal latencies in the tail flick test2,34. These antinociceptive effects in the formalin test were blocked by pretreatment of nAb with the dopamine receptor antagonist raclopride1. Correspondingly, neurotoxic lesions of dopamine neurons in VTA blocked the suppression of paw-licking in the formalin test produced by systemic administration of morphine or amphetamine, but did not alter the increase in tail flick latencies generated by these drug treatments39. These findings indicated that activation of mesoaccumbal dopamine projections selectively suppresses nociceptive processing at supraspinal levels of the neuraxis.
The capacity of VTA-administered carbachol to elevate vocalization thresholds is limited to its action within VTA. Unilateral injections of the highest dose of carbachol (4 μg) into sites surrounding the VTA failed to elevate VDS threshold and produced a greatly attenuated increase in VAD threshold. Muscarinic mediation of the antinociceptive actions of carbachol was revealed by its dose-dependent antagonism with the intra-VTA administration of atropine. Atropine is a non-selective competitive muscarinic receptor antagonist with very limited affinity for nicotinic receptors55. As atropine has nearly equivalent binding affinity for the 5 known muscarinic receptor subtypes17, the receptor subtype that mediates the increase in vocalization thresholds following carbachol treatment cannot be ascertained from results of the present study. However, only M5 mRNA is localized to cell bodies of dopamine neurons in the VTA53 and this receptor subtype mediates the sustained increase in accumbal dopamine levels generated by stimulation of LDTg or intra-VTA administration of carbachol20. We therefore speculate that activation of VTA dopamine neurons via M5 muscarinic receptors underlies the antinociceptive action of carbachol observed in the present study.
The preferential increase in VAD threshold following intra-VTA carbachol administration reflects suppression of the affective reaction to noxious stimulation. As noted earlier, previous research in this laboratory validated VADs as a rodent model of pain affect. Systemically administered drug treatments that preferentially suppress the affective reaction of humans to pain24,47 also preferentially suppress production of VADs11. Generation of VADs is suppressed by damage of or drug treatments into forebrain sites known to contribute to production of the affective response of humans to clinical and experimental pain8,14,26,27,36,40,51. Additionally, the capacity of noxious tailshock to support fear conditioning is directly related to its production of VADs5,6,9,14.
The present findings, therefore, support the hypothesis of an overlap between the neuronal circuits that underlie reinforcement and suppression of the affective reaction to pain21. Although we did not evaluate the reinforcing properties of carbachol administered into the VTA, previous studies demonstrated that unilateral administration of similar doses of carbachol (1 μg or 3 μg) supported development of conditioned place preference59. Additionally, rats learn to self-administer carbachol into the VTA30. As noted earlier, the reinforcing properties of intra-VTA administered carbachol are mediated by activation of mesoaccumbal dopamine projections.
Dopamine neurons in the VTA also project to forebrain sites (amygdala, anterior cingulate cortex) that receive nociceptive afferents and contribute to elaboration of affective behaviors3,42,45. For example, stimulation of VTA suppresses neural activity in the anterior cingulate cortex elicited by noxious peripheral stimulation35, intra-VTA administration of carbachol increases extracellular dopamine in the anterior cingulate cortex54, and infusion of dopamine into the anterior cingulate cortex suppresses neuropathic pain in rats33. Future studies will evaluate the contribution of these dopaminergic projections to the forebrain in supporting the affective analgesia elicited from the VTA.
The VTA does not project directly to midbrain or medullary sites that contribute descending antinociceptive projections. However, the forebrain sites (nAb, anterior cingulate cortex, amygdala) that receive dopaminergic input from VTA project to the ventrolateral periaqueductal gray (vPAG) and rostral ventromedial medulla (RVM), and these projections contribute to the antinociceptive effects mediated by these forebrain sites23,28,31. As generation of VDSs reflects nociceptive processing within the RVM (i.e., nucleus reticularis gigantocellularis) suppression of this processing (either directly or via the vPAG) may contribute to the increases in VDS threshold in the present study. These descending projections to the medulla may also inhibit nociceptive throughput to the forebrain sites responsible for production of VADs. The dual inhibition of nociceptive processing at medullary and forebrain levels could account for the greater maximum effect of intra-VTA carbachol on VAD threshold8.
Altier and Stewart2 proposed that the relation between the rewarding and antinociceptive actions of analgesic drugs such as morphine and amphetamine can be understood from the perspective of a motivational continuum. This motivational continuum has poles of extreme negative and positive affect with normal affect located in the middle. When noxious stimulation generates a negative affective state, opiates and psychostimulants suppress pain affect by enhancing transmission in mesocorticolimbic dopamine neurons that originate in the VTA. This activation shifts the negative affective state to the middle of the motivational continuum producing affective analgesia. However, when opiates and psychostimulants are taken in the absence of pain-induced negative affect, then normal affect is shifted to the extreme positive pole of the motivational continuum. The shift to the positive pole of the continuum is speculated to underlie the addictive liability of opiates or psychostimulants.
The conceptualization of a motivational continuum may account for clinical observations that chronic pain patients who are given access to opiates for the relief of pain rarely become addicted or tolerant37. When opioids are used for the management of clinical pain, they may help the patient to achieve an affective state normally experienced when free of pain, but not the extreme positive affect that might support addiction. Supporting this view are reports that the capacity of morphine to serve as a reinforcer is attenuated when given to rats that are in a chronic/tonic pain state,50, and that morphine tolerance and dependence fails to develop in rats when administered during a pain state,52. The neurobiology that relates reinforcement with analgesia is of obvious clinical importance and warrants further study.
Acknowledgments
The authors thank Laura A. Bolter for assistance in data collection. This research was supported by Grant R01 NS045720 from the National Institute of Neurological Disorders and Stroke (NINDS) to GSB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Altier N, Stewart J. Dopamine receptor antagonists in the nucleus accumbens attenuate analgesia induced by ventral tegmental area substance P or morphine and by nucleus accumbens amphetamine. Journal of Pharmacology and Experimental Therapeutics. 1998;285:208–215. [PubMed] [Google Scholar]
- 2.Altier N, Stewart J. The role of dopamine in the nucleus accumbens in analgesia. Life Sciences. 1999;65:2269–2287. doi: 10.1016/s0024-3205(99)00298-2. [DOI] [PubMed] [Google Scholar]
- 3.Asan E. The catecholaminergic innervation of the rat amygdala. Advances in Anatomy, Embryology, and Cell Biology. 1998;142:1–118. doi: 10.1007/978-3-642-72085-7. [DOI] [PubMed] [Google Scholar]
- 4.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Deletion of the M5 muscarinic acetylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proceedings National Academy of Science. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borszcz GS. The capacity of motor reflex and vocalization thresholds to support avoidance conditioning in the rat. Behavioral Neuroscience. 1993;107:678–693. doi: 10.1037//0735-7044.107.4.678. [DOI] [PubMed] [Google Scholar]
- 6.Borszcz GS. Pavlovian conditional vocalizations of the rat: A model system for analyzing the fear of pain. Behavioral Neuroscience. 1995;109:648–662. doi: 10.1037//0735-7044.109.4.648. [DOI] [PubMed] [Google Scholar]
- 7.Borszcz GS. Increases in vocalization and motor reflex thresholds are influenced by the site of morphine microinjection: Comparisons following administration into the periaqueductal gray, ventral medulla, and spinal subarachnoid space. Behavioral Neuroscience. 1995;109:502–522. doi: 10.1037//0735-7044.109.3.502. [DOI] [PubMed] [Google Scholar]
- 8.Borszcz GS. Differential contributions of medullary, thalamic, and amygdaloid serotonin to the antinociceptive action of morphine administered into the periaqueductal gray: A model of morphine analgesia. Behavioral Neuroscience. 1999;113:612– 631. doi: 10.1037//0735-7044.113.3.612. [DOI] [PubMed] [Google Scholar]
- 9.Borszcz GS. Contribution of the ventromedial hypothalamus to generation of the affective dimension of pain. Pain. 2006;123:155–168. doi: 10.1016/j.pain.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borszcz GS, Johnson CP, Anderson ME, Young BJ. Characterization of tailshock elicited withdrawal reflexes in intact and spinal rats. Physiology & Behavior. 1992;52:1055–1062. doi: 10.1016/0031-9384(92)90459-f. [DOI] [PubMed] [Google Scholar]
- 11.Borszcz GS, Johnson CP, Fahey KA. Comparison of motor reflex and vocalization thresholds following systemically administered morphine, fentanyl, and diazepam in the rat: assessment of sensory and performance variables. Pharmacology Biochemistry & Behavior. 1994;49:827–834. doi: 10.1016/0091-3057(94)90230-5. [DOI] [PubMed] [Google Scholar]
- 12.Borszcz GS, Johnson CP, Thorp MV. The differential contribution of spinopetal projections to increases in vocalization and motor reflex thresholds generated by the microinjection of morphine into the periaqueductal gray. Behavioral Neuroscience. 1996;110:368–388. doi: 10.1037//0735-7044.110.2.368. [DOI] [PubMed] [Google Scholar]
- 13.Borszcz GS, Johnson CP, Williams DH. Increases in vocalization and motor reflex thresholds generated by the intrathecal administration of serotonin or norepinephrine. Behavioral Neuroscience. 1996;110:809 – 822. doi: 10.1037//0735-7044.110.4.809. [DOI] [PubMed] [Google Scholar]
- 14.Borszcz GS, Leaton RN. The effect of amygdala lesions on conditional and unconditional vocalizations in rats. Neurobiology of Learning and Memory. 2003;79:212–225. doi: 10.1016/s1074-7427(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 15.Calabresi P, Lacey MG, North RA. Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. British Journal of Pharmacology. 1989;98:135–140. doi: 10.1111/j.1476-5381.1989.tb16873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll MN, Lim KS. Observations on the neuropharmacology of morphine and morphinelike analgesia. Archives Internationales de Pharmacodynamie et de Therapie. 1960;125:383–403. [PubMed] [Google Scholar]
- 17.Eglen RM, Choppin A, Watson N. Therapeutic opportunities from muscarinic receptor research. Trends Pharmacological Science. 2001;22:409–414. doi: 10.1016/s0165-6147(00)01737-5. [DOI] [PubMed] [Google Scholar]
- 18.Fanselow MS. Odors released by stressed rats produce opioid analgesia in unstressed rats. Behavioral Neuroscience. 1985;99:589–592. doi: 10.1037//0735-7044.99.3.589. [DOI] [PubMed] [Google Scholar]
- 19.Forster GL, Blaha CD. Laterodorsal tegmental stimulation elicits dopamine efflux in the rat nucleus accumbens by activation of acetylcholine and glutamate receptors in the ventral tegmental area. European Journal of Neuroscience. 2000;12:3596–3604. doi: 10.1046/j.1460-9568.2000.00250.x. [DOI] [PubMed] [Google Scholar]
- 20.Forster GL, Yeomans JS, Takeuchi J, Blaha CD. M5 muscarinic receptors are required for prolonged accumbal dopamine release after electrical stimulation of the pons in mice. Journal of Neuroscience. 2002;22:RC190. doi: 10.1523/JNEUROSCI.22-01-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franklin KBJ. Analgesia and the neural substrate of reward. Neuroscience & Biobehavioral Reviews. 1989;13:149–154. doi: 10.1016/s0149-7634(89)80024-7. [DOI] [PubMed] [Google Scholar]
- 22.Franklin KBJ. Analgesia and abuse potential: An accidental association or a common substrate. Pharmacology, Biochemistry & Behavior. 1998;59:993–1002. doi: 10.1016/s0091-3057(97)00535-2. [DOI] [PubMed] [Google Scholar]
- 23.Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. Journal of Neuroscience. 1999;19:7175–7181. doi: 10.1523/JNEUROSCI.19-16-07175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gracely RH, McGrath P, Dubner R. Validity and sensitivity of ratio scales of sensory and affective verbal pain descriptors: manipulation of affect by diazepam. Pain. 1978;5:19–29. doi: 10.1016/0304-3959(78)90021-0. [DOI] [PubMed] [Google Scholar]
- 25.Gronier B, Rasmussen K. Activation of midbrain presumed dopaminergic neurons by muscarinic cholinergic receptors: an in vivo electrophysiological study in the rat. British Journal of Pharmacology. 1998;124:455–464. doi: 10.1038/sj.bjp.0701850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harte SE, Kender RG, Borszcz GS. Activation of 5-HT1A and 5-HT7 receptors in the parafascicular nucleus suppresses the affective reaction of rats to noxious stimulation. Pain. 2005;113:405–415. doi: 10.1016/j.pain.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 27.Harte SE, Lagman AL, Borszcz GS. Antinociceptive effects of morphine injected into the nucleus parafascicularis thalami of the rat. Brain Research. 2000;874:78–86. doi: 10.1016/s0006-8993(00)02583-x. [DOI] [PubMed] [Google Scholar]
- 28.Helmstetter FJ, Tershner SA, Poore LH, Bellgowan PS. Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Research. 1998;779:104–118. doi: 10.1016/s0006-8993(97)01104-9. [DOI] [PubMed] [Google Scholar]
- 29.Ikemoto S. Dopamine reward circuitry: Two projection systems from the ventral midbrain to the nucleus accumbens–olfactory tubercle complex. Brain Research Reviews. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikemoto S, Wise RA. Rewarding effects of the cholinergic agents carbachol and neostigmine in the posterior ventral tegmental area. Journal of Neuroscience. 2002;22:9895–9904. doi: 10.1523/JNEUROSCI.22-22-09895.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaBuda CJ, Fuchs PN. Attenuation of negative pain affect produced by unilateral spinal nerve injury in the rat following anterior cingulate cortex activation. Neuroscience. 2005;136:311–322. doi: 10.1016/j.neuroscience.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Lacey MG, Calabresi P, North RA. Muscarine depolarizes rat substantia nigra zona compacta and ventral tegmental neurons in vitro through M1-like receptors. Journal of Pharmacology and Experimental Therapeutics. 1990;253:395–400. [PubMed] [Google Scholar]
- 33.Lopez-Avila A, Coffeen U, Ortega-Legaspi JM, del Angel R, Pellicer F. Dopamine and NMDA systems modulate long-term nociception in the rat anterior cingulate cortex. Pain. 2004;111:136–143. doi: 10.1016/j.pain.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Manning BH, Morgan MJ, Franklin KBJ. Morphine analgesia in the formalin test: Evidence for forebrain and midbrain sites of action. Neuroscience. 1994;63:289–294. doi: 10.1016/0306-4522(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 35.Mantz J, Milla C, Glowinski J, Thierry AM. Differential effects of ascending neurons containing dopamine and noradrenaline in the control of spontaneous activity and of evoked responses in the rat prefrontal cortex. Neuroscience. 1988;27:517–526. doi: 10.1016/0306-4522(88)90285-0. [DOI] [PubMed] [Google Scholar]
- 36.Mark VH, Ervin FR, Yakovlev PI. The treatment of pain by stereotaxic methods. Confinia Neurologica. 1962;22:238–245. [Google Scholar]
- 37.Melzack R. The tragedy of needless pain. Scientific American. 1990;262 :27–33. doi: 10.1038/scientificamerican0290-27. [DOI] [PubMed] [Google Scholar]
- 38.Miller AD, Forster GL, Yeomans JS, Blaha CD. Midbrain muscarinic receptors modulate morphine-induced accumbal and striatal dopamine efflux in the rat. Neuroscience. 2005;136:531–538. doi: 10.1016/j.neuroscience.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 39.Morgan MJ, Franklin KBJ. 6-Hydroxydopamine lesions of the ventral tegmentum abolish D-amphetamine and morphine analgesia in the formalin test but not in the tail flick test. Brain Research. 1990;519:144–149. doi: 10.1016/0006-8993(90)90072-j. [DOI] [PubMed] [Google Scholar]
- 40.Nandigama P, Borszcz GS. Affective analgesia following the administration of morphine into the amygdala of rats. Brain Research. 2003;959:343–354. doi: 10.1016/s0006-8993(02)03884-2. [DOI] [PubMed] [Google Scholar]
- 41.Nisell M, Nomikos GG, Svensson TH. Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacology and Toxicology. 1994;75:348–352. doi: 10.1111/j.1600-0773.1994.tb00373.x. [DOI] [PubMed] [Google Scholar]
- 42.Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Research. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 43.Oberst F. Symposium: Can the euphoric, analgetic and physical dependence of drugs be separated? Federation of American Societies for Experimental Biology Proceedings. 1943;2:187 – 203. [Google Scholar]
- 44.Omelchenko N, Sesack SR. Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. Journal of Comparative Neurology. 2005;483:217–235. doi: 10.1002/cne.20417. [DOI] [PubMed] [Google Scholar]
- 45.Onn SP, Wang XB. Differential modulation of anterior cingulate cortical activity by afferents from ventral tegmental area and mediodorsal thalamus. European Journal of Neuroscience. 2005;21:2975–2992. doi: 10.1111/j.1460-9568.2005.04122.x. [DOI] [PubMed] [Google Scholar]
- 46.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1998. [Google Scholar]
- 47.Price DD, Von der Gruen A, Miller J, Rafii A, Price C. A psychophysical analysis of morphine analgesia. Pain. 1985;22:261–269. doi: 10.1016/0304-3959(85)90026-0. [DOI] [PubMed] [Google Scholar]
- 48.Rada PV, Mark GP, Yeomans JS, Hoebel BG. Acetylcholine release in ventral tegmental area by hypothalamic self-stimulation, eating, and drinking. Pharmacology Biochemistry & Behavior. 2000;65:375–379. doi: 10.1016/s0091-3057(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 49.Redgrave P, Horrell RI. Potentiation of central reward by localised perfusion of acetylcholine and 5-hydroxytryptamine. Nature. 1976;262:305–307. doi: 10.1038/262305a0. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki T, Kishimoto Y, Misawa M. Formalin- and carrageenan-induced inflammation attenuates place preferences produced by morphine, methamphetamine and cocaine. Life Sciences. 1996;59:1667–1674. doi: 10.1016/0024-3205(96)00498-5. [DOI] [PubMed] [Google Scholar]
- 51.Sweet WH. Central mechanisms of chronic pain (neuralgias and certain other neurogenic pain) In: Bonica JJ, editor. Pain. Raven Press; New York: 1980. pp. 287–303. [PubMed] [Google Scholar]
- 52.Vaccarino AL, Marek P, Kest B, Ben-Eliyahu S, Couret LC, Jr, Kao B, Liebeskind JC. Morphine fails to produce tolerance when administered in the presence of formalin pain in rats. Brain Research. 1993;627:287–290. doi: 10.1016/0006-8993(93)90332-h. [DOI] [PubMed] [Google Scholar]
- 53.Vilaro MT, Palacios JM, Mengod G. Localization of m5 muscarinic receptor mRNA in rat brain examined by in situ hybridization histochemistry. Neuroscience Letters. 1990;114:154–159. doi: 10.1016/0304-3940(90)90064-g. [DOI] [PubMed] [Google Scholar]
- 54.Westerink BH, Enrico P, Feimann J, deVries JB. The pharmacology of mesocortical dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and prefrontal cortex of the rat brain. Journal of Pharmacology and Experimental Therapeutics. 1998;285:143–154. [PubMed] [Google Scholar]
- 55.Williams M, Robinson JL. Nicotinic receptors in mammalian brain. Progress in Neuropsychopharmacology and Biological Psychiatry. 1984;8:769–772. doi: 10.1016/0278-5846(84)90056-3. [DOI] [PubMed] [Google Scholar]
- 56.Wise RA, Rompre PP. Brain dopamine and reward, Annual Review of Psychology. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 57.Yeomans JS, Baptista M. Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain-stimulation reward. Pharmacology Biochemistry & Behavior. 1997;57:915–921. doi: 10.1016/s0091-3057(96)00467-4. [DOI] [PubMed] [Google Scholar]
- 58.Yeomans JS, Forster G, Blaha C. M5 muscarinic receptors are needed for slow activation of dopamine neurons and for rewarding brain stimulation. Life Sciences. 2001;68:2449–2456. doi: 10.1016/s0024-3205(01)01038-4. [DOI] [PubMed] [Google Scholar]
- 59.Yeomans JS, Kofman O, McFarlane V. Cholinergic involvement in lateral hypothalamic rewarding brain stimulation. Brain Research. 1985;329:19–26. doi: 10.1016/0006-8993(85)90508-6. [DOI] [PubMed] [Google Scholar]