

Abstract
Background and purpose:
Resveratrol (RES) has been shown to prolong lifespan and prevent cancer formation. At present, the precise cellular mechanisms of RES actions are still not clearly understood, and this is the focus of this study.
Experimental approach:
Using human hepatocellular carcinoma-derived HepG2 cells as a model, we studied RES-induced changes in cell growth, cell cycle progression and apoptosis.
Key results:
RES at lower concentrations induced a strong but reversible S-phase delay and mild DNA synthesis inhibition, yet without causing apoptotic or necrotic cell death. At high concentrations, RES induced apoptosis, which is mainly mediated by the mitochondrial pathway. Overall, RES was a relatively weak apoptotic agent. Mechanistically, MEK inhibition was identified as an important early signalling event for RES-induced apoptosis. In comparison, activation of CDK2 and checkpoint kinase 2, and inhibition of phosphatidylinositol 3′-kinase/Akt signalling pathway contributed to the induction by RES of a reversible, non-cytotoxic S-phase delay.
Conclusion and implications:
It is hypothesized that the induction of a non-cytotoxic S-phase delay may represent a useful mechanistic strategy for lifespan prolongation and cancer prevention. When cell cycles are selectively slowed down in the S phase, it would cumulatively increase the total lifespan of an organism if the total numbers of cell divisions of a given organism are assumed to remain basically constant. Likewise, when cells proceed through the cell cycles at a reduced pace during DNA replication, it may allow cells more time to repair the damaged DNA, and thereby reduce the chances for mutagenesis and tumour initiation.
Keywords: resveratrol, S-phase arrest, cell cycle regulation, lifespan prolongation, cancer prevention
Introduction
Resveratrol (trans-3,4′,5-trihydroxystilbene; RES), a well-known polyphenolic compound, is highly enriched in our daily food components, such as grapes, peanuts and red wine (Kopp, 1998; Sobolev and Cole, 1999; Roldán et al., 2003). This chemical has attracted enormous research interest in recent years partly because of the intriguing earlier suggestion that its diverse beneficial biological effects may explain the so-called French paradox, which refers to observations that less French people die of heart attack compared to Americans despite high intake of dietary cholesterol and saturated fat of the former (Constant, 1997; Ferrieres, 2004). In addition to its protective effect on the cardiovascular system, studies have also shown that RES has antiproliferative and chemopreventive properties in cultured cancer cells (Jang et al., 1997; Aggarwal et al., 2004) and laboratory animals (Carbóet al., 1999; Zhou et al., 2005; Garvin et al., 2006). Furthermore, RES was recently found to increase lifespan in yeast, metazoans and laboratory animals (Baur et al., 2006; Valenzano et al., 2006). These studies have created considerable excitement about the possibility of developing RES and/or its structural analogs as concept compounds that may prevent cancer while improving longevity. At present, the precise cellular mechanisms by which RES acts to exert some of its unique beneficial effects are still not clearly understood, although a few earlier studies have suggested that the activation of protein deacetylase SIRT1 may be, in part, responsible for some of these effects (Howitz et al., 2003; Borra et al., 2005; Kaeberlein et al., 2005; Yang et al., 2007; Mayers et al., 2008; Oberdoerffer et al., 2008; Pearson et al., 2008). A better understanding of the underlying mechanism(s) of its actions will undoubtedly be helpful to the ongoing effort to develop RES or other similar compounds for human use. This was precisely the purpose of this study as well as some of the earlier studies by others (Ahmad et al., 2001; She et al., 2001; Joe et al., 2002; Kuo et al., 2002; Aquilano et al., 2009).
A number of recent studies have begun to reveal that the development of cancer is a long-term process that is associated with alterations in various cell cycle events and/or apoptosis. It has become an attractive hypothesis in cancer therapy and prevention by designing and developing agents that can alter cell cycle events and/or induce apoptosis (Jacks and Weinberg, 1996; Kastan and Bartek, 2004; Sebolt-Leopold and Herrera, 2004; Schwartz and Shah, 2005; Malumbres and Barbacid, 2007; Roberts and Der, 2007). In eukaryotes, cell cycle progression is controlled, in part, by a family of protein kinase complexes, which include cyclin-dependent kinases and their activating partners, cyclins. They are regulated by cell cycle-inhibiting proteins (e.g. p21Waf1/Cip1, p27Kip1 and members of INK family proteins) (Jacks and Weinberg, 1996). In addition, the mitogen-activated protein kinases (MAPKs) are a family of protein serine/threonine kinases, including ERK1/2, the c-Jun NH2-terminal kinase (JNK) and p38 MAPKs (Widmann et al., 1999). These kinases play a crucial role in regulation of cell growth and apoptosis. In addition, the phosphatidylinositol 3′-kinase (PI3K)/Akt is another important regulatory pathway that also regulates cell cycle and survival (Chang et al., 2003; Franke et al., 2003; Ahn et al., 2004; Fresno Vara et al., 2004; Horvath et al., 2007).
In the present study, we have sought to evaluate further the effect of RES on cell growth and apoptosis in HepG2 cells, with an emphasis on its regulation of cell cycle progression. The selection of the HepG2 human hepatocellular carcinoma-derived cell line for our study was partly based on an earlier study suggesting that liver and kidney are two main target organs for RES actions in vivo (Aggarwal et al., 2004). Our data show that at very high concentrations (50 µM), RES can induce detectable apoptosis in HepG2 cells through up-regulation of the pro-apoptotic Bcl-2 family proteins; but at pharmacologically relevant lower concentrations, RES induces modest growth inhibition by selectively inducing a strong but reversible S-phase delay, yet without causing apoptosis and necrosis. We believe this unique effect of RES may partially contribute to its well-known effect of lifespan prolongation and cancer protection (discussed later). To better understand the mechanisms of RES concentration-dependent effects on S-phase delay and apoptosis, we also analysed the regulation by RES of a number of intracellular signalling molecules that regulate cell cycle progression and/or apoptosis.
Methods
Chemicals and reagents
RES (>99% purity), 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT), paraformaldehyde, N,N-dimethyl formamide, sodium dodecyl sulphate (SDS), propidium iodide (PI), RNase A and 3,3′-dihexyloxacarbocyanine iodide (DiOC6) were obtained from Sigma-Aldrich (St Louis, MO). The antibodies against phospho-ERK, phospho-Raf, phospho-PDK-1, Akt, CDK4/6, cyclin D1, cyclin D3, p15 INK4B, phospho-CDK2, CDK2, cyclin E, cyclin A, p21Waf1/Cip1, phospho-checkpoint kinase 2 (Chk2), caspase-3, caspase-7, PARP, Bax, Bim, Puma and β-actin; the peroxidase-conjugated anti-rabbit or anti-mouse IgGs; and the inhibitors (U0126 and LY294002) were purchased from Cell Signaling Technology (Beverly, MA). The p21–siRNA (catalog no. sc-29427), Chk2–siRNA (catalog no. sc-29271) and siRNA negative control (catalog no. sc-37007) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Lipofectamine 2000 was obtained from Invitrogen (San Diego, CA).
Cell culture
Human HepG2 cells and immortalized non-cancerous murine cell lines (C3H/10T1/2 fibroblasts and 3T3-L1 preadipocytes) were obtained from American Type Culture Collection (Manassas, VA). HepG2 cells and C3H/10T1/2 cells were cultured in the Eagle's minimum essential medium (Sigma) supplemented with 2.2 g·L−1 sodium bicarbonate, 100 mg·L−1 pyruvic acid, 292.2 mg·L−1l-glutamine, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 10% fetal bovine serum (FBS). HT22 murine immortalized hippocampal neuronal cells (a gift from Dr David Schubert, Salk Institute, La Jolla, CA) and 3T3-L1 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 0.11 g·L−1 sodium pyruvate, 1.083 g·L−1l-glutamine, 3.7 g·L−1 sodium bicarbonate, 4.5 g·L−1d-glucose, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 10% FBS. All cells were cultured at 37°C under 5% CO2 in a humidified incubator. During the experiments, all cells were incubated with RES in the complete medium supplemented with 10% FBS.
Assay of cell viability and growth
The effect of RES on HepG2 cell viability was determined by MTT assay. Briefly, the cells were seeded at 1 × 104 cells per well and first cultured in triplicate for 24 h in the presence of different concentrations of RES (12.5–100 µM) in 96-well plates at 37°C. The cells in the control group were treated with vehicle only. MTT (at 5 mg·mL−1) was added 1 h before the termination of the cell culture, and then the cells were lysed with buffer containing 10% SDS and 50% N,N-dimethyl formamide (pH 7.2). The O.D. values were read at a wavelength of 560 nm. When the inhibitors, U0126 (at 2.5 µM) and LY294002 (at 5 µM), were included in the cell cultures, the cells were pretreated with an inhibitor first for 3 h, and then followed by addition of RES.
Small interfering RNA (siRNA) treatment
To study the role of p21 and Chk2 in mediating the effect of RES on cell cycle regulation, p21–siRNA or Chk2–siRNA was used to selectively knock down the expression of p21 or Chk2 in HepG2 cells. The cells were seeded 12 h before transfection and reached a density of 30–50% confluence at the time of transfection. Then, 40 nmol of p21–siRNA, Chk2–siRNA or negative control–siRNA was used for transfection using Lipofectamine 2000 according to the manufacturer's instructions. The transfected cells were maintained in culture for 24 h before harvesting and further analyses. The efficiency of the siRNA knockdown of target protein expression was determined by Western blot analysis with specific antibodies.
Assay of DNA synthesis
HepG2 cells (seeded at 1 × 104 cells per well) were cultured in triplicate for 24 h with different concentrations of RES (12.5–100 µM) in 96-well plates. The cells were pulsed with [methyl-3H]thymidine (2–5 nM, 0.1–1 µCi·per well, PerkinElmer, Boston, MA) for 6 h (or as indicated). The radiolabelled thymidine was added along with fresh replenishing medium (100 µL). The cells were then harvested onto glass fibre filters, and the incorporated radioactivity was counted using a β-scintillation counter (MicroBeta Trilux, PerkinElmer Life Sciences).
Western immunoblotting
HepG2 cells were treated with RES for the indicated length of time. The cells were harvested and suspended in the lysis buffer (20 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, supplemented with a protease inhibitor mix). The lysates were centrifuged at 14 000 rpm for 5 min at 4°C, and the supernatants were boiled in the SDS sample buffer [50 mM Tris–HCl (pH 6.8), 2% SDS, 10% glycerol, 1.2% 2-mercaptoethanol and 0.02% bromphenol blue] for 5 min at 100°C. Equal amounts of proteins (approximately 20–30 µg per lane) were electrophoresed in 10% polyacrylamide gel and then transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). The membranes were treated with 10% non-fat milk for 1 h to block non-specific binding, then rinsed and incubated with various antibodies. The membranes were then treated with 1:2000 dilution of horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG for 1 h. Immune complexes were detected with a chemiluminescence substrate and exposed to an X-ray film (MIDSCI, St Louis, MO).
Flow cytometry analysis of cell cycle
The HepG2 cells were cultured with or without RES for the indicated length of time in six-well plates. The cells were then harvested and washed with phosphate-buffered saline (PBS), fixed in cool 70% ethanol at 4°C overnight. For PI staining, the cells were washed with cold PBS, and incubated with the PI/RNase A solution for 20 min at 37°C. The cells were then analysed on a BD FACS (BD LSR II, BD Biosciences, San Jose, CA).
Detection of the mitochondrial membrane potential
3,3′-Dihexyloxacarbocyanine iodide (DiOC6) is a dye commonly used to measure mitochondrial membrane potential. In brief, cells were treated with RES for the indicated length of time in six-well plates. The cells were then harvested and resuspended in culture medium. DiOC6 (50 nM) was added and incubated with cells for 15 min at 37°C in a humidified incubator. After centrifugation, the cells were suspended in PBS, and fluorescence intensity of DiOC6 was analysed using a flow cytometer.
Assay of DNA fragmentation
HepG2 cells were first cultured with or without RES for the indicated length of time in six-well plates, and were then harvested and washed with PBS. Total DNA was extracted using the DNeasy Mini kit (Qiagen, Valencia, CA). DNA was eluted with TE buffer and visualized by electrophoresis in 1% agarose gel containing ethidium bromide.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay
For TUNEL assays, HepG2 cells were first cultured with or without RES for the indicated length of time in six-well plates, and were then harvested, washed with PBS and fixed in 1% paraformaldehyde. TUNEL staining was performed according to the manufacturer's instructions (Chemicon International, Temecula, CA).
Reproducibility of experiments and statistical analysis
All the data and experiments described in this study were repeated multiple times (over three times for most experiments), and one set of representative data is presented. Some of the quantitative data were presented as mean ± SD as indicated. One-way analysis of variance followed by Student's t-test was used to determine the difference between two groups wherever appropriate. A P value of less than 0.05 was considered statistically significant.
Results
Effect of RES on cell growth, cell cycle progression and apoptosis
Inhibition of cell growth and DNA synthesis
Treatment of cells with 12.5–100 µM of RES for 24 h decreased cell viability (MTT assay) in a concentration-dependent manner (Figure 1A). RES also strongly suppressed DNA synthesis in these cells (based on the cumulative 3H-thymidine incorporation for the last 6 h), with an IC50 value of approximately 25 µM (Figure 1B). In an additional experiment, we further studied the effect of relatively lower concentrations of RES (at 6.25, 12.5 and 25 µM) on the rate of cumulative 3H-thymidine incorporation at different time points (data shown in Figure 1C). In this experiment, 2 nM 3H-thymidine was added into the replenishing medium. The untreated control cells incorporated 3H-thymidine in a linear fashion at earlier time-points (up to 3 h), and then they quickly reached a plateau. In comparison, cells treated with 6.25 µM RES had a reduced rate of 3H-thymidine incorporation at several earlier time-points (1–3 h), whereas at later time-points (6 and 9 h), the cumulative 3H-thymidine incorporation of the RES-treated cells reached the same plateau as the untreated cells. At higher concentrations of RES (12.5 and 25 µM), the slow down in the rate of 3H-thymidine incorporation became more pronounced (Figure 1C). These data suggest that RES slowed down the rate of 3H-thymidine incorporation when it was present at as low as 6.25 µM concentrations.
Figure 1.
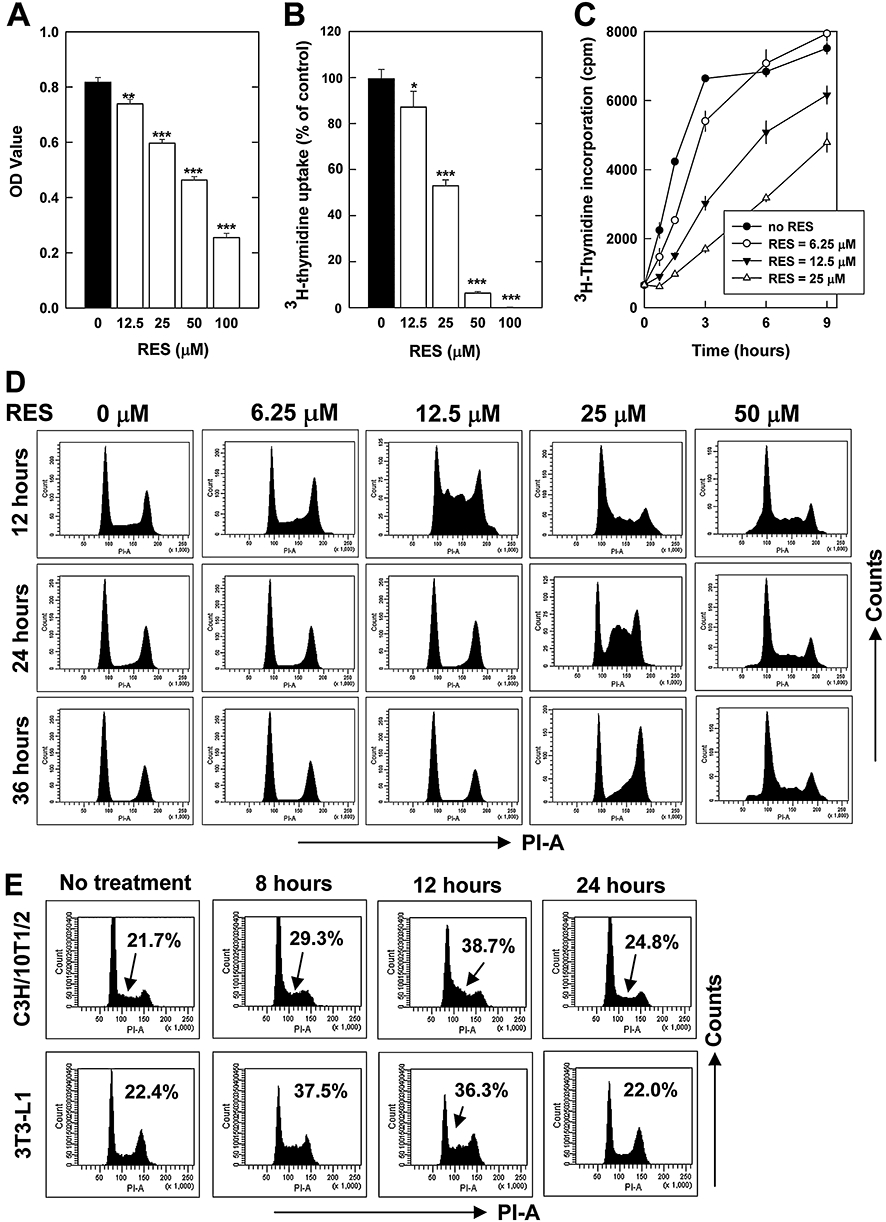
Effect of resveratrol (RES) on cell viability (A), 3H-thymidine incorporation (B, C) and cell cycle distribution in HepG2 cells (D) and in representative non-cancerous immortalized cell lines (E). For determining cell growth and DNA synthesis (A–C), HepG2 cells were treated with RES for 24 h in 96-well plates. Cell viability was determined by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assay, and the DNA synthesis was determined by using 3H-thymidine incorporation assay (described in the Methods section). Each value is mean ± SD of triplicate measurements. *P < 0.05; **P < 0.01; ***P < 0.001 versus respective controls. For determining cell cycle distribution (D, E), HepG2, C3H/10T1/2 and 3T3-L1 cells were treated with RES for varying length of time as indicated, and then they were harvested and stained with propidium iodide, followed by flow cytometry analysis. These analyses were repeated multiple times, and similar observations were made. A representative data set was shown.
Notably, similar concentration-dependent inhibition of cell viability by RES was also observed in MCF-7 and MDA-MB-435s breast cancer cells, as well as in several immortalized non-cancerous murine cell lines (HT22 hippocampal cells, C3H/10T1/2 fibroblasts and 3T3-L1 preadipocytes) (data not shown).
Induction of a reversible, non-cytotoxic S-phase delay
To characterize RES-induced inhibition of cell growth and DNA synthesis, cell cycle distribution was analysed. A significant accumulation of cells at the S-phase of the cell cycle was observed when HepG2 cells were treated with relatively low concentrations (such as 12.5 and 25 µM) of RES (Figure 1D). However, no significant cytotoxicity or cell death (apoptotic or necrotic) was observed in these cells. RES-induced S-phase accumulation had a unique dose–response pattern. At 50 µM, RES only caused a very small increase in S-phase accumulation, and the predominant effect seen at this high concentration was an increase of the sub-G1 fraction, suggesting an increased cell death. Notably, RES at relatively lower concentrations (such as 12.5 and 25 µM) actually induced a more rapid and more pronounced S-phase accumulation compared to higher concentrations of RES (50 or 100 µM). RES-induced accumulation of S-phase cells peaked at 12–24 h after treatment, and afterwards the cells slowly progressed through the cell cycle just as the control cells (only treated with vehicle). This observation suggests that RES only induced a temporary, non-cytotoxic S-phase delay. This unique phenomenon was repeated four times in total and at a wider range of RES concentrations, and highly reproducible results were obtained.
A similar time-dependent induction of S-phase delay was observed in several immortalized non-cancerous murine cell lines (HT22 hippocampal cells, C3H/10T1/2 fibroblasts and 3T3-L1 preadipocytes) when they were treated with 10 µM RES. Some of the data are summarized in Figure 1E.
Induction of apoptosis
At the 50 µM concentration, RES induced cell death as evidenced by an increase in the sub-G1 cell population (Figure 1D) in addition to a strong growth inhibition. To assess the relative contribution of apoptosis to the overall growth inhibition and cell death seen at different concentrations of RES, we studied RES-induced apoptosis. Treatment with RES (at 25 and 50 µM) for 12, 24 and 36 h caused mitochondrial membrane potential loss (DiOC6 staining) in a time- and concentration-dependent manner (Figure 2A). A small increase in TUNEL-positive cells and DNA fragmentation was detected at 24 h after treatment with 50 µM RES, and these changes became more visible at 36 and 48 h, respectively (Figure 2B,C). In comparison, RES at relatively lower concentrations (≤25 µM) did not noticeably increase DNA fragmentation (Figure 2C) or the number of TUNEL-positive cells (data not presented). Taken together, these results suggest that the significant growth inhibition seen at relatively lower concentrations of RES (≤25 µM; Figure 1A,B) was essentially not attributable to RES-induced apoptosis. However, at higher concentrations (50 and 100 µM), RES can also induce apoptosis, and the apoptotic effect of RES adds to the overall growth inhibition under this treatment condition.
Figure 2.
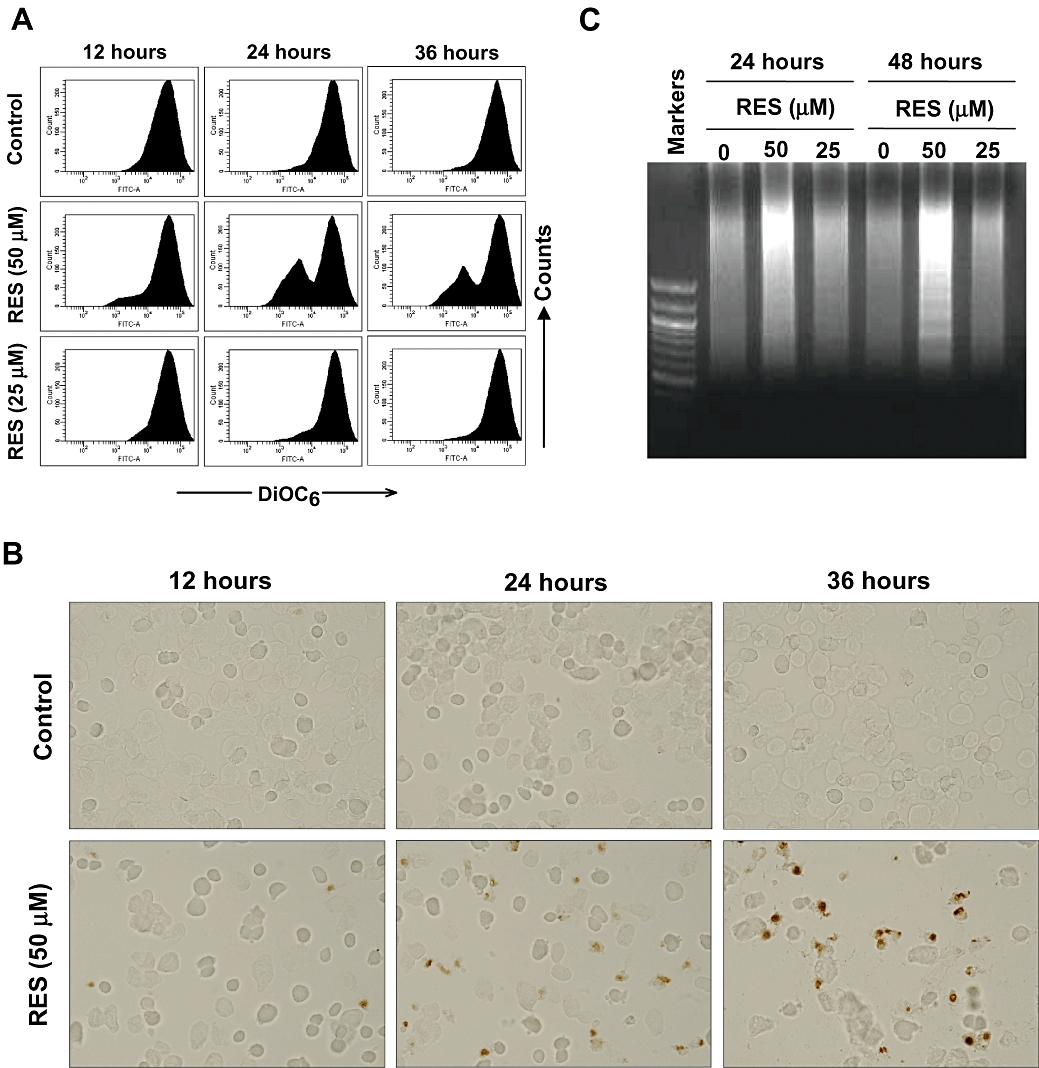
Effect of resveratrol (RES) on mitochondrial membrane potential (A), DNA integrity (B) and DNA fragmentation pattern (C). HepG2 cells were treated with 25 or 50 µM RES for 12, 24 and 36 h, or as indicated. The mitochondrial membrane potential was determined using flow cytometry analysis (DiOC6 staining). DNA integrity was determined using the terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling assay (original magnification, ×40) and DNA fragmentation (formation of DNA ladders).
Effect of RES on signalling pathways that regulate apoptosis and cell cycle events
Cell cycle regulatory proteins
To assess the role of various cell cycle regulatory proteins in mediating RES-induced S-phase delay, we examined their protein levels in HepG2 cells following treatment with RES (at 6.25, 12.5, 25 and 50 µM) for 12 and 24 h. One set of the representative data is shown in Figure 3.
Figure 3.

Effect of resveratrol (RES) on the cell cycle regulatory proteins. HepG2 cells were treated with RES (at 0, 6.25, 12.5, 25 and 50 µM) for 12 and 24 h, then cells were harvested, and the whole cell lysates were analysed by Western immunoblotting. The blots were also probed with anti-β-actin for the confirmation of equal loading of proteins in each lane.
It is known that CDK4/6 and cyclin D1/D3 are mostly involved in regulating the G1 phase of the cell cycle. We found that the protein levels of CDK4 and CDK6 were only marginally reduced when cells were treated with higher concentrations of RES (25 and 50 µM). Cyclin D1 levels were reduced in a concentration-dependent manner, and its reduction was >90% when 50 µM RES was present. In contrast, the level of cyclin D3 was markedly increased at 24 h in a concentration-dependent manner (but not at 12 h). The level of p15INK4B (one of the CDK4 inhibitors) was essentially not altered. Since the most notable change of the G1 cell cycle proteins involves a marked decrease of cyclin D1 levels after treatment with a high concentration (50 µM) of RES, this change may partially contribute to its growth inhibition seen at higher concentrations (≥50 µM).
While CDK2/cyclin E is known to play an important role in driving the cells in G1 phase to enter the S phase (Woo and Poon, 2003; Kaldis and Aleem, 2005), the CDK2-cyclin A is more involved in leading cells in the late S phase to enter the G2 phase. We found that while the total protein levels of CDK2 remained largely unchanged in cells treated with RES for 12 or 24 h, its active form (phospho-CDK2) was increased by treatment with RES. Notably, the increase of phospho-CDK2 was significant after treatment with relatively low concentrations of RES (6.25 and 12.5 µM) for 12 h, and it reached a peak at 25 µM. When the cells were treated with 6.25 µM RES for 24 h, the phospho-CDK2 level was somewhat decreased compared to the control, but its level was still significantly elevated when the cells were treated with 25 µM RES for 24 h. These concentration- and treatment time-dependent changes in the phospho-CDK2 level matched nicely the concentration- and time-dependent accumulation of S-phase cells as shown in Figure 1D. Certainly, this observation is in agreement with the known function of activated CDK2 (phospo-CDK2) in driving cells in late G1 phase into the S-phase. The cyclin E expression was also increased by treatment with 25 or 50 µM of RES for either 12 or 24 h. The levels of cyclin A expression were not significantly changed after treatment with RES for 12 h, but after treatment for 24 h with 12.5 or 25 µM RES, it was increased.
Moreover, we have also determined the levels of p21Waf1/Cip1 (p21) and Chk2 in RES-treated cells. Chk2 is a multifunctional enzyme that is involved in the induction of cell cycle arrest and apoptosis (Ahn et al., 2004). While the total level of Chk2 was only slightly increased by RES, its phosphorylated form (the active form) was increased in a concentration-dependent manner after RES treatment. The increase became very pronounced at higher concentrations. Similarly, the p21 protein levels were significantly increased by treatment with RES at higher concentrations (25 and 50 µM; Figure 3).
Since earlier studies showed that Chk2 plays an important role in the induction of S-phase arrest under certain conditions (Falck et al., 2001), we further probed the role of Chk2 in the induction of S-phase delay by using RNA interference (RNAi). In addition, the role of p21 was also studied for comparison. As shown in Figure 4A, gene silencing with Chk2–siRNA in HepG2 cells partially inhibited RES-induced S-phase delay, whereas silencing of the p21 gene did not show any effect on RES-induced S-phase arrest. Western blot analysis (Figure 4B) confirmed that treatment of HepG2 cells with specific siRNAs decreased the protein levels of Chk2 and phospho-Chk2 by over 80%. Similarly, the level of p21 protein was also decreased by approximately 80%. These data confirmed that Chk2 activation contributes to the induction of S-phase delay.
Figure 4.

Effect of p21 and checkpoint kinase 2 (Chk2) knockdown on resveratrol (RES)-induced S-phase delay. HepG2 cells were transfected with control small interfering RNA (siRNA, p21 siRNA or Chk2 siRNA as described in the Methods section). Twenty-four-hour posttransfection, the transfectants were subsequently treated with 25 or 50 µM of RES for 24 h, and then they were harvested and stained with propidium iodide, followed by flow cytometry analysis. The experiments were repeated twice, and similar observations were made. A representative data set was shown.
Apoptotic proteins
To elucidate the mechanism of RES-induced apoptosis, changes in the levels of representative apoptotic proteins were also determined in HepG2 cells treated with 25 or 50 µM RES for 12, 24 and 36 h (data shown in Figure 5A,B). RES at 50 µM increased the cleaved form of both caspase-7 and PARP, and it slightly decreased the levels of pro-caspase-7 and pro-PARP. Whereas RES also slightly decreased the level of pro-caspase-3, the level of its cleaved form (i.e. the active form) was not significantly changed (data not shown). This experiment was repeated several times, and similar observation was made. In addition, RES also increased the levels of pro-apoptotic members of the Bcl-2 family proteins, including Bax, Bim and Puma, and the increase of Puma was most pronounced (Figure 5).
Figure 5.
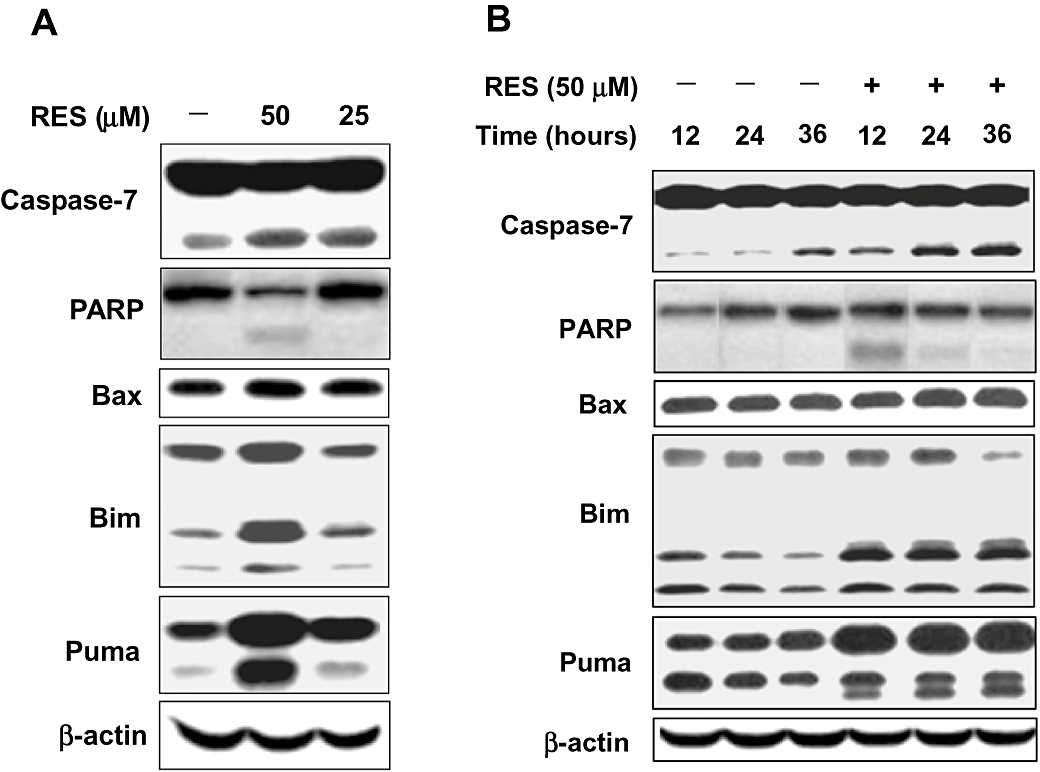
Dose- and time-dependent effect of resveratrol (RES) on the levels of apoptotic proteins. HepG2 cells were treated with 25 or 50 µM RES for 24 h (A) or with 50 µM RES for 12, 24 or 36 h (B), and then the cells were harvested, and the whole cell lysates were analysed using Western immunoblotting. The blots were also probed with anti-β-actin for the confirmation of equal loading of proteins in each lane.
Role of MAPKs and PI3K in mediating RES actions
MAPKs (JNK, p38, and ERK) and PI3K are important early upstream signals that mediate cell survival and growth, and the effect of RES on their regulation was thus also probed as part of our mechanistic study. We found that untreated HepG2 cells had barely detectable levels of JNK and p38, and treatment with 50 µM RES for 24 h did not noticeably affect the levels of the inactive forms of these two proteins (data not shown). Also, the levels of the phospho-JNK and phospho-p38 (their active forms) were not appreciably altered by treatment with 50 µM RES (Figure 6A). In comparison, treatment of HepG2 cells with 50 µM RES for 24 h strongly decreased the levels of phospho-ERK and phospho-PDK-1, although the effect was much weaker when 25 µM RES was present. The changes in the levels of phospho-Raf and Akt in RES-treated cells were rather modest (Figure 6A). The levels of phospho-PTEN (an inhibitory regulator of the PI3K/Akt signalling pathway) were not significantly altered by RES treatment (data not shown).
Figure 6.

Role of mitogen-activated protein kinases (c-Jun NH2-terminal kinase, p38 and extracellular signal-regulated kinase) and phosphatidylinositol 3′-kinase/Akt signalling pathways in resveratrol (RES)-induced S-phase delay and cell death. (A) HepG2 cells were treated with RES (at 0, 25 or 50 µM) for 24 h, then cells were harvested and the whole cell lysates were analysed by Western immunoblotting. The blots were also probed with anti-β-actin for the confirmation of equal loading of proteins in each lane. (B) HepG2 cells were pretreated with 2.5 µM U0126 or 5 µM LY294002 for 3 h, and followed by treatment with 25 or 50 µM RES for additional 24 h. The cell cycle was determined by using flow cytometry.
To further probe the role of ERK and PI3K signalling pathways in RES-induced cell cycle change and apoptosis, we examined the effect of specific inhibitors of ERK (U0126, at 2.5 µM) and PI3K (LY294002, at 5 µM). Note that at the concentrations of U0126 and LY294002 used in this study, it was expected that the activity of ERK and PI3K would be markedly inhibited but probably not completely abolished. The cells were first treated with each of the inhibitors for 3 h, and then they were cultured for additional 24 h in the presence of 25 or 50 µM RES. At 25 µM, RES would induce S-phase arrest without inducing apoptosis, whereas at 50 µM, it would preferentially induce a moderate growth inhibition (along with detectable apoptosis; see Figures 1A,B,D and 2C).
As shown in Figure 6B, when the cells were treated with 25 µM RES + LY294002 (a PI3K inhibitor), there was a noticeable change in the S-phase pattern, with a shift of some cells closer to the G1 phase, suggesting an increase of cells in the early S phase. This increase was relatively small because the cells treated with 25 µM had already developed a pronounced S-phase delay. Interestingly, when LY294002 was combined with 50 µM RES (which did not induce S-phase delay), this phenomenon completely disappeared. This observation suggests that the inhibition of PI3K pathway partially contributed to the induction of S-phase delay in HepG2 cells. In comparison, combined treatment of cells with 25 or 50 µM RES + the ERK inhibitor U0126, the S-phase cell population was actually decreased, and there was a marked increase in the sub-G1 cell population, clearly suggesting that inhibition of ERK did not contribute to the development of S-phase accumulation, but it contributed importantly to increased cell death through apoptosis (more data are provided below).
When 50 µM RES was combined with U0126, there was a marked increase in the sub-G1 population (Figure 6B), along with a significant enhancement of RES-induced growth inhibition (Figure 7A). In addition, RES + U0126 also increased the loss of membrane potential (Figure 6C) and DNA fragmentation in these cells (Figure 6D), suggesting an increase in apoptotic cell death. Consistent with this observation, RES + U0126 increased PARP cleavage and the expression of the pro-apoptotic proteins Bax and Bim, while it also slightly decreased the expression of the anti-apoptotic protein Puma (Figure 7B). Together with the data in Figure 6A showing that RES at 50 µM markedly decreased the levels of phospho-ERK, these data unequivocally show that inhibition of ERK plays a crucial role in mediating RES-induced growth inhibition and apoptosis. Consistent with this observation, a recent study showed that RES at a 100 nM concentration induced growth inhibition in a gastric adenocarcinoma cell line by inactivating MEK1/2-ERK1/2-c-Jun signalling pathway (Aquilano et al., 2009). In comparison, combination of 50 µM RES with the PI3K inhibitor LY294002 did not markedly enhance the apoptotic effect of RES (Figure 6B) or its growth inhibitory effect (Figure 7A). Similarly, the apoptotic proteins were also not appreciably affected by the combination of RES with LY294002 (Figure 7B). These data suggest that PI3K does not play an important role in the development of apoptotic cell death induced by high concentrations of RES.
Figure 7.

Modulation by U0126 and LY294002 of resveratrol (RES)-induced changes in cell growth (A), apoptotic proteins (B), mitochondrial membrane potential (C) and DNA fragmentation pattern (D). HepG2 cells were pretreated with 2.5 µM U0126 or 5 µM LY294002 for 3 h, and followed by treatment with 25 or 50 µM RES for additional 24 h (A–C) or 50 µM RES as indicated (D). Cell growth was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assay; cell cycle and mitochondrial membrane potential were determined using flow cytometry; the levels of apoptotic proteins were determined by using Western immunoblotting; and DNA fragmentation patterns were determined by assaying the DNA ladder formation.
Discussion
Studies in recent years have revealed that RES can exert unique biological actions, resulting in lifespan prolongation and cancer protection. However, the cellular and molecular mechanisms of these unique actions are still not clear despite many studies. Based on the findings described in a number of recent studies concerning the modulatory effect of RES on cell growth and particularly on apoptosis (Ahmad et al., 2001; She et al., 2001; Joe et al., 2002; Kuo et al., 2002; Aquilano et al., 2009), we sought to further investigate the effect of RES on cell cycle progression, in an effort to shed lights on the cellular mechanisms of some of its unique biological actions. Our results showed that the most notable effect of RES at relatively low concentrations (6.25–25 µM) in cultured HepG2 cells is a strong but reversible induction of the S-phase delay along with a mild DNA synthesis inhibition, which is accompanied by a temporary slow down of cell cycle progression, yet without causing cytotoxicity or increasing cell death (apoptosis or necrosis). Notably, the induction of S-phase delay by 12.5 and 25 µM RES actually occurred more rapidly and the effect was more pronounced compared to cells treated with higher concentrations (e.g. 50 µM) of RES. Following a prolonged stay at the S phase, cells treated with RES at 12.5 or 25 µM (but not at 50 µM) slowly progressed through the S phase and then entered the normal cell cycle just as the vehicle-treated control cells. No significant increase in apoptotic and/or necrotic cell death was detected during the process. This observation suggests that RES at relatively lower concentrations appears to selectively slow down the pace of cell cycle progression by prolonging the S-phase stay, yet without increasing cytotoxicity or cell death. Interestingly, this effect was mostly disappeared when higher concentrations of RES were present.
Based on this observation, we believe this effect may represent a rather general mechanism for some of the unique biological actions (such as lifespan prolongation and cancer prevention) of RES as well as other similar compounds. Understandably, when each cell cycle is slowed down at the S phase, it would cumulatively increase the total lifespan of an organism assuming that the total numbers of cell divisions of a given organism remain the same. Similarly, when cells proceed through cell cycles at a slightly reduced pace during the process of DNA replication (which occurs in the S phase), it likely would be beneficial to the cells by allowing them more time to repair the damaged DNA and thereby reduce the chances for mutagenesis and tumorigenesis.
It is known that CDK2 is a catalytic subunit of the cyclin-dependent protein kinase complex, whose activity is essential for cell cycle G1/S-phase transition (Falck et al., 2001; Bartek and Lukas, 2003; Woo and Poon, 2003; Kaldis and Aleem 2005; Tyagi et al., 2005). This protein is associated with and regulated by the regulatory subunits of a complex which includes cyclin E, and the CDK inhibitors p21 and p27. The results of our present study show that RES increases CDK2 activity, and this change helps drive cells to enter the S phase. Consistent with the suggested role of CDK2 in RES-induced S-phase delay, additional studies using RNAi also showed that knockdown of Chk2 expression partially diminished RES-induced S-phase delay.
A number of earlier studies have reported that RES can biochemically inhibit PI3K, consequently resulting in the inactivation of its downstream target PKB (and ultimately glucose utilization) in some of the cancer cell lines (Faber et al., 2006; Sexton et al., 2006; Fröjdöet al., 2007). The results of our present study also suggest that inhibition of PI3K pathway may be an early signalling event partly involved in the induction of RES-induced S-phase delay. This suggestion is based on the following two observations made in the present study. First, moderate changes in the PDK-1 activation, total Akt protein levels and Raf activation were observed in cells treated with RES, which suggest a partial blockade of the PI3K signalling cascade. Second, concomitant treatment of cells with RES + a PI3K inhibitor further increased cell accumulation in the early S-phase. Notably, this enhancing effect of a PI3K inhibitor was only seen when it was combined with 25 µM RES (a concentration suitable for inducing S-phase delay), but the effect was not seen when combined with 50 µM RES (which did not induce S-phase delay). This piece of data with the PI3K inhibitor suggests that PI3K is an early initiator involved in RES-induced S-phase delay.
Interestingly, studies in recent years using yeasts have shown that RES as well as some of its synthetic analogs can activate Sir2 histone deacetylase (Howitz et al., 2003; Yang et al., 2007; Oberdoerffer et al., 2008; Pearson et al., 2008), and this effect has been suggested to be a possible mechanism for increasing yeast replicative lifespan. Similarly, there were also studies showing that RES (at relatively high concentrations, >50 µM) may activate the rodent and human homologs of Sir2 (Borra et al., 2005; Kaeberlein et al., 2005; Mayers et al., 2008). It will be of interest to examine whether SIRT1 activation by RES is also involved in its induction of the non-cytotoxic S-phase delay in mammalian cells as described in this study.
There are many studies in the literature reporting the apoptosis-inducing effect of RES (Ahmad et al., 2001; She et al., 2001; Joe et al., 2002; Kuo et al., 2002). Our data also show that at high concentrations (≥50 µM), RES can induce apoptosis, but interestingly S-phase accumulation was mostly disappeared at these high concentrations. Based on our data, it is estimated that the contribution of RES-induced apoptosis to the overall cell growth inhibition is relatively small because RES is a relatively weak apoptotic agent even at high concentrations. We found that RES-induced apoptosis is preceded by the breakdown of mitochondrial transmembrane potential and DNA fragmentation. The observed increase in the levels of pro-apoptotic Bcl-2 family members, such as Bax, Bim and Puma, may contribute to the depolarization of the mitochondrial membrane and the release of cytochrome c. The latter may further lead to the activation of caspases and ultimately apoptotic cell death. Mechanistically, our results clearly showed that the inhibition of ERK activation by RES is an important upstream event that contributes in a major way to the growth inhibition and apoptosis induced by high concentrations of RES (50 or 100 µM), but it does not play an appreciable role in the development of S-phase arrest. In comparison, while inhibition of PI3K signalling pathway by RES does not appear to play a role in the development of apoptosis, it may be partially involved in the induction of the non-cytotoxic S-phase delay.
Lastly, it is worth noting that the concentrations of RES that were added to the culture medium in this study were relatively high because the cultured HepG2 hepatocytes have a high capacity to rapidly metabolize RES. It was observed in this study that over 50% of RES was disappeared only after 3 h (data not shown). If a more stable level of RES can be maintained in the culture medium, we believe the S-phase delay can be induced by RES at lower concentrations.
In summary, although a number of earlier studies have already reported that RES can induce apoptosis in several cell lines (Ahmad et al., 2001; She et al., 2001; Joe et al., 2002; Kuo et al., 2002), the results of our present study show that the major effect of RES seen at pharmacologically more relevant lower concentrations is the induction of a strong but reversible S-phase delay and a mild DNA synthesis inhibition, yet without the induction of apoptotic or necrotic cell death. We believe the observed effect of RES may represent a novel mechanism that may explain some of the beneficial biological actions of RES, such as the prolongation of lifespan in organisms and protection against early development of cancer. At high concentrations, RES can induce a rather modest level of apoptosis, which is mainly mediated by the mitochondrial pathway. While MEK inhibition and Chk2 activation are important early signalling events responsible RES-induced apoptosis, activation of p-CDK2 and cyclin A is believed to play a role in the induction of a reversible, non-cytotoxic S-phase delay.
Acknowledgments
This study was supported in part by a National Institutes of Health (NIH) grant (ES015242). The real-time PCR set-up and also the image analysis system used in the present study are part of a core facility that is supported, in part, by the NIH Grant P20RR021940 from the National Center for Research Resources.
Glossary
Abbreviations:
- CDK
cyclin-dependent kinase
- Chk
checkpoint kinase
- ERK
extracellular signal-regulated kinase
- MEK
mitogen-activated protein kinase kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide
- PI
propidium iodide
- PI3K
phosphatidylinositol 3′-kinase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling
Conflicts of interest
None.
References
- Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–2840. [PubMed] [Google Scholar]
- Ahmad N, Adhami VM, Afaq F, Feyes DK, Mukhtar H. Resveratrol causes WAF-1/p21-mediated G1-phase arrest of cell cycle and induction of apoptosis in human epidermoid carcinoma A431 cells. Clin Cancer Res. 2001;7:1466–1473. [PubMed] [Google Scholar]
- Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004;3:1039–1047. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Aquilano K, Baldelli S, Rotilio G, Ciriolo MR. trans-Resveratrol inhibits H2O2-induced adenocarcinoma gastric cells proliferation via inactivation of MEK1/2-ERK1/2-c-Jun signalling axis. Biochem Pharmacol. 2009;77:337–347. doi: 10.1016/j.bcp.2008.10.034. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Carbó N, Costelli P, Baccino FM, López-Soriano FJ, Argilés JM. Resveratrol, a natural product present in wine, decreases tumour growth in a rat tumour model. Biochem Biophys Res Commun. 1999;254:739–743. doi: 10.1006/bbrc.1998.9916. [DOI] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Constant J. Alcohol, ischemic heart disease, and the French paradox. Coron Artery Dis. 1997;8:645–649. doi: 10.1097/00019501-199710000-00007. [DOI] [PubMed] [Google Scholar]
- Faber AC, Dufort FJ, Blair D, Wagner D, Roberts MF, Chiles TC. Inhibition of phosphatidylinositol 3-kinase-mediated glucose metabolism coincides with resveratrol-induced cell cycle arrest in human diffuse large B-cell lymphomas. Biochem Pharmacol. 2006;72:1246–1256. doi: 10.1016/j.bcp.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- Ferrieres J. The French paradox: lessons for other countries. Heart. 2004;90:107–111. doi: 10.1136/heart.90.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Fröjdö S, Cozzone D, Vidal H, Pirola L. Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochem J. 2007;406:511–518. doi: 10.1042/BJ20070236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvin S, Ollinger K, Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006;231:113–122. doi: 10.1016/j.canlet.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Horvath Z, Marihart-Fazekas S, Saiko P, Grusch M, Ozsüy M, Harik M, et al. Novel resveratrol derivatives induce apoptosis and cause cell cycle arrest in prostate cancer cell lines. Anticancer Res. 2007;27:3459–3464. [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Jacks T, Weinberg RA. Cell-cycle control and its watchman. Nature. 1996;381:643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Joe AK, Liu H, Suzui M, Vural ME, Xiao D, Weinstein IB. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res. 2002;8:893–903. [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Kaldis P, Aleem E. Cell cycle sibling rivalry: Cdc2 vs. Cdk2. Cell Cycle. 2005;4:1491–1494. doi: 10.4161/cc.4.11.2124. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kopp P. Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur J Endocrinol. 1998;138:619–620. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]
- Kuo PL, Chiang LC, Lin CC. Resveratrol-induced apoptosis is mediated by p53-dependent pathway in Hep G2 cells. Life Sci. 2002;72:23–34. doi: 10.1016/s0024-3205(02)02177-x. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007;17:60–65. doi: 10.1016/j.gde.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Mayers JR, Iliff BW, Swoap SJ. Resveratrol treatment in mice does not elicit the bradycardia and hypothermia associated with calorie restriction. FASEB J. 2008;23:1032–1040. doi: 10.1096/fj.08-115923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Roldán A, Palacios V, Caro I, Pérez L. Resveratrol content of Palomino fino grapes: influence of vintage and fungal infection. J Agric Food Chem. 2003;51:1464–1468. doi: 10.1021/jf020774u. [DOI] [PubMed] [Google Scholar]
- Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23:9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Sexton E, Van Themsche C, Leblanc K, Parent S, Lemoine P, Asselin E. Resveratrol interferes with AKT activity and triggers apoptosis in human uterine cancer cells. Mol Cancer. 2006;5:45. doi: 10.1186/1476-4598-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61:1604–1610. [PubMed] [Google Scholar]
- Sobolev VS, Cole RJ. trans-Resveratrol content in commercial peanuts and peanut products. J Agric Food Chem. 1999;47:1435–1439. doi: 10.1021/jf9809885. [DOI] [PubMed] [Google Scholar]
- Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R. Resveratrol causes Cdc2-tyr15 phosphorylation via ATM/ATR-Chk1/2-Cdc25C pathway as a central mechanism for S phase arrest in human ovarian carcinoma Ovcar-3 cells. Carcinogenesis. 2005;26:1978–1987. doi: 10.1093/carcin/bgi165. [DOI] [PubMed] [Google Scholar]
- Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–324. [PubMed] [Google Scholar]
- Yang H, Baur JA, Chen A, Miller C, Adams JK, Kisielewski A, et al. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell. 2007;6:35–43. doi: 10.1111/j.1474-9726.2006.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HB, Chen JJ, Wang WX, Cai JT, Du Q. Anticancer activity of resveratrol on implanted human primary gastric carcinoma cells in nude mice. World J Gastroenterol. 2005;11:280–284. doi: 10.3748/wjg.v11.i2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]