

Abstract
Dyslipidaemias, particularly those characterized by the ‘atherogenic profile’ of high low-density lipoprotein-cholesterol and triglycerides and low high-density lipoprotein-cholesterol, are the major modifiable risk factor for atherosclerosis. The search for drugs to favourably alter such lipid profiles, reducing the associated morbidity and mortality, remains a major research focus. Niacin (nicotinic acid) is the most effective agent available for increasing high-density lipoprotein-cholesterol, but its use is associated with side effects that negatively affect patient compliance: these appear to arise largely as a result of production of prostaglandin D2 and its subsequent activation of the DP1 receptor. Desire to reduce the side effects (and improve pharmacokinetic parameters) has led to the development of a number of agonists that have differing effects, both in terms of clinical potency and the severity of adverse effects. The recent discovery of the niacin G-protein-coupled receptor HM74A (GPR109A) has clarified the distinction between the mechanism whereby niacin exerts its therapeutic effects and the mechanisms responsible for the generation of side effects. This has allowed the development of new drugs that show great potential for the treatment of dyslipidaemia. However, recent advances in understanding of the contribution of prostaglandin metabolism to vascular wall health suggest that some of the beneficial effects of niacin may well result from activation of the same pathways responsible for the adverse reactions. The purpose of this review is to emphasize that the search for agonists that show higher tolerability must take into account all aspects of signalling through this receptor.
Keywords: niacin, HM74A, dyslipidaemia, PPAR, nicotinamide, acipimox, acifran, prostaglandin D2
Background
Niacin (nicotinic acid; vitamin B3) is enjoying something of a renaissance as a treatment for abnormal blood lipids and the associated pathology. As Wise et al. (2003) describe, treatment with niacin results in ‘a desirable normalization of a range of cardiovascular risk factors’. However, use of this agent is associated with severe side effects affecting compliance, reducing its clinical usefulness. The recent discovery of the niacin receptor (Soga et al., 2003; Pike and Wise, 2004; Benyo et al., 2005; Richman et al., 2007) coupled with a greater understanding of the mechanism responsible for the side effects (Benyo et al., 2006; Maciejewski-Lenoir et al., 2006; Papaliodis et al., 2008) has therefore opened the door for the development of effective, tolerable drugs for the treatment of dyslipidaemia and the associated cardiovascular disease. The beneficial effects of niacin are wide-ranging and appear to result from its impact on a number of cell signalling pathways. There are several excellent reviews covering the interaction of niacin with its receptor (Offermanns, 2006; Kamanna and Kashyap, 2007; Bodor and Offermanns, 2008; Gille et al., 2008), and so it is not the purpose of this review to cover this topic in detail, but more to consider the signalling pathways activated by the niacin receptor, particularly those that are relevant to the underlying pathology of atherosclerosis. Because some of these signalling pathways also mediate the undesirable effects of niacin, the need to consider balancing the desirable versus the undesirable consequences of niacin receptor agonists in drug design is discussed.
Lipid profiles and cardiovascular disease
Abnormal blood lipids are the major modifiable risk factor underlying the development of cardiovascular disease and the identification of drugs to treat this condition remains a major pharmaceutical focus. The term ‘dyslipidaemia’ covers a range of lipid abnormalities, but of greatest concern is the so-called ‘atherogenic profile’ (often referred to as ‘the triad’) of low high-density lipoprotein-cholesterol (HDL-C), elevated triglycerides (TGs) and increased levels of small, dense low-density lipoprotein-cholesterol (LDL-C) particles (Despres, 2006). This profile is associated with an elevated risk of coronary heart disease, particularly in individuals with the metabolic syndrome, or type 2 diabetes (Nesto, 2005). LDL-C has generally been considered the major individual risk factor as it represents the transport and distribution of cholesterol-containing lipoprotein particles to peripheral tissues, including the vascular intima. However, even aggressive reduction of LDL-C only reduces the occurrence of cardiac events by 25–40%, and it is now generally accepted that low levels of HDL-C are more significant in predicting risk (Bodor and Offermanns, 2008).
Lipid storage and metabolism
Excess blood lipid has a serious impact on whole-body metabolic function. Such excess can result from too high an intake of dietary fats, but it may also result from reduced fat utilization by skeletal muscle tissue, or a combination of both (Bajaj et al., 2007), leading to an increased accumulation of lipid within skeletal muscle cells. Lipids may be stored as triacylglycerol, diacylglycerol, fatty acid acyl CoA and ceramides, all of which have a profound effect on insulin signalling within the muscle tissue. For example, diacylglycerol and fatty acid acyl CoA have been shown to promote serine phosphorylation of insulin receptor substrate-1 (Powell et al., 2004), while ceramides may activate phosphatases that inactivate Akt, and simultaneously protein kinase C (Schubert et al., 2000; Teruel et al., 2001). Furthermore, lipid accumulation is associated with mitochondrial dysfunction, which appears to be primarily mediated by a down-regulation of expression of the transcriptional co-activator PGC1 (Patti et al., 2003). The combination of these events is resistance of skeletal muscle to circulating insulin, thus affecting glucose utilization in such tissues. Indeed, it has been shown that experimental elevation of free fatty acids (FFAs) causes inhibition of glucose disposal stimulation by insulin (Gormsen et al., 2007). Disorders of glucose metabolism such as insulin resistance and diabetes greatly accelerate the development of atherosclerosis.
As well as the effects on skeletal muscle, circulating free FFAs have a profound effect on whole-body storage and utilization of fat. For example, elevated FFA levels affect the function of the hypothalamic-pituitary-adrenal axis, leading to an increased release of ACTH (Kok et al., 2004b), and leptin production. Leptin is produced by adipocytes and acts on the hypothlamus to induce feelings of satiety, but it also has peripheral effects, essentially promoting fuel utilization rather than storage (Watson et al., 2000; Worm et al., 2000). Some studies have suggested that leptin may cause peripheral insulin resistance: it has been shown to inhibit insulin-stimulated glucose uptake and disposal in adipocytes, fibroblasts and hepatocytes (Cohen et al., 1996; Muller et al., 1997), and indeed high levels of circulating leptin are seen in obesity, a condition closely associated with the development of insulin resistance (Paz-Filho et al., 2008). In obese patients, it has been suggested that the leptin : adiponectin ratio can be used as an index of insulin resistance (Oda et al., 2008). In contrast, obesity and insulin resistance have been associated with a depressed regulatory function of leptin, whereby increased activity of normal leptin signalling pathways is associated with increased insulin sensitivity (Wang-Fisher et al., 2002).
Lipids and atherosclerosis
While abnormal blood lipids are a key risk factor in the development of cardiovascular disease, it is only part of the story. Vascular disease is caused by the development of atherosclerotic plaques, which are thought to form as a response to endothelial injury [reviewed by Newby (2000)]. Inflammation therefore plays a pivotal role in both the initiation and development of lesions. As part of this inflammatory response, macrophages enter the intimal layer of the blood vessel, where they become activated and take up large amounts of oxidized LDL – a process mediated by elevated expression of the macrophage scavenger receptors CD36 and SRA (Moore et al., 2001). The resultant foam cells form the major cellular component of the plaque. Cytokine signals produced by these cells trigger further monocyte recruitment, as well as activating smooth muscle cells from the underlying media, which proliferate and migrate into the plaque, ingesting lipid, also taking on a foamy appearance. Foam cells of smooth muscle origin are important with regard to the clinical significance of plaque formation because they secrete extracellular matrix proteins (most notably collagen) forming a fibrous cap over the plaque, acting to stabilize the whole structure (Stoll and Bendszus, 2006). Destabilization of the plaque may lead to rupture, precipitating clot formation, which in the case of the coronary arteries is the event underlying myocardial infarction (MI) (Badimon et al., 2001).
Plaque formation is not the only consequence of abnormal lipid profiles; there are numerous other changes that significantly effect cardiovascular function. Lipid-induced vascular smooth muscle cell proliferation results in an increase in intima–media thickness, a measurement recognized as a surrogate marker for clinical coronary events (Taylor et al., 2007; Thoenes et al., 2007). Increased media thickness is associated with a decreased vascular compliance, which, combined with direct inflammatory effects of abnormal blood lipids on the myocardium itself, leads to impaired ventricular function. Ventricular function is likely to be further compromised in the event that dyslipidaemia results in an acute MI following plaque rupture in the coronary arteries. Furthermore, atherosclerosis is associated with heightened oxidative stress, some of it caused by increased myeloperoxidase production by activated macrophages and neutrophils (Boudjeltia et al., 2006; Meuwese et al., 2007). Production of reactive oxygen species (ROS) may also result from altered homocysteine metabolism: elevated levels of this amino acid are associated with increased atherosclerosis and a heightened risk of cardiovascular events (Ciaccio et al., 2008; Potter et al., 2008). Abnormal oxidative metabolism is now known to be important both in the oxidation of LDL and in eliciting the inflammatory response that is characteristic of atherosclerosis (Alexander, 1995; 2002;).
Niacin as a lipid-lowering agent
Niacin is the most effective drug treatment currently available for increasing HDL-C, an effect that is accompanied by a reduction in total cholesterol, LDL-C, TG and very low-density lipoproteins (VLDLs), as well as lipoprotein a [Lp(a])], a known independent risk factor for coronary vascular disease (Mahboubi et al., 2006; Bodor and Offermanns, 2008). It is particularly useful, therefore, in treating the subgroup of individuals who, despite statin therapy, still have low levels of HDL-C.
Many of the effects of niacin are considered to result from its action on adipose tissue: this acts as a store of excess energy, most of which is stored in the form of TGs. Adipocytes also accumulate cholesterol and collectively contain the largest cholesterol pool within the body (Zhao et al., 2008). Plasma lipoprotein concentrations exist in a state of ‘dynamic equilibrium’ with the lipids and cholesterol stored within adipocytes, and it is thought that adipose tissue acts as a buffer for excess cholesterol. This may explain why even moderate weight reductions can improve blood lipid profiles in obese patients by facilitating greater adipocyte-buffering capacity (Medina-Gomez et al., 2007). Following administration, niacin rapidly inhibits adipocyte lipolysis, and this is accompanied by a similarly rapid drop in plasma levels of FFAs. It is hypothesized that this results in a reduced substrate supply for synthesis of TGs and VLDLs by the liver (Mahboubi et al., 2006), and that the consequent attenuation of VLDL production limits the cholesterol ester transfer protein-mediated exchange of: (i) cholesterol from HDL to VLDL; (ii) of TG from VLDL to HDL; and (iii) of cholesterol between HDL and LDL (Bodor and Offermanns, 2008; Gille et al., 2008). The net effect is a reduced catabolism of HDL and a decreased accumulation of cholesterol esters in LDL particles. It has also been suggested that niacin may directly inhibit the uptake and catabolism of ApoA1-containing HDL particles, thus acting to further increase plasma levels of HDL (Lamon-Fava et al., 2008; Zhao et al., 2008).
Niacin receptors
Niacin agonists have been shown to impact on all aspects of metabolic control, and extensive clinical trials indicate that these effects translate into a reduced morbidity and mortality from cardiovascular disease (Taylor et al., 2004; 2007; Ballantyne et al., 2008; Green et al., 2008; Poldermans et al., 2008). Many of these effects are now known to be the result of activation of specific niacin receptors, which has led to the need to re-visit the mechanisms underlying their beneficial effects.
Two niacin receptors have been identified in humans: HM74 (GPR109b) and HM74A (GPR109a) (Soga et al., 2003; Pike and Wise, 2004; Tunaru et al., 2005). These are not polymorphic variants, but products of distinct genes on chromosome 12q24.31. HM74A is the orthologue of the murine PUMA-G and is thought to be responsible for the majority of the clinical effects of niacin, especially the effects on plasma lipids. HM74 appears to have arisen as a result of late gene duplication, and at the nucleotide level, there is a 96% sequence identity, with an amino acid identity of 89%. The differences result in a 1000× reduction in affinity of HM74 for niacin. Both HM74 and HM74A are G-protein-coupled receptors (GPCR).
HM74A shows a very specific tissue distribution. It is highly expressed in adipocytes, and its activation in these cells accounts for many of the effects of agonists of the receptor. However, it is also found in keratinocytes and certain immune cells, namely mature neutrophils (where it appears to be a marker of differentiation), macrophages, dendritic and Langerhans cells, but not monocytes (Cusi et al., 2006). It is noteworthy that, despite the profound effects that HM74A signalling has on glucose metabolism (discussed later in this review), there is no detectable expression of the receptor in the tissues most closely involved with glucose handling, such as hepatocytes, skeletal muscle and pancreatic β-cells (Cusi et al., 2006).
HM74A is coupled to a Gi subunit and thus activation results in decreased adenylate cyclase activity with a subsequent reduction in the intracellular concentration of cAMP. cAMP is the principal mediator of adipocyte lipolysis, largely because subsequent protein kinase A activation phosphorylates and activates hormone sensitive lipase. Inhibition of cAMP therefore results in reduced release of adipocyte FFAs into the plasma (Tang et al., 2006; Bodor and Offermanns, 2008).
Structure activity relationships and the niacin receptor
There are numerous molecules that are capable of binding to and activating HM74A, and the characteristic feature of such agonists is the presence of a carboxylic acid group. Like other GPCRs, HM74A is a ‘seven transmembrane receptor’, possessing seven membrane-spanning α-helices, separated by interhelical loops, an extracellular N-terminal region and an intracellular C-terminal region. Using a combination of site-directed mutagenesis and structural modelling to analyse HM74A chimeras, Tunaru et al. (2005) characterized the ligand binding pocket for niacin.
Site-directed mutagenesis revealed Arg111 (located in the third transmembrane helix, TMH3) to be essential, because it permits the formation of an important salt cross bridge between itself and the carboxylic acid group of niacin. Mutation of Arg111 to Ala was sufficient to completely prevent the binding of niacin, and this fits with the observation that compounds not containing a carboxylic acid group (such as nicotinamide) are not able to activate the receptor.
Other residues shown to be important in the binding include Asn86 and Trp91, at the junction of the second transmembrane helix (TMH2) and the first of the extracellularly located interhelical loops (ECL1). ECL2 contains another amino acid – Ser 178 – that also appears to be significant in terms of ligand binding. Within the HM74A binding pocket there are a number of aromatic residues, and mutagenesis revealed Phe276 and Tyr284 to be critical, acting as the main aromatic anchors. When the receptor is bound, the pyridine ring of nicotinic acid is embedded between Trp91 and Phe276, and Tunaru et al. suggest that these two residues act as a ‘gateway’, facilitating access to the binding pocket.
Although there is great similarity between HM74A and HM74, most of the differences are to be found in the region of the binding pocket, and it is likely that this accounts for the ligand selectivity between the two.
Niacin receptor agonists
β-Hydroxybutyrate
The fact that the plasma concentrations required to elicit effects at HM74A are far higher than those achieved physiologically indicated that niacin was unlikely to be the endogenous ligand, and the receptor remained an orphan until Taggart et al. (2005) identified β-hydroxybutyrate as the likely candidate. β-hydroxybutyrate is a small water-soluble carboxylic acid, released into the blood by the liver following fatty acid β-oxidation and was shown to bind and fully activate HM74A at plasma concentrations well within the range that could be achieved under certain circumstances such as fasting, when it is used as an alternative energy source. It is also produced in insulin-deficient states where glucose cannot be used as the primary energy source, and is one of three ketone bodies produced by the liver (the others being acetone and acetoacetate). Interestingly, it has antilipolytic effects that are not shared by the others. This suppression of adipocyte lipolysis decreases the availability of serum precursors for hepatic ketogenesis, thus limiting ketogenesis, having the combined effects of protecting against potentially fatal ketoacidosis and ensuring that fat stores are used efficiently. There are, however, some differences in the effects elicited by β-hydroxybutyrate when compared with niacin, particularly with regard to glucose metabolism. There is considerable evidence to indicate that niacin can have apparently undesirable effects on glucose metabolism (Poynten et al., 2003; Chang et al., 2006; Vittone et al., 2007) – it has been associated with a loss of glycaemic control in type 2 diabetic patients, but also with impairment of glucose tolerance in non-diabetic individuals. As a result, its use for treating dyslipidaemias linked with failed insulin signalling has been questioned, although clinical trials have indicated that the overall effect of niacin treatment in patients suffering from either type 2 diabetes or the metabolic syndrome is favourable (Grundy et al., 2002).The effect of niacin on insulin sensitivity appears to be related to the ‘rebound’ in plasma non-esterified fatty acids observed following the acute drop observed immediately after treatment. The elevation in plasma non-esterified fatty acids is a physiological burden equivalent to that seen in obese insulin-resistant individuals. In contrast to niacin, β-hydroxybutyrate has been shown to have a potentially beneficial effect with regard to insulin resistance and impaired glucose tolerance. Green et al. (1984) demonstrated that this ketone was able to enhance insulin sensitivity (and increase binding to the insulin receptor) when insulin concentrations were sub-maximal. Whether this has any physiological relevance remains unclear – type 2 diabetes is almost invariably accompanied by hyperinsulinaemia, and therefore insulin concentrations are unlikely to be sub-maximal.
Acipimox
Acipimox (5-methyl-pyrazine-2-carboxylic acid-4-oxide; Olbetam) is a nicotinic acid-derived antilipolytic drug that is used clinically to treat atherogenic dyslipidaemias. It shows similarity to niacin in its receptor selectivity: both show much higher affinity for HM74A. Acipimox also inhibits lipolysis (Fucella et al., 1980), also as a result of an inhibition of hormone sensitive lipase (HSL) (Christie et al., 1996), and interestingly, the inhibition of lipolysis is sustained for some 9–12 h following administration, compared with less than 3 h for standard niacin preparations. Acipimox is clinically effective at lower doses than niacin (typically 250 mg three times a day), and this may account for the reduced side effects (and therefore greater patient tolerability) associated with its use (Hadigan et al., 2006).
In addition to its effects on plasma lipids, acipimox has been shown to cause regression of atherosclerosis (Stuyt et al., 1998), and it has also proved particularly useful in treating dyslipidaemias associated with high levels of LDL or TGs (Crepaldi et al., 1988; O'Kane et al., 1992). Like niacin, it is also effective in reducing circulating levels of Lp(a) (Stuyt et al., 1998).
Most of the effects of acipimox are similar to the parent compound, including an enhancement of both leptin release from adipocytes, and also increased leptin sensitivity in target tissues (Wang-Fisher et al., 2002; Dullart et al., 2005), but there are some significant differences that are worthy of exploration. Perhaps the most significant difference is with regard to insulin sensitivity. While niacin has been shown to have an adverse effect with regard to glucose tolerance and insulin resistance, acipimox treatment (at least in the short term) has been shown to increase glucose uptake by skeletal muscle cells, increasing insulin sensitivity and reducing plasma levels of both glucose and insulin (Worm et al., 2000; Blachere et al., 2001). Second, short-term exposure to acipimox has been shown to impair reverse cholesterol transport in both normal and type 2 diabetic patients (Dullart and van Tol, 2001), which may have implications for longer term treatment.
The pharmacokinetics, patient tolerability and the combined effects on lipid and glucose metabolism have meant that acipimox has a number of clinical uses in addition to the treatment of primary dyslipidaemias. Growth hormone (GH) deficiency is associated with metabolic derangements (Cordido et al., 2003), and GH therapy is known to negatively affect insulin sensitivity, resulting in an increased risk of developing type 2 diabetes (Vickers et al., 2006).These effects arise from the fact that GH is a lipolytic hormone and causes an increase in plasma FFAs. Acipimox is effective in controlling this, but also appears to enhance spontaneous release of GH (Kok et al., 2004a), as well as enhancing tissue sensitivity to GH (Pontiroli et al., 1996). It has also been used as an adjunct to treat the dyslipidaemic aspects of polycystic ovarian syndrome in obese women (Ciampelli et al., 2002). Furthermore, the drug has proved useful for assessing myocardial viability – an important predictor of morbidity and mortality – in patients with diabetes, where the associated metabolic abnormalities affect the quality of metabolic results produced using 18F fluorodeoxyglucose imaging (Bax et al., 1997; Schinkel et al., 2003). Finally, it has been suggested that acipimox may be useful for the treatment and prevention of gestational diabetes and its associated risk of complications in late pregnancy. Towards the end of pregnancy, lipolytic activity is increased, particularly in the liver, and tissues that have metabolic flexibility switch to fat burning to spare glucose for the rapidly growing foetus and those tissues that are unable to use fat. Increasing levels of plasma FFAs increase the risk of insulin resistance and eclampsia, effects that – in rats – have been shown to be reversible with acipimox treatment (Sanchez-Vera et al., 2002). However, acipimox crosses the placenta, and the effects on the foetus are as yet unclear.
Acifran
The lipid-lowering properties of acifran have been known for some time (Cayen et al., 1982; Kallai-Sanfacon et al., 1983), but recently it has been shown to bind and activate both HM74A and HM74. Acifran analogues have been shown not only to bind to HM74A, but also to activate ERK1/2, thus allowing full signalling through the receptor (Mahboubi et al., 2006). Early clinical trials showed that beneficial effects on lipids could be seen with doses as low as 100 mg, and although there were some cutaneous side effects (such as flushing and pruritis), these were not sufficient to affect compliance. Such trials indicated that acifran was suitable for treating type II and IIa hyperlipoproteinaemia (Hunninghake et al., 1985; LaRosa et al., 1987). Like niacin, acifran is well absorbed – within 1–2 h of administration. Unlike niacin it undergoes no biotransformation. It is excretion is almost entirely renal (Cayen et al., 1986; 1990;).
Anthranilic acid derivatives
Niacin is not strictly speaking a vitamin – it can be synthesized hepatically from the precursor tryptophan, although this is slow, and requires vitamin B6 as a co-factor, meaning that many people are, in fact, niacin-deficient. Anthranilic acid derivatives such as furosemide have long been used as pharmaceuticals, and as anthranilic acid is a prescursor of tryptophan, it seemed reasonable to hypothesize that derivatives may impact on niacin pathways. Shen et al. (2007) investigated the ability of anthranilic acid derivatives to interact with HM74A, measuring binding affinity and receptor activation. They found that an anthranilic acid derivative with a methoxyphenyl group had moderate activity in both assays, but that this activity could be improved to full agonist levels by replacing the methoxyphenyl side chain with a napthyl group. Further modifications showed that this napthyl group could be replaced with a biphenyl structure without any apparent loss of activity. Biphenyl anthranilides therefore appeared to be promising niacin agonists. However, consideration of ADME characteristics revealed there to be a number of problems: these included poor bioavailability and high clearance. Furthermore, they were also shown to be potent inhibitors of the enzymes CYP2C8 and 2C9, enzymes are involved in the conversion of arachidonic acid to epoxyeicosatetraenoic acid, and known to play an important role in the maintenance of cardiovascular homeostasis. Collectively, these molecules are referred to as ‘endothelium derived hyperpolarising factors’ that act on the underlying vascular smooth muscle, preventing depolarization and therefore contraction. The effect of blocking these enzymes in vivo is unclear – while it might seem that inhibition of a vasorelaxant factor would be pro-atherogenic, there is evidence to suggest that sulfaphenazole (a specific 2C9 inhibitor) reduces lesion size in experimentally induced MI, an effect that may be due to inhibition of reperfusion-induced damage (Doggrell, 2004). However, such side effects are likely to be undesirable, and as they appeared to be related to the presence of the biphenyl group, a series of systematic modifications were carried out and examined for binding, activity and the potency of inhibition of CYP2C8/9. Replacement of either the terminal phenol with a hydroxypyridine moiety, or substitution of the inner phenol group with a thiazole significantly reduced CYP inhibition, while a compound bearing both substitutions had even less inhibitory activity, but also had increased affinity for HM74A. Substitution of the inner phenyl ring with either pyrazole or imidazole moieties further increased affinity for HM74A. Based on the results of these assays, many of these compounds appeared to have excellent potential as niacin agonists, and the ability to reduce plasma FFAs in a murine in vivo model was therefore investigated. The results of this were considered alongside measurements of blood perfusion, assayed using laser Doppler flowmetry in an established mouse ear model. The inner pyrazole substituted biaryl anthranilide appeared particularly interesting in this regard in that it was able (at micromolar doses) to elicit a similar reduction in plasma FFAs to 10 mg·kg−1 doses of niacin. Furthermore, pharmacokinetic characteristics were favourable, and the compound did not induce flushing, making such agonists very promising anti-hyperlipidaemics.
Pleiotropic effects of niacin
Although inhibition of adipocyte lipolysis has been suggested as the mechanism accounting for the improvement in plasma lipid profiles, this cannot be the only explanation: although niacin treatment results in a rapid drop in plasma FFAs, this effect is transient – some 2–3 h after administration, a ‘rebound’ effect is observed, with levels of FFAs climbing to above baseline (Dhalla et al., 2007). Additionally, it is becoming increasingly apparent that many of the beneficial effects of niacin on the cardiovascular system are independent of its lipid-lowering effects (Figure 1). As we gain a greater insight as to the downstream signalling events following HM74A activation we are acquiring a greater appreciation of the probability that niacin may directly exert pleiotropic effects that influence events in the development of cardiovascular disorders.
Figure 1.
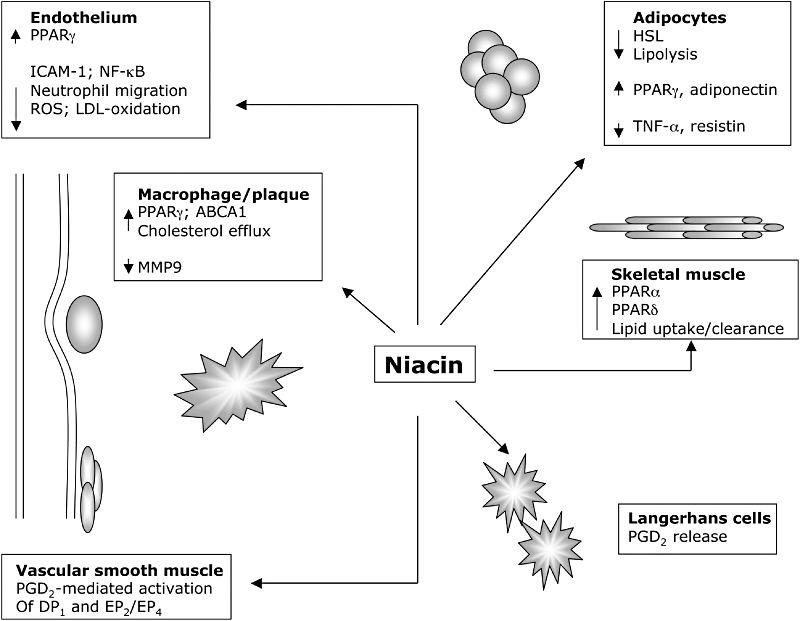
Actions of niacin modulate a multitude of processes that are protective against the development of atherosclerosis. The direct effects of niacin mediated through HM74A signalling inhibit adipocyte lipolysis and thus have a beneficial effect on blood lipids. Activation of nuclear receptor and prostanoid signalling pathways alters gene expression profiles in a number of cell types, eliciting profound anti-inflammatory effects. LDL, low-density lipoprotein; PGD2, prostaglandin D2; PPAR, peroxisome proliferator-activated receptor; ROS, reactive oxygen species.
Generation of NAD+
Structurally, niacin consists of a pyridine ring with a carboxylic acid group in the 3-position. As discussed previously, this acid group is particularly important for activity: substitutions are generally poorly tolerated, as exemplified by nicotinamide, which has an amide group at the 3-position and is almost inactive at the high-affinity HM74A receptor.
Niacin is administered orally, after which it undergoes extensive first pass metabolism in a dose-rate-specific manner. At the concentrations required to elicit lipid lowering, this process is saturable. In humans, there are two main pathways for niacin metabolism. The first is a low-affinity, high-capacity pathway involving a simple conjugation with glycine to form nicotinuric acid, which is also active with regard to HM74A (Meyers et al., 2007). The second pathway is high-affinity and low-capacity, and leads to the generation of nicotinamide and nicotinamide adenine dinucleotide (NAD+) and pyrimidine metabolites. NAD+ activity (as an acceptor of hydride equivalents) is essential for all living cells, and high doses of niacin or nicotinamide thus increase cellular NAD+ pools. Although it is thought that this is unrelated to its effects on lipid profiles, there is evidence to suggest that increased intracellular NAD+ may be beneficial with respect to cardiovascular disease in a number of ways. For example, niacin has been shown to inhibit ROS production in endothelial cells, an effect that is accompanied by a reduction in NF-κB activity, with subsequent down-regulation of a number of cytokines and adhesion molecules that are known to be involved in the development and progression of atherosclerosis, such as C-reactive protein, vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1, as well as enzymes involved in the oxidation of LDL. By inhibiting ROS production and LDL oxidation in this manner, niacin-induced increases in NAD+ may directly interfere with the cellular and molecular processes underlying atherosclerosis (Ganji et al., 2009). However, effects on ROS production notwithstanding, treatment with niacin has also been associated with increases in plasma homocysteine levels, which may offset the antioxidant effects to some degree (Basu et al., 2002; DeSouza et al., 2002).
There is also evidence to suggest that maintaining high concentrations of NAD+ may offer protection against damage caused by ischaemia/reperfusion following MI. DNA damage resulting from tissue damage initiates the PARP-1 activation cascade that depletes cells of NAD+ thus leaving surviving cells with a diminished capacity to deal with oxidative stress (Sauve, 2008). Nicotinamide has been shown to protect against this. Interestingly, nicotinamide undergoes further metabolism to produce 1-methynicotinamide, which has been shown in vivo to have an antithrombotic effect mediated by COX-2-derived prostacyclin (Choplicki et al., 2007).
Induction of prostanoid synthesis
One of the major downstream effects of signalling through the niacin receptor is the generation of eicosanoids, a phenomenon known to account for the side effect of severe cutaneous flushing associated with niacin use (Benyo et al., 2005; Papaliodis et al., 2008) (see below). However, these bioactive lipids are also capable of inducing further signalling events, which potentially account for at least some of the beneficial effects of niacin observed within the cardiovascular system.
Generally, GPCR activation is associated with the liberation of arachidonic acid from the cell membrane, resulting from PKC or MAPK phosphorylation of the cytosolic form of phospholipase A2, which then undergoes a calcium-dependent translocation to the membrane, where it catalyses the release of arachidonic acid from membrane phospholipids (Lin and Chen, 1998). Gq-coupled receptors are particularly effective at this as they also activate phospholipase C, generating IP3 and DAG, which mobilize intracellular calcium stores and thus activate PKC. In most cell types, Gi-coupled receptors do not normally stimulate release of physiologically relevant amounts of arachidonic acid, and HM74A is typical in this regard. However, HM74A has been shown to potentiate the effects of other stimuli on release, and niacin has been shown to cause an increase in intracellular Ca2+ concentration in mouse macrophages expressing PUMA-G (the murine orthologue of HM74A), suggesting synergism between Gi- and Gq-coupled receptors, although the precise mechanism through which this occurs is unknown (Knowles et al., 2006; Tang et al., 2006).
There is however compelling evidence from a number of sources that indicate that not only is significant eicosanoid production associated with HM74A signalling, but that it is the production of prostaglandins D2 and E2 (PGD2 and PGE2) that are responsible for the cutaneous flushing associated with its use (Morrow et al., 1989; Benyo et al., 2005). Specifically, flushing has been shown to result (at least partly) from the production of PGD2 and PGE2 by dermal/epidermal immune cells, which then exerts vasodilatory effects via the DP1 and EP2/EP4 receptors. This backs up the long-standing clinical observation that niacin-induced flushing in patients can be attenuated by co-administration with cyclo-oxygenase inhibitors such as acetylsalicylic acid, although a recent study by Papaliodis et al. (2008) suggests that the flushing response is also contributed to by the release of serotonin from platelets, and that complete resolution of flushing symptoms may require blocking of both pathways. The role of HM75A in mediating skin flushing was demonstrated in a series of elegant experiments by Benyo et al. (2005), who showed that PUMA-G knockout mice did not show skin flushing in response to treatment with niacin. Moreover, mice with normal PUMA-G expression, but lacking either the enzyme cyclo-oxygenase, the PGD2 receptor or the PGE2 receptor showed markedly decreased skin flushing, indicating the involvement of arachidonic acid signalling in this phenomenon. It seems likely that it is Langerhans cells, uniquely located within the skin, that are responsible for the cutaneous hyperaemia associated with niacin use (Benyo et al., 2006), and it has been shown that PGD2 synthase is abundantly expressed in these immune cells. Indeed, stimulation of Langerhans cells with niacin is capable of eliciting significant release of PGD2 (Maciejewski-Lenoir et al., 2006). The notion that the production of PGD2 is a local response is supported by the observation that, following niacin treatment, PGD2 levels are elevated in the venous circulation, but not so in the arterial (Morrow et al., 1992). Stimulation of Langerhans cells with niacin induces a transient increase in intracellular calcium concentration, suggesting that the HM74A/Gi effects result in a Gβγ-mediated activation of PLC (and subsequent IP3- and DAG-mediated calcium release) – a classic immune cell response (Exton, 1996; Rhee, 2001; Benyo et al., 2005). There is also evidence to suggest that it is ERK1/2 signalling may contribute to the enhanced arachidonic acid release by Langerhans cells associated with niacin treatment (Tang et al., 2006).
Activation of peroxisome proliferator-activated receptors (PPARs)
Many of the clinical effects of niacin are similar to those observed with PPARγ agonists (namely the glitazone class of insulin-sensitizing drugs), and it has been suggested that at least some of the effects of niacin result, in fact, from PPAR-mediated transcriptional regulation. PPARs are ligand-activated transcription factors that control lipid metabolism at the molecular level. There are three isoforms of these receptors: α, γ and δ. PPARα is expressed in liver, heart and skeletal muscle, where it controls the expression of a subset of genes involved in fatty acid oxidation, and thus activation enhances clearance of lipids through the liver. Because many inflammatory mediators (such as leukotriene B4) are lipids themselves, PPARα activation also enhances their clearance, and thus plays a major role in limiting the duration of the inflammatory response (Devchand et al., 1996). The fibrate class of drugs, known to be particularly effective in reducing serum levels of TGs, as well as mildly raising HDL, are agonists of PPARα.
PPARγ is one of the major regulators of adipocyte development and controls genes involved in adipocyte differentiation and fat storage, as well as inhibiting genes involved in lipolysis and release of FFA from adipose tissue (Brun and Spiegelman, 1997; Spiegelman, 1998). PPARγ activation also enhances insulin sensitivity and, although the precise mechanism is unclear, it appears that it is due (at least in part) to simultaneous inhibition of the release from adipose tissue of signalling molecules (e.g. TNFα and resistin) that promote insulin resistance, and production of adiponectin that has the opposite effect. PPARγ agonists (such as rosiglitazone) have been used with some success to enhance insulin sensitivity in the treatment of type 2 diabetes (The DREAM Trial Investigators, 2006). PPARγ activity may also be beneficial in atherosclerotic vascular disease because of its inhibitory effect on matrix metalloproteinase production, which favours plaque stability (Zhang et al., 2007).
The role of PPARδ in lipid metabolism is less clear – agonists have been shown to have a favourable effect on plasma lipid profiles (Oliver et al., 2001), and the combined up-regulation of both PPARα and δ in skeletal muscle appears to be at least partly responsible for the beneficial effects of exercise on the cardiovascular system (Watt et al., 2004). It seems that PPARδ is activated in response to VLDL and switches on genes that are involved in fatty acid transport to (and oxidation within) the mitochondrion, thus enhancing lipid clearance (Lee et al., 2006; Furnsinn et al., 2007; Takahashi et al., 2007). However, at the level of the macrophage, PPARδ activation has been shown to be pro-inflammatory and to enhance lipid accumulation that has the potential to cause (or exacerbate) atherosclerosis (Vosper et al., 2001). Such PPARδ-mediated lipid accumulation has also been observed in smooth muscle cells in vascular lesions, which may also be significant with regard to atherosclerosis (Zhang et al., 2002). Interestingly, the efficacy of rosiglitazone in the treatment of insulin resistance has recently been brought into question following reports that there is an increased risk of death from major cardiovascular events such as MI (Nissen and Wolski, 2007; Psaty and Furberg, 2007). It has been suggested that that the underlying cause is that clinical doses are sufficiently high to activate PPARδ, and thus may enhance progression of atherosclerosis (Hall and McDonnell, 2007).
An effect on PPARs has been suggested as a mechanism by which niacin exerts at least some of its beneficial effects. Signalling downstream from the niacin receptor has been shown to involve induction and activation of PPARγ expression in both adipocytes and macrophages, while activation of HM74A has been shown to induce both expression and transcriptional activation of PPARγ in human macrophages (Zhao et al., 2008). Interestingly, niacin has also been shown to up-regulate expression of PPARα and δ in skeletal muscle, an effect similar to that observed with chronic low-intensity exercise (Watt et al., 2004), although paradoxically, niacin appears to attenuate the beneficial effects of exercise in terms of lowering plasma TGs (Plaisance et al., 2008). Niacin has also been shown to up-regulate both transcription and nuclear translocation of PPARγ in both liver and monocytoid cells, and to increase the transcription of two known PPARγ targets; CD36, a scavenger receptor known to be involved in the uptake of modified lipids by macrophages, and ABCA1, one of the major reverse cholesterol transporters in macrophages, responsible for the ApoA1-mediated transfer of intracellular cholesterol to HDL particles (Nicholson, 2004;Rubic et al., 2004). While one might expect niacin-induced increased expression of CD36 to be pro-atherogenic, in animal studies the elevation in CD36 transcription following treatment with PPARγ agonists has been shown to be accompanied by an increase in transcription of ABCA1, which enhances cholesterol clearance (Chawla et al., 2001).
The prostanoid-PPAR loop
As explained above, the PGD2-mediated effects of niacin have been associated with the undesirable flushing response. However, the major breakdown product of PGD2 (15-deoxy-Δ12,14-prostaglandin J2, 15d-PGJ2), has been identified as a potent endogenous ligand of the transcription factor, PPARγ (Scher and Pillinger, 2005). Moreover, the niacin-induced up-regulation of CD36 can be inhibited by treatment with cyclo-oxygenase inhibitors, which supports the notion that a cyclo-oxygenase product is involved. In contrast, such inhibitors have no effect on niacin-induced up-regulation of ABCA1, suggesting different mechanisms of action for niacin-mediated activation of PPARγ. This suggests that an exquisite relationship exists between the ability of niacin to activate PPARs directly, or indirectly through prostanoid production.
In addition to the effects on whole-body lipid metabolism, there is substantial evidence to indicate that PPARγ activation by 15d-PGJ2 is anti-inflammatory by a mechanism involving repression of transcription of a number of inflammatory genes, including COX-2 and TNFα (Ricote et al., 1998; 1999;). In support of an anti-inflammatory role for PPARγ, treatment with rosiglitazone has been shown to be effective in attenuating ischaemia/reperfusion injury in the gut (Cuzzocrea et al., 2003), most likely mediated (at least in part) by the PPARγ-mediated up-regulation of iNOS, as 15d-PGJ2-induced NO release has been shown to inhibit neutrophil migration as result of down-regulated ICAM-1 expression cells in the vessels of the mesenteric microcirculation (Napimoga et al., 2008). Synthetic PPARγ agonists such as rosiglitazone have been shown to have a similar effect on neutrophil migration in experimentally induced pancreatitis (Cuzzocrea et al., 2004). The effects of PPARγ activation are unlikely to be restricted to the gut and pancreas: there is also increasing evidence to suggest that PPARγ agonists have anti-inflammatory effects at the level of the vessel wall, and as such it has been suggested that this receptor may represent a therapeutic target for the prevention and treatment of ischaemic stroke (Culman et al., 2007). The anti-inflammatory effects may well result from a PPARγ-induced inhibition of NF-κB activity, resulting in a down-regulation in many inflammatory gene targets (Culman et al., 2007; Napimoga et al., 2008). There is also evidence to suggest that niacin may enhance neutrophil apoptosis, although this is likely to result from HM74A-mediated down-regulation of cAMP, which will enhance the activity of the pro-apototic protein bad, a member of the bcl-2 family (Kostylina et al., 2008). It is becoming increasingly clear that neutrophils are involved as part of the cellular component of the atherosclerotic plaque (Paulsson et al., 2006; van Leeuwen et al., 2008), and thus enhanced neutrophil apoptosis is likely to be beneficial.
Overcoming the side effects of niacin
Niacin treatment is associated with a number of side effects, including headache, itching and gastrointestinal disturbances, but these are generally mild. The most severe of the side effects is flushing, and this is sufficiently severe to negatively affect compliance. As discussed earlier, this flushing is caused by the production of PGD2, which also gives rise to endogenous ligands for PPARγ therefore generating effects that are beneficial with respect to the cardiovascular system. This presents a potential dilemma and has prompted further investigation into the wider effects of niacin to inform the development of effective drugs with improved tolerability.
Niacin: modified release formulations
Flushing is probably the most severe of the side effects associated with niacin use, but, in addition, niacin also has a short duration of action, which means that frequent dosing is required. To address this, sustained and extended release formulations have been developed, but the pharmacokinetics are markedly different for all three types of preparation (McKenney, 2004). Sustained release preparations are not approved for clinical use and are generally sold over the counter as food supplements. Niacin is usually completely absorbed with 1–2 h, compared with over 12 h for a typical sustained release preparation. Extended release preparations (such as Niaspan™) have been developed for clinical use, and the time taken for complete absorption represents a mid-ground between the immediate and sustained release formulations. The rate of absorption will affect the amount of drug metabolized via the pathways discussed above. Immediate release formulations rapidly saturate the nicotinamide pathway, with the result that HM74A active metabolites are produced in large concentrations. This means that immediate release preparations have the best effects on lipid parameters, but are also associated with more severe flushing. Sustained release formulations reduce the relative amount of metabolism through the conjugation pathway, thus reducing flushing (but also potentially reducing the potency of the drug in terms of lipid modulation) as well as increasing the duration of action. However, there is also increased risk of hepatotoxicity. Extended release formulations balance the metabolism between the two pathways, and this is the preferred way of administering the drug (McKenney, 2004), but it is possible that reduced activity of the conjugation pathway may mean a reduction in both the direct and indirect benefits resulting from HM74A signalling.
Partial agonists and agonists that do not activate ERK signalling
Attempts to find drugs that yield the beneficial effects of niacin (in terms of lipid lowering) but without the side effects of flushing led to the discovery of partial agonists: partial agonists are frequently shown to demonstrate tissue selectivity, and it seemed possible that such compounds may act on adipoctytes and the liver to reproduce the effects on lipid profile without causing the cutaneous effects. Van Herk et al. (2003) described a series of pyrazole derivatives that demonstrated partial agonist activity at the HM74A receptor. However, subsequent work by Richman et al. (2007) have shown that the effects of such compounds are not due to tissue selectivity, but rather due to a phenomenon described as ‘agonist directed trafficking of receptor signals’. They looked at a number of compounds demonstrating activity at the receptor, including a number of the ‘pyrazole’ partial agonists described by van Herk et al. (2003), as well as a number of full agonists. In terms of the physiological effects elicited, these compounds could be divided into those that induced flushing and those that did not. Full agonists of the HM74A receptor were found in both categories, suggesting that it is not the phenomenon of partial agonism that accounts for the differing response. Further investigation revealed that compounds that elicit flushing were those compounds that activated ERK1/2 MAP kinase and receptor internalization – an effect not observed with the ‘flush-free’ counterparts, although these drugs were still capable of stimulating the Gi-mediated inhibition of adenylyl cyclase and thus reducing plasma lipids. The notion that agonists at receptors may trigger only a subset of downstream signalling events has been proposed for a number of GPCRs: it may simply be that differing efficacies of agonists at the receptor result modulate the strength of the resulting signal, and that this signal may or may not be large enough to induced further events in the pathway. This may well result from the differing abilities of the binding agents to elicit conformational changes in the receptor, which in turn impacts on further signalling events. Similar modes of action have been hypothesized for other receptors, including 5HT2, α2a-adrenergic and adenosine A1 receptors. It may be that partial agonists are more likely than the full agonists to elicit different conformational effects, and this combined with tissue selectivity may account for the specific characteristics of each drug.
Combination drugs
The realization that cutaneous flushing is mediated through a PGD2/DP1 receptor effect, led to the development of dual drugs comprising extended release niacin with a DP1 antagonist. Merck's Tredaptive™ (Niaspan combined with the DP1 receptor antagonist laropiprant) has recently been licensed in Europe for the treatment of hyperlipidaemia. This combination has similar clinical efficacy to Niaspan™, but without the associated flushing and the reduced compliance associated with this side effect. However, in 2008, the FDA declined approval for the drug (known as Cordaptive™ in the US), and to some extent, this may have been fuelled by recent controversies surrounding the use of drugs such as the glitazones.
It is possible that adverse effects may arise in the long term from use of drugs that involve DP1 blockade. PGD2 is an inflammatory mediator, up-regulating expression of pro-inflammatory cytokines such as IL8 (Fu et al., 2002), but there is increasing evidence to suggest that in certain tissues it may have an anti-inflammatory role (Sadig et al., 2007), and is involved (along with PGE2) in controlling both the onset and resolution of inflammation (Rajakariar et al., 2007). These effects are mediated partly by the actions of PGD2 breakdown products – primarily 15d-PGJ2– catalysed by the enzyme PGD synthase, but also through direct DP1 activation (Hata and Breyer, 2004) leading to both the generation of anti-inflammatory cytokines as well as the inhibition of pro-inflammatory mediators (Bellows et al., 2005). Interestingly, polymorphisms in the gene encoding for PGDS are associated with cardiovascular disease such as carotid atherosclerosis (Miwa et al., 2004), while obese PGD synthase knockout mice develop insulin resistance, nephropathy and show thickening of the aortic intima, an effect that can be reversed by treatment with exogenous PGDS. In patients, elevated serum levels of PGDS predict a decreased occurrence of restenosis, a process caused by vascular smooth muscle cell proliferation (Ragiola et al., 2005). While some of these anti-inflammatory actions can be attributed to PPARγ activation, other effects have been shown to be mediated by binding of the DP1 receptor: in fact there is even evidence to suggest that activation of this receptor may enhance clearance of macrophages from sites of chronic inflammation such as atherosclerotic lesions. In contrast to the anti-inflammatory role of this PGD2, PGE2 has been shown to be pro-inflammatory: in particular, a PGE-dependent mechanism is responsible for an NF-κB-mediated increase in MMP-9. Because MMP-9 activity is known to be associated with plaque collagen breakdown, it is possible that instability may result from changes in the balance of PGES and PGDS in atherosclerotic lesions (Cipollone et al., 2004).
In addition to such dual drugs, niacin has also been co-administered with statins (Vittone et al., 2007) to very good effect. However, it has been shown that the changes in ApoA1 and HDL in response to such combinations are blunted if the patient is also taking antioxidant supplements (Cheung et al., 2001).
Conclusion
The profound improvements (in terms of lipid profile) seen during treatment means that HM74A agonists are some of the most promising drugs available for treating metabolic disorders that lead to vascular disease. Such agonists are also providing the scientific community with useful tools for studying the mechanisms underlying whole-body glucose and lipid metabolism. Side effects associated with niacin treatment are sufficiently severe to affect compliance, and thus limit the clinical usefulness of the drug. Because HM74A agonism is one of the most effective ways of treating atherogenic lipid profiles, it is imperative that the search continues for drugs that are free from adverse effects. Recent evidence has revealed that there are distinct differences between lipid-lowering mechanisms and those through which adverse effects are mediated, which has led to the development of new agonists, which try to block or reduce signalling through eicosanoid pathways. However, it is becoming increasingly clear that signalling through these pathways elicits at least some of the favourable effects associated with niacin treatment, and it is essential that this is borne in mind if the lipid-lowering effects of new drugs are to translate into a clinical benefit in terms of a reduction in cardiovascular risk.
Drug and receptor nomenclature conforms to the British Journal of Pharmacology's guide to receptors and channels (Alexander et al., 2008).
Glossary
Abbreviations:
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- FFA
free fatty acids
- GH
growth hormone
- GPCR
G-protein-coupled receptor
- HDL-C
high-density lipoprotein-cholesterol
- LDL-C
low-density lipoprotein-cholesterol
- Lp(a)
lipoprotein a
- MI
myocardial infarction
- PGD
prostaglandin D
- PGE
prostaglandin E
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- TG
triglyceride
- VLDL
very low-density lipoprotein
References
- Alexander RW. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995;25:155–161. doi: 10.1161/01.hyp.25.2.155. [DOI] [PubMed] [Google Scholar]
- Alexander RW. Oxidised LDL autoantibodies, endothelial dysfunction and transplant-associated atheroscelerosis. Arterioscler Thromb Vasc Biol. 2002;22:1950–1951. doi: 10.1161/01.atv.0000047863.88603.ce. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) 3rd Edition (2008 Revision) Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon L, Vilahur G, Sanchez S, Duran X. Atherosclerosis plaque formation and thrombogenesis: formation, risk factors and therapeutic approaches. Eur Heart J. 2001;3:I16–I22. [Google Scholar]
- Bajaj M, Medina-Navarro R, Suraamornkul S, Meyer C, DeFronzo RA, Mandarino LJ. Paradoxical changes in muscle gene expression in insulin-resistant subjects after sustained reduction in plasma free fatty acid concentration. Diabetes. 2007;56:743–752. doi: 10.2337/db06-0840. [DOI] [PubMed] [Google Scholar]
- Ballantyne CM, Davidson MH, McKenney J, Keller LH, Bajorunas DR, Karas RH. Comparison of the saftey and efficacy of a combination tablet of niacin extended release and simvastatin vs simvastatin monotherapy in patients with increased non-HDL cholesterol (from the SEACOAST-1 study) Am J Cardiol. 2008;101:1428–1436. doi: 10.1016/j.amjcard.2008.02.092. [DOI] [PubMed] [Google Scholar]
- Basu TK, Makhani N, Sedgwick G. Niacin (nicotinic acid) in non-physiological doses cause hyperhomocysteineaemia in Sprague-Dawley rats. Br J Nutr. 2002;87:115–119. doi: 10.1079/BJN2001486. [DOI] [PubMed] [Google Scholar]
- Bax JJ, Veening MA, Visser FC, Heine RJ, Cornel JH, Visser CA. Optimal metabolic conditions during fluorine-18 fluorodeoxyglucose imaging: a comparative study using different protocols. Eur J Nucl Med. 1997;24:35–41. doi: 10.1007/BF01728306. [DOI] [PubMed] [Google Scholar]
- Bellows CF, Alder A, Wludyka P, Jaffe BM. Modulation of macrophage nitric oxide production by prostaglandin D2. J Surg Res. 2005;132:92–97. doi: 10.1016/j.jss.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Benyo Z, Gille A, Kero J, Csiky M, Suchankova MC, Nusing RM, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest. 2005;115:3634–3640. doi: 10.1172/JCI23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyo Z, Gille A, Bennett CL, Clausen BE, Offermanns S. Nicotinic acid-induced flushing is mediated by activation of epidermal Langerhans cells. Mol Pharmacol. 2006;70:1844–1849. doi: 10.1124/mol.106.030833. [DOI] [PubMed] [Google Scholar]
- Blachere J-C, Perusse F, Bukowiecki LJ. Lowering plasma free fatty acids with acipimox mimics the antidiabetic effects of the β3-adrenergic agonist CL-16243 in obese Zucker diabetic fatty rats. Metabolism. 2001;50:945–951. doi: 10.1053/meta.2001.24868. [DOI] [PubMed] [Google Scholar]
- Bodor ET, Offermanns S. Nicotinic acid: an old drug with a promising future. Br J Pharmacol. 2008;153:S68–S75. doi: 10.1038/sj.bjp.0707528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudjeltia K, Legssyer I, Van Antwerpen P, Kisoka R, Babar S, Moguilevsky N, et al. Triggering of inflammatory response by myeloperoxidase-oxidized LDL. Biochem Cell Biol. 2006;84:805–812. doi: 10.1139/o06-061. [DOI] [PubMed] [Google Scholar]
- Brun RP, Spiegelman BM. PPARgamma and the molecular control of adipogenesis. J Endocrinol. 1997;155:217–218. doi: 10.1677/joe.0.1550217. [DOI] [PubMed] [Google Scholar]
- Cayen MN, Kallai-Sanfacon MA, Dubuc J, Greselin E, Dvornik D. Evaluation of the lipid-lowering activity of AY-25,712 in rats. Atherosclerosis. 1982;45:267–279. doi: 10.1016/0021-9150(82)90228-3. [DOI] [PubMed] [Google Scholar]
- Cayen MN, Gonzalez R, Ferdinandi ES, Greselin E, Hicks DR, Kraml M, et al. The metabolic disposition of acifran, a new antihyperlipidaemic agent, in rats and dogs. Xenobiotica. 1986;16:251–263. doi: 10.3109/00498258609043528. [DOI] [PubMed] [Google Scholar]
- Cayen MN, Ferdinandi ES, Hicks DR, Gonzalez R, Cosyns L, Dubuc J, et al. Pharmacokinetics and disposition of the lipid-lowering drug acifran in normal subjects and in patients with renal failure. Clin Pharmacol Ther. 1990;47:50–56. doi: 10.1038/clpt.1990.7. [DOI] [PubMed] [Google Scholar]
- Chang AM, Smith MJ, Galecki AT, Bloem CJ, Halter JB. Impaired β-cell function in human aging: response to nicotinic acid-induced insulin resistance. J Clin Endocrinol Metab. 2006;91:3303–3309. doi: 10.1210/jc.2006-0913. [DOI] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- Cheung MC, Zhao X-Q, Chait A, Albers JJ, Brown G. Antioxidant supplements block the response of HDL to simvastatin-niacin therapy in patients with coronary artery disease and low HDL. Arterioscler Thromb Vasc Biol. 2001;21:120–1326. doi: 10.1161/hq0801.095151. [DOI] [PubMed] [Google Scholar]
- Choplicki S, Swies J, Mogielnicki A, Buczko W, Bartus M, Lomnicka M, et al. 1-methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br J Pharmacol. 2007;152:230–239. doi: 10.1038/sj.bjp.0707383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie AW, McCormick DK, Emmison N, Kraemer FB, Alberti KG, Yeaman SJ. Mechanism of anti-lipolytic action of acipimox in isolated rat adipocytes. Diabetologia. 1996;39:45–53. doi: 10.1007/BF00400412. [DOI] [PubMed] [Google Scholar]
- Ciaccio M, Bivona G, Bellia C. Therapeutical approach to plasma homocysteine and cardiovascular risk reduction. Ther Clin Risk Manag. 2008;4:219–224. doi: 10.2147/tcrm.s1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampelli M, Leoni F, Lattanzi F, Guido M, Apa R, Lanzone A. A pilot study of the long-term effects of acipimox in polycystic ovary syndrome. Hum Reprod. 2002;17:647–653. doi: 10.1093/humrep/17.3.647. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Fazia M, Iezzi A, Ciabattoni G, Pini B, Cuccurullo C, et al. Balance between PGD synthase and PGE synthase is a major determinant of atherosclerotic plaque instability in humans. Arterioscler Thromb Vasc Biol. 2004;24:1259–1265. doi: 10.1161/01.ATV.0000133192.39901.be. [DOI] [PubMed] [Google Scholar]
- Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274:1185–1188. doi: 10.1126/science.274.5290.1185. [DOI] [PubMed] [Google Scholar]
- Cordido F, Alvarez-Castro P, Isidro ML, Casanueva FF, Dieguez C. Comparison between insulin tolerance test, growth hormone (GH)-releasing hormone (GHRH), GHRH plus acipimox and GHRH plus GH-releasing peptide-6 for the diagnosis of adult GH deficiency in normal subjects, obese and hypopituitary patients. Eur J Endocrinol. 2003;149:117–122. doi: 10.1530/eje.0.1490117. [DOI] [PubMed] [Google Scholar]
- Crepaldi G, Avogaro P, Descovich GC, Di Perri T, Postiglione A, Sirtori CR, et al. Plasma lipid lowering activity of acipimox in patients with type 2 and type 4 hyperlipoproteinaemia. Atherosclerosis. 1988;70:115–121. doi: 10.1016/0021-9150(88)90105-0. [DOI] [PubMed] [Google Scholar]
- Culman J, Zhao Y, Gohlke P, Herdegen P. PPAR gamma: a therapeutic target in ischaemic stroke. Trends Pharmacol Sci. 2007;28:244–249. doi: 10.1016/j.tips.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Cusi K, Kashyap S, Gastaldelli A, Bajaj M, Cersosimo E. Effects on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects genetically prediposed to type 2 diabetes. Am J Physiol Endocrinol Metab. 2006;292:E1775–E1781. doi: 10.1152/ajpendo.00624.2006. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Patel NS, Di Paola R, et al. Rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion of the gut. Br J Pharmacol. 2003;140:366–376. doi: 10.1038/sj.bjp.0705419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Britti D, Patel NS, et al. Rosiglitazone, a ligand of the peroxisome proliferator-actvated receptor-γ, reduces acute pancreatitis induced by cerulein. Intensive Care Med. 2004;30:951–956. doi: 10.1007/s00134-004-2180-1. [DOI] [PubMed] [Google Scholar]
- DeSouza C, Keebler M, McNamara DB, Fonseca V. Drugs affecting homocysteine metabolism: impact on cardiovascular risk. Drugs. 2002;62:605–616. doi: 10.2165/00003495-200262040-00005. [DOI] [PubMed] [Google Scholar]
- Despres J-P. Abdominal obesity: the most prevalent cause of the metabolic syndrome and related cardiometabolic risk. Eur Heart J. 2006;8:B4–B12. [Google Scholar]
- Devchand P, Keller H, Peters J, Vazquez M, Gonzalez F, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Dhalla AK, Santikul M, Smith M, Wong M-Y, Shryock JC, Belardinelli L. Antilipolytic activity of a novel partial A1 adenosine receptor agonist devoid of cardiovascular effects: comparison with nicotinic acid. J Pharmacol Exp Ther. 2007;321:327–333. doi: 10.1124/jpet.106.114421. [DOI] [PubMed] [Google Scholar]
- Doggrell SA. Inhibition of cardiac cytochrome P450: a new approach to cardiac ischaemia-reperfusion damage. Expert Opin Ther Targets. 2004;8:491–493. doi: 10.1517/14728222.8.5.491. [DOI] [PubMed] [Google Scholar]
- Dullart RP, van Tol A. Short-term acipimox decreases the ability of plasma from Type 2 diabetic patients and healthy subjects to stimulate cellular cholesterol efflux: a potentially adverse effect on reverse cholesterol transport. Diabet Med. 2001;18:509–513. doi: 10.1046/j.1464-5491.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- Dullart RP, Riemens SC, Meinardi JR, Wolffenbuttel BH, Sluiter WJ. Plasma adiponectin is modestly decreased during 24-hour insulin infusion but not after inhibition of lipolysis by Acipimox. Scand J Clin Lab Invest. 2005;65:523–531. doi: 10.1080/00365510500209090. [DOI] [PubMed] [Google Scholar]
- Exton JH. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- Fu Y, Luo N, Lopes-Virella MF. Upregulation of interleukin-8 expression by prostaglandin D2 metabolite 15-deoxy-delta12, 14 prostaglandin J2 (15d-PGJ2) in human THP-1 macrophages. Atherosclerosis. 2002;160:11–20. doi: 10.1016/s0021-9150(01)00541-x. [DOI] [PubMed] [Google Scholar]
- Fucella LM, Goldaniga G, Lovisolo P, Maggi E, Musatti L, Mandelli V, et al. Inhibition of lipolysis by nicotinic acid and by acipimox. Clin Pharmacol Ther. 1980;28:790–795. doi: 10.1038/clpt.1980.236. [DOI] [PubMed] [Google Scholar]
- Furnsinn C, Wilson TM, Brunmair B. Peroxisome proliferator-activated receptor-δ, a regulator of oxidative capacity, fuel switching and cholesterol transport. Diabetologia. 2007;50:8–17. doi: 10.1007/s00125-006-0492-0. [DOI] [PubMed] [Google Scholar]
- Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML. Niacin inhibits vascular oxidative stress, redox–sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis. 2009;202:68–75. doi: 10.1016/j.atherosclerosis.2008.04.044. [DOI] [PubMed] [Google Scholar]
- Gille A, Bodor ET, Ahmed K, Offermanns S. Nicotinic acid: pharmacological effects and mechanisms of action. Annu Rev Pharmacol Toxicol. 2008;48:79–106. doi: 10.1146/annurev.pharmtox.48.113006.094746. [DOI] [PubMed] [Google Scholar]
- Gormsen LC, Jessen N, Gjedsted J, Gjedde S, Norrelund H, Lund S, et al. Dose-response effects of free fatty acids on glucose and lipid metabolism during somatostatin blockade of growth hormone and insulin in humans. J Clin Endocrinol Metab. 2007;92:1834–1842. doi: 10.1210/jc.2006-2659. [DOI] [PubMed] [Google Scholar]
- Green A, Bustillos DP, Misbin RI. Beta-hydroxybutyrate increases the insulin sensitivity of adipocyte glucose transport at a post-receptor level. Diabetes. 1984;33:1045–1050. doi: 10.2337/diab.33.11.1045. [DOI] [PubMed] [Google Scholar]
- Green PJ, Vaiser T, Pennathur S, Kulstad JJ, Moore AB, Marcovina S, et al. Combined statin and niacin therapy remodels the high density lipoprotein proteome. Circulation. 2008;118:1259–1267. doi: 10.1161/CIRCULATIONAHA.108.770669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Vega GL, McGovern ME, Tulloch BR, Kendal DM, Fitz-Patrick D, et al. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes. Arch Intern Med. 2002;162:1568–1576. doi: 10.1001/archinte.162.14.1568. [DOI] [PubMed] [Google Scholar]
- Hadigan C, Liebau J, Torriani M, Andersen R, Grinspoon S. Improved triglycerides and insulin sensitivity with 3 months of acipimox in human immunodeficiency virus-infected patients with hypertriglyceridemia. J Clin Endocrinol Metab. 2006;91:4438–4444. doi: 10.1210/jc.2006-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, McDonnell DP. The molecular mechanisms underlying the proinflammatory action of thiazolidinediones in human macrophages. Mol Endocrinol. 2007;21:1756–1768. doi: 10.1210/me.2007-0060. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- van Herk T, Brussee J, van den Nieuwendijl AMCH, vander Klein PAM, Jzerman API, Stannek C, et al. Pyrazole derivatives as potent agonists for the nicotinic acid receptor. J Med Chem. 2003;46:3945–3951. doi: 10.1021/jm030888c. [DOI] [PubMed] [Google Scholar]
- Hunninghake DB, Edwards KD, Sopko GS, Tosiello RL. Controlled trial of acifran in type II hyperlipoproteinaemia. Clin Pharmacol Ther. 1985;38:313–317. doi: 10.1038/clpt.1985.177. [DOI] [PubMed] [Google Scholar]
- Kallai-Sanfacon MA, Cayen MN, Dubuc J, Greselin E, Dvornik D. Effect of AY-25,712 and other lipid-lowering agents on liver catalase and liver carnitine acetyltransferase in rats. Proc Soc Exp Biol Med. 1983;173:367–371. doi: 10.3181/00379727-173-41658. [DOI] [PubMed] [Google Scholar]
- Kamanna VS, Kashyap ML. Nicotinic acid (niacin) receptor agonists: will they be useful therapeutic agents? Am J Cardiol. 2007;100:S53–S61. doi: 10.1016/j.amjcard.2007.09.080. [DOI] [PubMed] [Google Scholar]
- Knowles HJ, Poole RT, Workman P, Harris AL. Niacin induces PPARgamma expression and transcriptional activation in macrophages via HM74 and HM74a-mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol. 2006;71:646–656. doi: 10.1016/j.bcp.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Kok P, Buijs M, Kok S, Van Ierssel I, Frolich M, Roelfsema F, et al. Acipmox enhances spontaneous growth hormone secretion in obese women. Am J Physiol Regul Integr Comp Physiol. 2004a;286:R693–R698. doi: 10.1152/ajpregu.00595.2003. [DOI] [PubMed] [Google Scholar]
- Kok P, Kok SW, Buijs MM, Westenberg JJM, Roelfsema F, Frolich M, et al. Enhanced circadian ACTH release in obese premenopausal women: reversal by short-term acipimox treatment. Am J Physiol Endocrinol Metab. 2004b;287:E848–E856. doi: 10.1152/ajpendo.00254.2004. [DOI] [PubMed] [Google Scholar]
- Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU. Neutrophil apoptosis is mediated by nicotinic acid receptors (GP109A) Cell Death Differ. 2008;15:134–142. doi: 10.1038/sj.cdd.4402238. [DOI] [PubMed] [Google Scholar]
- LaRosa JC, Miller VT, Edwards KD, DeBovis MR, Stoy DB. Acifran: a double-blind, randomized, placebo-controlled efficacy study in type IIa hyperlipoproteinemic patients. Artery. 1987;14:338–350. [PubMed] [Google Scholar]
- Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Nyaku M, Horvath KV, et al. Extended release niacin alters the metabolism of plasma apolipoprotein (Apo)-A1 and Apo-B-containing lipoproteins. Arterioscler Thromb Vasc Biol. 2008;28:1672–1678. doi: 10.1161/ATVBAHA.108.164541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong L-W, et al. Peroxisome proliferator-activated receptor δ promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci USA. 2006;10:2434–2439. doi: 10.1073/pnas.0510815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen M, Gijbels MJJ, Duijvestijn A, Smook M, van der Gaar MJ, Heeringa P, et al. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler Thromb Vasc Biol. 2008;28:84–89. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- Lin W-W, Chen BC. Distinct PKC isoforms mediate the activation of cPLA2 and adenylyl cyclase by phorbol ester in RAW264.7 macrophages. Br J Pharmacol. 1998;125:1601–1609. doi: 10.1038/sj.bjp.0702219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahboubi K, Witman-Jones T, Adamus JE, Letsinger JT, Whitehous D, Moorman AR, et al. Triglyceride modulation by acifran analogs: activity towards the niacin high and low affinity G-protein coupled receptors HM74A and HM74. Biochem Biophys Res Commun. 2006;340:482–490. doi: 10.1016/j.bbrc.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Maciejewski-Lenoir D, Richman JG, Hakak Y, Gaidarov I, Behan DP, Connolly DT. Langerhans cells release prostaglandin D2 in response to nicotinic acid. J Invest Dermatol. 2006;126:2637–2646. doi: 10.1038/sj.jid.5700586. [DOI] [PubMed] [Google Scholar]
- McKenney J. New perspectives on the use of niacin in the treatment of lipid disorders. Arch Intern Med. 2004;164:697–705. doi: 10.1001/archinte.164.7.697. [DOI] [PubMed] [Google Scholar]
- Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007;3:e64. doi: 10.1371/journal.pgen.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwese MC, Stroes ESG, Hazen SL, van Miert JN, Kuivenhoven JA, Schaub RG, et al. Serum myeloperoxidase levels are associated with the future coronary artery disease in apparently healthy individuals. J Am Coll Cardiol. 2007;50:159–165. doi: 10.1016/j.jacc.2007.03.033. [DOI] [PubMed] [Google Scholar]
- Meyers CD, Liu P, Kamanna VS, Kashyap ML. Nicotinic acid induces secretion of prostaglandin D2 in human macrophages: an in vitro model of the niacin flush. Atherosclerosis. 2007;192:25–258. doi: 10.1016/j.atherosclerosis.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Miwa Y, Takiuchi S, Kamide K, Yoshii M, Horio T, Tanaka C, et al. Identification of lipocalin-type prostaglandin D synthase and its association with carotid atherosclerosis in Japanese hypertensive patients. Biochem Biophys Res Commun. 2004;322:428–433. doi: 10.1016/j.bbrc.2004.07.143. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, et al. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Parsons WG, Roberts LJ. Release of markedly increased quantities of prostaglandin D2 in vivo in humans following administration of nicotinic acid. Prostaglandins. 1989;38:263–274. doi: 10.1016/0090-6980(89)90088-9. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Awad JA, Oates JA, Roberts LJ. Identification of skin as a major site of prostaglandin D2 release follwoing oral administration of niacin in humans. J Invest Dermatol. 1992;98:812–815. doi: 10.1111/1523-1747.ep12499963. [DOI] [PubMed] [Google Scholar]
- Muller G, Ertl J, Gerl M, Preibisch G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. J Biol Chem. 1997;272:10585–10593. doi: 10.1074/jbc.272.16.10585. [DOI] [PubMed] [Google Scholar]
- Napimoga MH, Viera SM, Dal-Secco D, Freitas A, Sonto FO, Mestringer FL, et al. Peroxisome proliferator-activated receptor-γ ligand, 15-deoxy-Δ12–14– Prostaglandin JZ, reduces neubrophil migration via a nitric oxide pathway. J Immunol. 2008;180:609–617. doi: 10.4049/jimmunol.180.1.609. [DOI] [PubMed] [Google Scholar]
- Nesto RW. Beyond low-density lipoprotein: addressing the atherogenic lipid triad in type 2 diabetes mellitus and the metabolic syndrome. Am J Cardiovasc Drugs. 2005;5:379–387. doi: 10.2165/00129784-200505060-00005. [DOI] [PubMed] [Google Scholar]
- Newby AC. An overview of the vascular response to injury: a tribute to the late Russell Ross. Toxicol Lett. 2000;112(113):519–529. doi: 10.1016/s0378-4274(99)00212-x. [DOI] [PubMed] [Google Scholar]
- Nicholson AC. Expression of CD36 in macrophages and atherosclerosis. The role of lipid regulation of PPARγ signaling. Trends Cardiovasc Med. 2004;14:8–12. doi: 10.1016/j.tcm.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- O'Kane MJ, Trinick TR, Tynan MB, Trimble ER, Nicholls DP. A comparison of acipmox and nicotinic acid in type b hyperlipidaemia. Br J Clin Pharmac. 1992;33:451–453. doi: 10.1111/j.1365-2125.1992.tb04067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda N, Imamura S, Fujita T, Uchida Y, Inagaki K, Kakizawa H, et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism. 2008;57:268–273. doi: 10.1016/j.metabol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Offermanns S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol Sci. 2006;27:384–390. doi: 10.1016/j.tips.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, et al. A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaliodis D, Boucher W, Kemperaj D, Michaelian M, Wolfberg A, House M, et al. Niacin-induced ‘flush’ involves release of PGD2 from mast cells and serotonin from platelets: evidence from human cells in vitro and an animal model. J Pharmacol Exp Ther. 2008;327:665–672. doi: 10.1124/jpet.108.141333. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Kusi K, Berria R, Kashyap S, et al. Co-ordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson J, Dadfar E, Held C, Jacobson SH, Lundahl J. Activation of peripheral and in vivo transmigrated neutrophils in patients with stable coronary artery disease. Atherosclerosis. 2006;192:328–334. doi: 10.1016/j.atherosclerosis.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Paz-Filho G, Eposito K, Hurwitz BE, Sharma A, Dong C, Andreev VP, et al. Changes in insulin sensitivity during leptin replacement therapy in leptin-deficient patients. Am J Physiol Endocrinol Metab. 2008;295:E1401–E1408. doi: 10.1152/ajpendo.90450.2008. Oct 14 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike NB, Wise A. Identification of a nicotinic acid receptor: is this the molecular target for the oldest lipid-lowering drug? Curr Opin Investig Drugs. 2004;5:271–275. [PubMed] [Google Scholar]
- Plaisance EP, Mestek ML, Mahurin AJ, Taylor JK, Moncada-Jimenez J, Grandjean PW. Postprandial triglyceride responses to aerobic exercise and extended-release niacin. Am J Clin Nutr. 2008;88:30–37. doi: 10.1093/ajcn/88.1.30. [DOI] [PubMed] [Google Scholar]
- Poldermans D, Dunkelgrun M, Schouten O, Hostalek U. Niaspan-induced HDL elevation for optimising risk control (NEMO) study. Eur Surg Res. 2008;41:313–318. doi: 10.1159/000155896. [DOI] [PubMed] [Google Scholar]
- Pontiroli A, Manzoni M, Malighetti M, Lanzi R. Restoration of growth hormone (GH) response to GH-releasing hormone in elderly and obses subjects by acute pharmacological reduction of plasma free fatty acids. J Clin Endocrinol Metab. 1996;81:3998–4001. doi: 10.1210/jcem.81.11.8923850. [DOI] [PubMed] [Google Scholar]
- Potter K, Hankey GJ, Green DJ, Eikelboom J, Jamrosik K, Arnolda LF. The effect of long-term homocysteine-lowering on carotid intima-media thickness and flow-mediated vasodilation in stroke patients: a randomised controlled trial and meta-analysis. BMC Cardiovasc Disord. 2008;8:24. doi: 10.1186/1471-2261-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DJ, Turban S, Gray A, Hajducjh E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J. 2004;382:619–629. doi: 10.1042/BJ20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynten AM, Gan SK, Kriketos AD, O'Sullivan A, Kelly JJ, Ellis BA, et al. Nicotinic acid-induced insulin resistance is related to increasing circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism. 2003;52:699–704. doi: 10.1016/s0026-0495(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Psaty BM, Furberg CD. The record on rosiglitazone and the risk of myocardial infarction. N Engl J Med. 2007;357:67–69. doi: 10.1056/NEJMe078116. [DOI] [PubMed] [Google Scholar]
- Ragiola L, Palaia T, Hall CE, Maesaka JK, Eguchi N, Urade Y. Accelerated glucose intolerance, nephropathy and atherosclerosis in prostaglandin D2 synthase knock-out mice. J Biol Chem. 2005;280:29946–29955. doi: 10.1074/jbc.M502927200. [DOI] [PubMed] [Google Scholar]
- Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyΔ12–14PGJ2. Proc Natl Acad Sci USA. 2007;104:20979–20984. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholiase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman JG, Kanemitsu-Parks M, Gaidarov I, Cameron JS, Griffin P, Zheng H, et al. Nicotinic acid receptor agonists differentially activate downstream effect. J Biol Chem. 2007;282:18028–18036. doi: 10.1074/jbc.M701866200. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK. The peroxisome proliferator-activated receptor (PPAR-γ) as a regulator of monocyte/macrophage function. J Leukocyte Biol. 1999;66:733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- Rubic T, Trottmann M, Lorenz RL. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol. 2004;67:411–419. doi: 10.1016/j.bcp.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Sadig H, Pease JE, Saroe I. Contrary prostaglandins: the opposing roles of PGD2 and its metabolites in leukocyte function. J Leukoc Biol. 2007;81:372–382. doi: 10.1189/jlb.0706424. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vera I, Bonet B, Viana M, Herrera E, Indart A. Effect of acipimox on plasma lipids and glucose/insulin in pregnant rats. Int J Exp Diabetes Res. 2002;3:233–239. doi: 10.1080/15604280214938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008;324:883–893. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114:100–109. doi: 10.1016/j.clim.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Schinkel AFL, Bax JJ, Valkema R, Elhendy A, van Domburg RT, Vourvouri EC, et al. Effect of diabetes mellitus on myocardial 18F-FDG SPECT using acipimox for the assessment of myocardial viability. J Nucl Med. 2003;44:877–883. [PubMed] [Google Scholar]
- Schubert K, Sheid M, Duronio V. Ceramide inhibits protein kinase B/Akt signalling by promoting dephosphorylation of serine 473. J Biol Chem. 2000;275:13330–13335. doi: 10.1074/jbc.275.18.13330. [DOI] [PubMed] [Google Scholar]
- Shen HC, Ding F-X, Luell S, Forrest MJ, Carballo-Jane E, Wu KK, et al. Discovery of biaryl anthranilides as full agonists for the high affinity niacin receptor. J Med Chem. 2007;50:6303–6306. doi: 10.1021/jm700942d. [DOI] [PubMed] [Google Scholar]
- Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, et al. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303:364–369. doi: 10.1016/s0006-291x(03)00342-5. [DOI] [PubMed] [Google Scholar]
- Spiegelman B. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- Stuyt PMJ, Kleinjans HAJ, Stalenhoef AFH. Tolerability and effects of high doses acipimox as additional lipid-lowering therapy in familial hypercholesterolemia. Netherlands. J Med. 1998;53:228–233. doi: 10.1016/s0300-2977(98)00076-x. [DOI] [PubMed] [Google Scholar]
- Taggart AKP, Kero J, Gan X, Cai T-Q, Cheng K, Ippolito M, et al. D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Tanaka T, Sakai J. New therapeutic target for metabolic syndrome: PPARδ. Endocr J. 2007;54:347–357. doi: 10.1507/endocrj.kr-99. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhou L, Gunnet JW, Wines PG, Cryan EV, Demarest KT. Enhancement of arachidonic acid signalling pathway by nicotinic acid receptor HM74A. Biochem Biophys Res Commun. 2006;345:29–37. doi: 10.1016/j.bbrc.2006.04.051. [DOI] [PubMed] [Google Scholar]
- Taylor AJ, Sullenberger LE, Lee HJ, Grace KA. Arterial biology for the investigation of the treatment effects of reducing cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–3517. doi: 10.1161/01.CIR.0000148955.19792.8D. [DOI] [PubMed] [Google Scholar]
- Taylor AJ, Zhu D, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Relationship between glycemic status and progression of carotid intima-media thickness during treatment with combined statin and extended-release niacin in ARBITER 2. Vasc Health Risk Manag. 2007;3:159–164. [PMC free article] [PubMed] [Google Scholar]
- Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-α in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- The DREAM (Diabetes REduction Assessment with ramipril and rosiglitazone Medication) Trial Investigators. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- Thoenes M, Oguchi A, Nagamia S, Vaccari CS, Hammoud R, Umpierrez GE, et al. The effects of extended-release niacin on carotid intimal thickness, endothelial function and inflammatory markers in patients with the metabolic syndrome. Int J Clin Pract. 2007;61:1942–1948. doi: 10.1111/j.1742-1241.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- Tunaru S, Lattig J, Kero J, Krause G, Offermanns S. Characterization of determinants of ligand binding to the nicotinic acid receptor GPR109A (HM74A/PUMA-G) Mol Pharmacol. 2005;68:1271–1280. doi: 10.1124/mol.105.015750. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Hofman PL, Gluckman PD, Lobie PE, Cutfield WS. Combination therapy with acipimox enhances the effect of growth hormone treatment on linear body growth in the normal and small-for-gestational age rat. Am J Physiol Endocrinol Metab. 2006;291:E1212–E1219. doi: 10.1152/ajpendo.00614.2005. [DOI] [PubMed] [Google Scholar]
- Vittone F, Chait A, Morse JS, Fish B, Brown G, Zhao X-Q. Niacin plus simvastatin reduces coronary stenosis progression among patients with metabolic syndrome despite a modest increase in insulin resistance: a subgroup analysis of the HDL-atherosclerosis treatment study (HATS) J Clin Lipidol. 2007;1:203–210. doi: 10.1016/j.jacl.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosper H, Patel L, Graham TL, Khoudoli GA, Hill A, Macphee CH, et al. The peroxisome proliferators-activated receptor delta promotes lipid accumulation in human macrophages. J Biol Chem. 2001;276:44258–44265. doi: 10.1074/jbc.M108482200. [DOI] [PubMed] [Google Scholar]
- Wang-Fisher Y-L, Han J, Guo W. Acipimox stimulates leptin production from isolated rat adipocytes. J Endocrinol. 2002;174:267–272. doi: 10.1677/joe.0.1740267. [DOI] [PubMed] [Google Scholar]
- Watson PM, Commins SP, Beiler RJ, Hatcher HC, Gettys TW. Differential regulation of leptin expression and function in A/J vs C57BL/6J mice during diet-induced obesity. Am J Physiol Endocrinol Metab. 2000;279:E356–E365. doi: 10.1152/ajpendo.2000.279.2.E356. [DOI] [PubMed] [Google Scholar]
- Watt MJ, Southgate RJ, Holmes AG, Febbraio MA. Suppression of plasma free fatty acids upregulates peroxisome proliferator-activated receptor (PPAR) alpha and delta and PPAR coactivator 1alpha in human skeletal muscle, but not lipid regulatory genes. J Mol Endocrinol. 2004;33:533–544. doi: 10.1677/jme.1.01499. [DOI] [PubMed] [Google Scholar]
- Wise A, Foord JM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, et al. Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem. 2003;278:9869–9874. doi: 10.1074/jbc.M210695200. [DOI] [PubMed] [Google Scholar]
- Worm D, Vinten J, Vaag A, Henriksen JE, Beck-Nielsen H. The nicotinic acid analogue acipimox increases plasma leptin and decreases free fatty acids in Type 2 diabetic patients. Eur J Endocrinol. 2000;143:389–395. doi: 10.1530/eje.0.1430389. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fu M, Zhu X, Xiao Y, Mou Y, Zheng H, et al. Peroxisome proliferator-activated receptor δ is up-regulated during vascular lesion formation and promotes post-confluent cell proliferation in vascular smooth muscle cells. J Biol Chem. 2002;277:11505–11512. doi: 10.1074/jbc.M110580200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ge H, Wang CQ, Guo TB, He Q, Shao Q, et al. Inhibitory effect of PPAR on the expression of EMMPRIN in macrophages and foam cells. Int J Cardiol. 2007;117:373–380. doi: 10.1016/j.ijcard.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Zhao SP, Yang J, Li J, Dong SZ, Wu ZH. Effect of niacin on LXRalpha and PPARgamma expression in HDL-induced cholesterol efflux in adipocytes of hypercholesterolemic rabbits. Int J Cardiol. 2008;124:172–178. doi: 10.1016/j.ijcard.2006.12.032. [DOI] [PubMed] [Google Scholar]