

Abstract
Background and purpose:
The soluble leptin receptor (SLR) is the major, circulating, leptin-binding protein and, in vitro, the SLR inhibits leptin-binding to cell surface receptors. Here we assessed the effects of the SLR on physiological responses to leptin, in vivo.
Experimental approach:
SLR and leptin were given as a single injection (intracerebroventricularly, i.c.v.) or by central (i.c.v.) and peripheral (s.c.) infusion to normal adult F344XBN rats. Phosphorylation of hypothalamic STAT3 (Western blot), food intake and body weight, and the thermogenic response in brown adipose tissue (BAT) were measured.
Key results:
Acute central co-administration of SLR (13.5 µg) and leptin (90 ng) blocked the threefold increase in hypothalamic STAT3 phosphorylation induced by leptin alone, 1 h after the injections. Peripheral leptin infusion (0.1 mg·day−1 for 7 days; s.c.) induced a significant reduction in food intake and body weight, which were partially blocked with a simultaneous central infusion of SLR (4.3 µg·day−1; i.c.v.). In a second experiment, SLR central infusion alone (5.5 µg·day−1) increased food intake and body weight, suggesting that the SLR was able to neutralize endogenous leptin in the brain. This dose of SLR, infused together with a lower dose of peripheral leptin (0.05 mg·day−1), abolished the thermogenic response in BAT, but the anorexic responses and weight reduction were only partially attenuated.
Conclusions:
These results provide direct evidence that the SLR neutralizes leptin, endogenous or exogenous, in vivo. By neutralizing leptin, the SLR may play a regulatory role in energy homeostasis.
Keywords: leptin signalling, STAT3, UCP1, leptin neutralization, energy homeostasis, soluble receptor
Introduction
Leptin is a key peptide hormone in energy homeostasis. Produced in white adipose tissue (WAT), leptin is secreted into the circulation and transported across the blood brain barrier (BBB) via a saturatable transport system (Banks et al., 1996). Leptin acts within several sites in the brain, including the satiety centre in the hypothalamus, leading to reduced appetite and increased energy expenditure (Friedman and Halaas, 1998). Leptin levels in the circulation are generally in proportion to whole body fat mass (Maffei et al., 1995; Considine et al., 1996).
There are multiple isoforms of leptin receptor as a result of alternative splicing (Lee et al., 1996). The full-length leptin receptor, Ob-Rb, consists of an extracellular domain, a transmembrane domain and a cytoplasmic tail with full signalling capacity. Ob-Rb is most abundantly expressed in the hypothalamus. Ob-Ra, also called the short-form leptin receptor, shares the same sequence with Ob-Rb, except for a truncated cytoplasmic tail, and thus, devoid of signalling capacity (Friedman and Halaas, 1998). Ob-Ra is widely expressed in various tissues, including the choroid plexus, a part of the BBB, and this form of the receptor is proposed to serve as a leptin transporter across the BBB (Hileman et al., 2002). A third isoform, Ob-Re, or the soluble leptin receptor (SLR), is the major leptin-binding protein in the circulation (Gavrilova et al., 1997). Consisting of only the extracellular domain of the full-length receptor, it is generated by ectodomain shedding of membrane-anchored leptin receptors, and in rodents, it can also be generated by alternative splicing (Li et al., 1998; Maamra et al., 2001; Ge et al., 2002). The SLR binds leptin with an affinity similar to that of the full-length Ob-Rb, and thus regulates the bioavailability of leptin (Liu et al., 1997). Studies in Zucker rats indicate that the SLR prolongs the half-life of leptin in the circulation, presumably by protecting it from clearance (Huang et al., 2001). Mice studies using I125-leptin demonstrated leptin transport across the BBB was inhibited by SLR (Tu et al., 2008). Cell culture studies indicate that SLR prevents leptin-binding to its membrane receptor, thus effectively inhibiting leptin-mediated signalling (Yang et al., 2004).
Most soluble forms of receptors act as physiological antagonists by binding the hormone, thus preventing interaction with the native receptor. Leptin is a member of the class I family of cytokine receptors, several of which have soluble forms of their receptors that inhibit ligand action. These include the soluble forms of the growth hormone receptor and various interleukin receptors (Fisker, 2006).
Collectively, the previous studies with SLR and known actions of other soluble forms of receptors predict that the SLR will also act as a negative regulator of leptin's physiological function, although direct evidence of this function by SLR is lacking, in vivo. Therefore, in the present study, we examined whether SLR is able to neutralize leptin-mediated signal transducer and activator of transcription 3 (STAT3) signalling and leptin-induced anorexic responses and elevated energy expenditure in normal, leptin-responsive, rats.
To this end, we used a recombinant murine SLR – Fc chimera to assess if the SLR could block leptin-mediated energy regulation. First, we established the effectiveness of the SLR in blocking central leptin-mediated STAT3 signalling in the hypothalamus. Then, we used two doses of the SLR to counter a peripheral leptin infusion and examined the consequent anorexic responses, weight reduction and thermogenesis in brown adipose tissue (BAT).
Methods
Experimental animals
Animals were cared for, and experimental procedures were, in accordance with the principles of the Guide to the Care and Use of Experimental Animals. Three-month-old male F344 x Brown Norway (F344xBN) rats were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Upon arrival, rats were examined and remained in quarantine for 1 week. Animals were housed individually with a 12:12 h light: dark cycle (07:00 to 19:00 h).
Experimental design
This study comprised three experiments:
Experiment 1
Central injection of SLR and leptin. Rats were given either murine leptin (90 ng), leptin (90 ng) plus SLR (13.5 µg), SLR alone or artificial cerebrospinal fluid (ACSF; see Materials for composition) by intracerebroventricular (i.c.v.) injection into the third ventricle. Rats were killed 1 h later and hypothalamic leptin signalling assessed by STAT3 phosphorylation levels.
Experiment 2
Central infusion of SLR coupled with peripheral infusion of leptin. A cannula was implanted into the lateral ventricle of each animal and connected to a subcutaneous osmotic minipump filled with vehicle (ACSF). Two weeks after full recovery from the surgery, the minipump was replaced with a pump containing either ACSF or SLR (4.3 µg·day−1, lateral ventricle) for an additional 7 days. At the same time as the pump replacement, a second subcutaneous minipump was implanted containing leptin (0.1 mg·day−1) for a simultaneous 7-day peripheral infusion. Rats infused with ACSF (lateral ventricle) and saline (subcutaneous) were included as controls. Food intake and body weight were recorded daily and animals were killed at day 7 for tissue analysis.
Experiment 3
Effects of central SLR and peripheral leptin infusions. Rats were implanted with cannulas and vehicle-filled minipumps as described above. At day 0, the minipumps were replaced with those containing either ACSF or SLR (5.5 µg·day−1, lateral ventricle) for an additional 7 days. At the same time as the pump replacement, a second subcutaneous minipump was implanted containing either saline or leptin (0.05 mg·day−1) for a simultaneous 7-day peripheral infusion. This yielded four groups: saline-ACSF, saline-SLR, leptin-ACSF and leptin-SLR. Food intake and body weight were recorded daily and animals were killed at day 7 for tissue analysis.
Acute leptin and SLR injection
Under anaesthesia (ketamine, 75 mg·kg−1 and xylazine, 7 mg·kg−1), the animal's head was prepared for surgery and the animal placed into a stereotaxic frame. A small incision (1.5 cm) was made over the midline of the skull to expose the landmarks of the cranium (Bregma and Lamda). The coordinates for injection into the third cerebral ventricle are 1.3 mm anterior to Bregma, 9.6 mm ventral from the skull surface, at an angle of 20 degrees anterior to posterior. A small hole was drilled through the skull and a 23 gauge stainless steel guide cannula inserted followed by an injection cannula. Using a 10-µL syringe, a 4-µL volume was delivered over a 5-min period.
Central SLR infusion
The animal was prepared for surgery as previously described (Zhang et al., 2007). A cannula (Durect Corporation, Cupertino, CA) was placed into the lateral ventricle using the following coordinates: 1.3 mm posterior to bregma, 1.9 mm lateral to the mid-sagittal suture, and to a depth of 3.5 mm. The cannula was anchored to the skull using acrylic dental cement. A subcutaneous pocket on the dorsal surface was created by blunt dissection and an osmotic minipump (Alzet model 2001, 1 µL·h−1 for 7 days, Durect Corporation, Cupertino, CA) was inserted. A catheter tube was employed to connect the cannula to the osmotic minipump flow moderator. Animals were infused with control vehicle for 14 days to ensure full recovery from surgery. At day 0, the control minipump was replaced by a new minipump filled with SLR solution or control vehicle.
Subcutaneous leptin infusion
The animal's back was prepared for surgery and a small incision was made over the midline on the dorsal surface. An osmotic minipump (Alzet model 2001) was inserted into a subcutaneous pocket created by blunt dissection.
Body composition measurement
Body composition was determined by time domain-nuclear magnetic resonance (TD-NMR) analyser (Minispec, Bruker Optics, The Woodlands, TX). The MiniSpec measures whole body fat mass on restrained but conscious animals in approximately 2 min. Validation of TD-NMR methodology has been provided (Tinsley et al., 2004).
Tissue harvesting and preparation
Rats were killed by cervical thoracotomy under anaesthesia (5% isoflurane). Blood samples were collected by cardiac puncture and serum was prepared by a 10-min centrifugation in serum separator tubes. The circulatory system was perfused with 30 mL of cold saline. Epididymal white adipose tissues (EWAT), BAT and the hypothalamus were excised. The hypothalamus was removed and sonicated as described previously (Zhang et al., 2007). Protein concentrations were determined using the DC Bradford assay kit (Bio-Rad, Hercules, CA). BAT samples prepared similarly were filtered through a 0.45 µm syringe filter (Whatman, Clifton, NJ) to remove lipid particles prior to protein measurement.
Western blot analysis
Methods for STAT3 and phosphorylated STAT3 assay were described in detail previously (Li et al., 1997). Briefly, protein homogenate (20 µg) was boiled and separated on a Tris-HCl polyacrylamide gel (BioRad, Hercules, CA) and electrotransferred to nitrocellulose membranes (BioRad). Immunoreactivity was assessed with antibodies specific to Tyr 705-phosphorylated STAT3, total STAT3 (Cell Signaling, Danvers, MA) or uncoupling protein 1 (UCP1) (Linco Research, St. Charles, MO). Immunoreactivity was visualized by ECL plus detection system (Amersham, Piscataway, NJ) and quantified by ImageQuant TL (Amersham, Piscataway, NJ).
RNA isolation and relative quantitative RT-PCR
Expression of leptin in EWAT and pre-opiomelanocortin (POMC), neuropeptide Y (NPY) and agouti-related protein (AgRP) in the hypothalamus were measured by relative quantitative RT-PCR using QuantumRNA 18S Internal Standards kit (Ambion, Austin, TX) as described previously (Li et al., 2002). Relative PCR was performed by multiplexing corresponding primers [leptin sense 5′-TGACACCAAAACCCTCATCA-3′, antisense 5′-TGAGCTATCTGCAGCACGTT-3′; primer sequences for POMC, NPY and AgRP were described previously (Li et al., 2005)], 18S primers and competimers and co-amplifying for 23 (leptin), 28 (POMC), 22 (NPY) or 24 (AgRP) cycles. The optimum ratio of 18S primer to competimer was 1:7 for leptin, 1:9 for POMC, 1:6 for NPY and 1:7 for AgRP.
Serum leptin
Serum leptin levels were measured using a rat radioimmunoassay kit (Linco Research).
Statistical analysis
All data are expressed as mean ± standard error of mean and were analysed by one-way or two-way anova. When the main effect was significant, a post hoc test was applied to determine individual differences between means. A value of P < 0.05 was considered significant.
Materials
The SLR used in these experiments is a recombinant chimera of 1-839 aa of murine leptin receptor fused to Fc fragment of human IgG (R&D systems Minneapolis, MN). Leptin was purchased from PeproTech, Rocky Hill, NJ, USA, isoflurane from Webster Veterinary, Sterling, MA, USA, ketamine and xylazine both from Phoenix Pharmaceutical, St. Joseph, Mo, USA. The composition of the ACSF is adapted from that described by Oka et al. (1996) and was (in mmol·L−1): NaCl, 129.91; KCl, 1.96; MgCl2·6H2O, 1.18; CaCl2·2H2O, 1.185; KH2PO4, 1.13; sodium lactate, 1.68; glucose, 3.99; NaHCO3 11; pH 7.4.
Results
Experiment 1
SLR neutralizes leptin signalling in vivo
Hypothalamic leptin signalling was assessed as leptin-induced STAT3 phosphorylation (p-STAT3) in response to leptin at a dose (90 ng, i.c.v.) previously determined to evoke sub-maximal signalling (Scarpace et al., 2001). This dose of leptin alone, in combination with SLR (13.5 µg), SLR alone (13.5 µg) or ACSF control were centrally injected and hypothalamic leptin signalling assessed 1 h later. As expected, leptin increased p-STAT3 by threefold over control rats. This increase in p-STAT3 was fully blocked by co-administration of the SLR. Injection of SLR alone did not alter p-STAT3 levels when compared with the controls (Figure 1).
Figure 1.
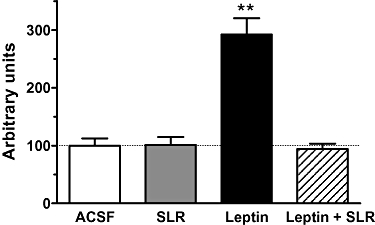
Experiment 1: SLR inhibition of leptin-mediated STAT3 phosphorylation in the hypothalamus 1 h after i.c.v. injection of control (ACSF), SLR (13.5 µg), leptin (90 ng) or leptin (90 ng) + SLR (13.5 µg). Results are expressed in arbitrary units µg−1 hypothalamic protein. STAT3 phosphorylation was normalized to total STAT3 and levels in control rats were set to 100, with SE adjusted proportionally. Values are means ± SE of six animals per group. **P < 0.01 versus control.
Experiment 2
SLR partially blocks leptin-induced anorexic effects
Rats were infused peripherally with leptin (0.1 mg·day−1) via s.c. minipumps for 7 days and simultaneously infused centrally with SLR (4.3 µg·day−1, lateral ventricle) or ACSF. This yielded two groups: leptin and leptin + SLR. A third group, infused with saline (s.c.) and ACSF (i.c.v.) served as the control. Food intake was significantly reduced in the leptin group from day 1, immediately after infusion. In the leptin + SLR group, the initial drop in food intake was similar to the leptin group. Subsequently, the anorexia was generally attenuated throughout the remaining infusion period (Figure 2A).
Figure 2.
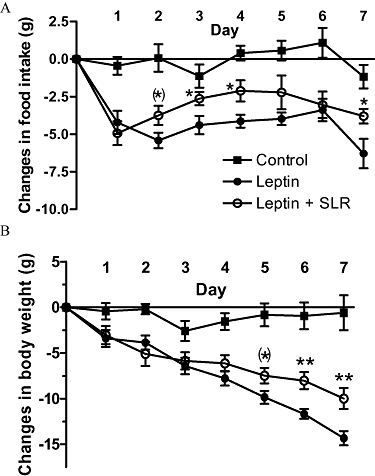
Experiment 2: Change in daily food intake (A) and change in body weight (B) in rats after 7 days of infusion of vehicle, leptin (0.1 mg·day−1, s.c.) or leptin (0.1 mg·day−1, s.c.) + SLR (4.3 µg·day−1, i.c.v.). Changes were calculated from average food intake before treatment (control, 16.7 ± 0.8 g; leptin, 17.7 ± 0.5 g; leptin + SLR, 17.1 ± 0.5 g) or from body weight before surgery (control, 309.2 ± 10.8 g; leptin, 312.0 ± 8.5 g; leptin + SLR, 308.3 ± 6.2 g). Values are mean ± SE of seven animals per group. (*) P < 0.05 versus leptin by one-tailed t-test; *P < 0.05, **P < 0.01 versus leptin by two-tailed t-test.
Moreover, the leptin group consumed 27.4 ± 2.7 g less food in total from day 2 to day 7 when compared with its own pre-infusion baseline. In comparison, the leptin + SLR group only consumed 17.5 ± 3.3 g less than its pre-infusion baseline, and this is significantly less than the leptin group (P < 0.05). In contrast, the control group consumed nearly the same amount of food (−0.2 ± 2.9 g less than pre-infusion consumption).
Body weight reduction was significant in both leptin and leptin + SLR groups starting from day 2. The slope of body weight reduction in leptin + SLR group was not as steep as the leptin group (leptin, −1.85 ± 0.11; leptin + SLR, −1.01 ± 0.10, P < 0.001) and decrease in body weight significantly diverged, beginning at day 5 continuing through day 7, in the leptin + SLR compared with the leptin group (Figure 2B).
Changes in whole body fat mass
Body composition was measured and the changes in whole body fat mass were compared after the 7-day leptin, leptin plus SLR, or control infusions (Table 1). The control group showed no significant change, while the leptin infusion reduced fat mass by nearly 10 g. Infusion with leptin plus SLR partially blocked this decrease (Table 1, upper half).
Table 1.
Energy homeostasis-related parameters following SLR central infusion
|
Experiment 2: Low dose SLR infusion (4.3 µg·day−1) |
||||
|---|---|---|---|---|
| Control | Leptin | Leptin + SLR | ||
| Change in whole body fat mass (g) | 2.8 ± 1.3 | −9.4 ± 0.7** | −5.1 ± 1.6**† | |
| Serum leptin, day 7 (ng·mL−1) | 3.73 ± 0.23 | 13.49 ± 0.90** | 11.84 ± 1.42** | |
|
Experiment 3: Higher dose SLR infusion (5.5 µg·day−1) |
||||
| Control | SLR | Leptin | Leptin + SLR | |
| Change in whole body fat mass (g) | 3.4 ± 2.0 | 7.0 ± 1.6 | −2.6 ± 1.0* | 2.7 ± 1.7† |
| Serum leptin, day 7 (ng·mL−1) | 2.53 ± 0.32 | 2.66 ± 0.31 | 4.89 ± 0.44** | 6.81 ± 0.73**† |
Values are means ± SE of 6–7 animals in each group.
P < 0.05
P < 0.01 versus control,
P < 0.05 versus leptin alone.
Leptin-induced BAT UCP1 is partially blocked by SLR
At death, UCP1 protein levels in BAT were assessed. Consistent with our previous finding (Scarpace et al., 1997), leptin infusion elevated UCP1 level in BAT by almost 40%, whereas simultaneous central SLR infusion partially prevented the increase in UCP1 protein (Figure 3).
Figure 3.
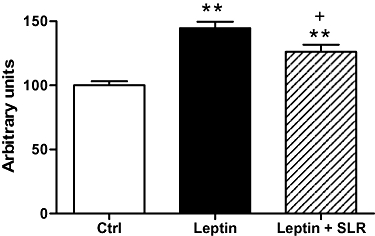
Experiment 2: UCP1 protein levels in BAT after 7 days of infusion of vehicle, leptin (0.1 mg·day−1, s.c.) or leptin (0.1 mg·day−1, s.c.) + SLR (4.3 µg·day−1, i.c.v.). Results are expressed in arbitrary units·µg−1 BAT protein and levels in control rats were set to 100, with SE adjusted proportionally. Values are means ± SE of 6–7 animals per group. **P < 0.01 versus control, †P < 0.05 versus leptin.
Serum leptin levels
Serum leptin was determined in seven randomly selected rats prior to any infusion, and the average serum leptin level was found to be 3.38 ± 0.18 ng·mL−1. At day 7, blood was collected from all the rats at death. Serum leptin in control rats was unchanged at day 7, whereas following leptin infusion, serum leptin was elevated by fourfold. Similarly, in rats infused with leptin plus SLR, there was a comparable increase in serum leptin (Table 1, upper half).
Leptin signalling markers at the end of 7-day infusion
At the end of the 7-day infusion, hypothalamic leptin signalling markers, including p-STAT3 and expression levels of POMC, NPY and AgRP were measured. Despite the significant physiological responses to the leptin infusion and partial blockade of these responses by SLR, no difference in STAT3 phosphorylation was detected among the three groups (Table 2). Similarly, expression levels of neuropeptides downstream of the leptin receptor – STAT3 signalling pathway were also unchanged (Table 2).
Table 2.
Experiment 2: Central leptin signalling parameters at day 7 following SLR central infusion (4.3 µg·day−1)
| Control | Leptin | Leptin + SLR | |
|---|---|---|---|
| p-STAT3 | 100.0 ± 8.7 | 97.5 ± 7.0 | 100.0 ± 7.9 |
| POMC expression | 100.0 ± 7.4 | 91.6 ± 6.2 | 99.13 ± 5.6 |
| NPY expression | 100.0 ± 2.5 | 97.0 ± 2.5 | 101.6 ± 3.9 |
| AgRP expression | 100.0 ± 2.6 | 100.0 ± 2.7 | 99.0 ± 3.1 |
Values are mean ± SE of 6–7 animals in each group, in arbitrary units. p-STAT3, phosphorylated signal transducer and activator of transcription 3; POMC, pre-opiomelanocortin; NPY, neuropeptide Y; AgRP, agouti-related protein. P > 0.05 among all groups.
Experiment 3
Higher dose of SLR infusion
In order to raise the ratio of SLR to exogenously infused leptin, we increased the dose of central SLR infusion to 5.5 µg·day−1 and coupled this with a lower dose of peripheral leptin (0.05 mg·day−1) infusion. This leptin dose was determined based on the dose-response study described previously (Judge et al., 2008), in which this dose induced significant anorexic and weight reducing effects. In addition, in this experiment, a fourth group was included, treatment with SLR alone.
When the SLR was infused centrally by itself, both food intake and especially, body weight were increased over the controls (Figure 4), suggesting the SLR blocked the action of endogenous leptin in the brain. Leptin infusion, at a dose lower than that used in experiment 2, significantly reduced food intake and body weight as expected. However, despite the higher SLR dose employed, SLR infusion still only partially blocked these leptin-induced responses (Figure 4).
Figure 4.
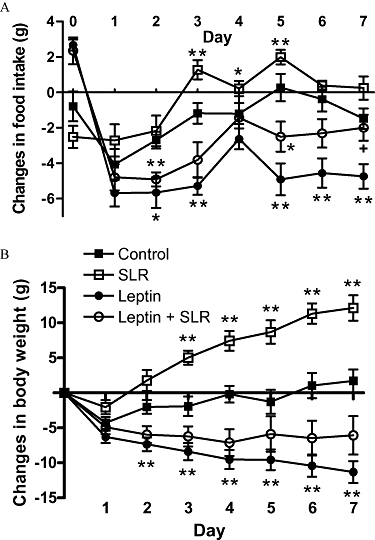
Experiment 3: Change in daily food intake (A) and change in body weight (B) in rats after 7 days of infusion of vehicle, leptin (0.05 mg·day−1, s.c.), SLR (5.5 µg·day−1, i.c.v.) or leptin (0.05 mg·day−1, s.c.) + SLR (5.5 µg·day−1, i.c.v.). Changes were calculated from average food intake before treatment (control, 17.9 ± 0.5 g; SLR, 18.9 ± 0.7 g; leptin, 18.2 ± 0.5 g; leptin + SLR, 17.4 ± 0.3 g) or from body weight before surgery (control, 306.3 ± 8.2 g; SLR, 299.3 ± 11.8 g; leptin, 304.1 ± 6.9 g; leptin + SLR, 302.2 ± 7.2 g). Values are mean ± SE of seven animals per group. *P < 0.05, **P < 0.01 versus control; †P < 0.05 versus leptin.
Whole body fat mass
Parallel to the body weight results, rats centrally infused with the higher dose of SLR demonstrated no significant increase in whole body fat mass over the controls (P > 0.05). Peripheral infusion of leptin alone significantly reduced fat mass, while this decrease was completely prevented by SLR infusion in the leptin + SLR group (P < 0.05 vs. leptin; Table 1, lower half).
UCP1 elevation in BAT is fully blocked by SLR central infusion
Despite the changes in body weight and food intake, central infusion of SLR alone (5.5 µg·day−1) did not change the basal UCP1 levels in the BAT. Peripheral leptin infusion increased UCP1 by almost 20%, which was completely prevented by SLR infusion (P < 0.05 vs. leptin, P > 0.05 vs. control; Figure 5A).
Figure 5.
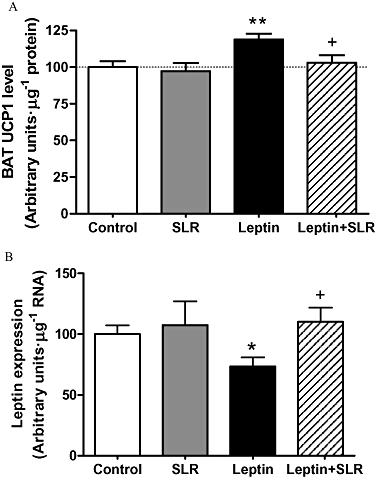
Experiment 3: UCP1 protein levels in BAT (A) and leptin expression levels in EWAT (B) after 7 days of infusion of vehicle, leptin (0.05 mg·day−1, s.c.), SLR (5.5 µg·day−1, i.c.v.) or leptin (0.05 mg·day−1, s.c.) + SLR (5.5 µg·day−1, i.c.v.). Results are expressed in arbitrary units·µg−1 of protein or total RNA and levels in control rats were set to 100, with SE adjusted proportionally. Values are means ± SE of 6–7 animals per group. *P < 0.05, **P < 0.01 versus control; †P < 0.05 versus leptin alone.
Inhibition of leptin expression in EWAT is fully blocked by SLR
Leptin expression levels in EWAT were measured at the end of the infusions. The SLR infusion alone did not change leptin expression compared with the controls. Consistent with previous data (Scarpace et al., 1998), leptin infusion inhibited leptin expression in the EWAT by 25%. Moreover, this inhibition was completely prevented by the simultaneous infusion with leptin plus SLR (Figure 5B).
Serum leptin levels
Serum levels were sampled from seven randomly selected rats from all animals before the infusion and the average was 3.23 ± 0.30 ng·mL−1. At day 7, leptin levels in rats infused with SLR alone were almost identical to the controls (Table 1, lower half), whereas in leptin-infused rats, serum leptin was almost twofold greater than control level (P < 0.01 vs. control). Interestingly, the leptin + SLR group had the highest serum leptin, 40% higher than the leptin group (P < 0.05 vs. leptin; Table 1, lower half).
Leptin signalling markers at the end of the 7-day infusion
As observed in the first set of infusions in experiment 2, there were no significant changes in STAT3 phosphorylation among the groups (control 100.0 ± 4.14, SLR 129.8 ± 29.8, leptin 106.8 ± 7.9, leptin + SLR 120.3 ± 14.5).
Discussion
Soluble receptors in various ligand-receptor systems have an important role in the regulation of ligand availability and subsequent receptor-mediated physiology (Heaney and Golde, 1993; Baumann, 2002; Rose-John et al., 2006). The SLR is the major leptin-binding protein in the circulation. It was previously demonstrated that the SLR inhibits leptin-induced signaling in vitro (Yang et al., 2004). In addition to this indirect antagonism of the leptin receptor, other studies have demonstrated potential roles for the SLR in leptin pharmacokinetics. The SLR prolongs the half-life of leptin in blood in Zucker rats, presumably by binding and thus protecting leptin from clearance (Huang et al., 2001). In addition, a study in mice demonstrated SLR inhibits leptin transport across the BBB, thus limiting the access of leptin to its receptors in the brain (Tu et al., 2008).
Leptin is an important peptide hormone in energy regulation. This is readily apparent in rodents with genetic mutations lacking leptin or with defective leptin receptors (Halaas et al., 1995). Thus, factors, such as the SLR, that have the potential to limit the access of leptin to its receptor may play an important role in energy homeostasis. However, the direct demonstration of the impact of the SLR on energy balance (food intake or energy expenditure) in normal animals is lacking. In this report, we used an available SLR, a chimera of murine SLR and Fc of human IgG, to examine the impact of SLR on leptin physiology in F344xBN rats.
First, the SLR could neutralize leptin signalling, in vivo, as the SLR inhibited leptin-mediated STAT3 signalling in the hypothalamus. Leptin-induced STAT3 phosphorylation was completely prevented by the simultaneous administration of SLR without pre-incubation, confirming the antagonistic features of SLR, in vivo. The rapid nature of this antagonism was consistent with the known high affinity binding between SLR and leptin (Liu et al., 1997).
Moreover, by co-infusion of leptin and SLR, we demonstrated that the SLR was able to counteract leptin's regulation of energy balance. Due to the high binding affinity, the SLR and leptin were infused by separate routes, avoiding formation of any leptin : SLR complexes prior to the individual compounds reaching the physiologically relevant regions of the brain. Leptin was infused peripherally, to mimic the elevation of serum leptin in physiological conditions, whereas the SLR was infused centrally in order to most efficiently block leptin action. At a dose of 4.3 µg·day−1, SLR infusion partially blocked leptin-induced anorexia and weight reduction. Although whole body energy expenditure was not measured, levels of the UCP1 protein, a reasonable marker for BAT thermogenesis, was elevated by leptin infusion. This increase was also partially inhibited by the SLR co-infusion. Collectively, these data indicate that the SLR was able to prevent leptin, at least partially, from acting in the CNS to regulate energy balance. The nature of the partial inhibition may simply be a matter of competition, i.e. there was insufficient SLR to fully bind the available leptin.
In an attempt to achieve a full blockade of leptin action, we increased the SLR infusion dose to 5.5 µg·day−1 while decreasing the dose of the peripherally infused leptin (0.05 mg·day−1). In addition, we included a group that received only central infusion of the SLR. Interestingly, the SLR alone increased food consumption and body weight significantly over the control, presumably by neutralizing endogenous CSF leptin. Moreover, the increase in body weight with this amount of SLR infusion paralleled the increase in body weight following full leptin receptor blockade with a leptin receptor antagonist (Zhang et al., 2007). In our earlier study, we determined the dose of a leptin receptor antagonist that achieved full leptin receptor blockade, and found that this dose increased body weight by 12.71 ± 10.36 g over a 7-day treatment period (Zhang et al., 2007). This increase in body weight is almost identical to that observed with the SLR in the present study and both studies used rats of same strain, age and body weight. Collectively, these data indicate the SLR effectively neutralizes endogenous central leptin-mediated actions.
However, when infusion of this higher SLR dose was coupled with the lower dose of leptin, this combination still resulted in only a partial blockade of leptin-induced anorexia and weight reduction. In contrast, the leptin-induced increase of UCP1 in BAT was completely prevented by the SLR. Similarly, the infusion of central SLR fully reversed the inhibition by exogenous leptin, of endogenous leptin expression in the EWAT. This leptin inhibition of adipose leptin expression is consistent with our previous finding that leptin inhibits leptin expression in WAT through a central mechanism (Shek and Scarpace, 2000). Complete prevention of these two markers of leptin action indicates that our higher dose of SLR coupled with the lower dose of leptin was able to achieve a more complete antagonism of leptin, although some anorexia and weight reduction remained.
There are several potential explanations for the incomplete blockade of responses to exogenous leptin. First, and most likely, is that neither dose of the SLR was sufficient to fully bind all the available leptin. Use of higher doses was impractical due to cost. Second, the separate routes of delivery may have allowed a temporal dissociation between the leptin and SLR reaching the CNS. The leptin-containing, subcutaneously implanted minipumps infused leptin directly into the subcutaneous tissues, where it was available for rapid uptake into the systemic circulation and hence into the brain. In contrast, each SLR-containing, subcutaneously implanted minipump was connected to a ventricular cannula via a 7.5 cm long piece of tubing. We calculated that that the SLR may require 28 h to traverse this tubing before reaching the lateral ventricle and it is likely that the leptin reached the CNS and activated leptin receptors before the first of the SLR made an appearance. This would allow an initial burst of leptin receptor activity that was unchallenged, and thus accounting for the partial blockade of leptin action. Third, and most intriguing is the possibility that the response due to central leptin action was fully blocked and that the residual activity was due to peripheral leptin action. This suggests a role for peripheral leptin action in the energy balance, although most studies, so far, have failed to identify a peripheral component of leptin action separate from centrally mediated leptin responses. Further experiments would be necessary to resolve this issue.
Despite the clear differences in physiological responses between the leptin and leptin + SLR groups, we were unable to relate these differential responses to an inhibition of leptin signalling at the end of the infusions. This was mainly because the peripheral leptin infusion failed to elevate hypothalamic leptin signalling on day 7 despite a significant reduction in food consumption and high serum leptin levels. In contrast, when a pharmacological dose of leptin was injected centrally, leptin signalling was significantly increased after 1 h and was fully inhibited by co-administration of the SLR. Similarly, when leptin is administered peripherally by intravenous injection, the increase in STAT3 phosphorylation is observed within 30 min, is maximal by 1 h and returns to basal level by 14 h post injection (Scarpace et al., 2000). These data suggest that either timing or dosing is responsible for the lack of an increase in STAT3 phosphorylation with a peripheral leptin infusion. For instance, the amount of leptin that reaches the brain after peripheral infusion may not be enough to activate leptin signalling, at least to a level detectable by our method of examining whole hypothalamic STAT3 phosphorylation, or the signalling is transient and has already returned to basal level by day 7. Nevertheless, it is apparent that the SLR can prevent leptin signalling due to an acute injection and infused leptin from exerting its full effect on food intake, body weight and BAT thermogenesis.
Obese animals and humans have elevated leptin whereas circulating SLR levels remain comparable to their lean counterparts (Wu et al., 2002). The inability to up-regulate SLR with the development of obesity results in an excessively high level of free leptin in the circulation. Whether the elevated leptin is simply secondary to the obesity or a causative factor in pathogenesis of obesity is still a matter for debate (Scarpace and Zhang, 2007). In dietary obese rats, leptin treatment exacerbates rather than inhibits further high-fat induced obesity (Scarpace et al., 2005). Elevated leptin in humans, independent of obesity, is a predictor of metabolic syndrome after 5 and 10 years (Franks et al., 2005). It is not clear if normalization of the elevated leptin associated with obesity is desirable and this manuscript provides no evidence that neutralization of leptin is beneficial. The present report indicates that binding of leptin to its soluble receptor prevents activation of cellular receptors and the subsequent physiological responses, thus demonstrating the feasibility of leptin normalization. The SLR, in addition to other agents, such as leptin synthesis blockers or leptin receptor antagonists, are potential approaches to reduce the consequences, if any, of hyperleptinemia.
In summary, the present report describes the physiological responses to the central infusion of the SLR. This isoform of the leptin receptor was able to block hypothalamic leptin signalling induced by an acute central injection of leptin. In addition, central infusion of the SLR significantly increased food intake and body weight effectively neutralizing endogenous central leptin. When infused centrally in conjunction with a peripheral leptin infusion, the SLR partially blocked leptin-induced anorexic responses and body weight reduction, and fully prevented the elevation in BAT UCP1 levels as well as the inhibition of leptin expression in EWAT. These data imply that the SLR is able to neutralize leptin action in vivo, suggesting the potential of a regulatory role in energy homeostasis.
Acknowledgments
This work was supported by National Institute on Aging grant AG-26159.
Glossary
Abbreviations:
- ACSF
artificial cerebrospinal fluid
- AgRP
agouti-related protein
- BBB
blood brain barrier
- i.c.v.
intracerebroventricular
- NPY
neuropeptide Y
- POMC
pre-opiomelanocortin
- SLR
soluble leptin receptor
- STAT3
signal transducer and activator of transcription 3
- UCP1
uncoupling protein 1
Conflicts of interest
None.
References
- Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305–311. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- Baumann G. Growth hormone binding protein. The soluble growth hormone receptor. Minerva Endocrinol. 2002;27:265–276. [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Fisker S. Physiology and pathophysiology of growth hormone-binding protein: methodological and clinical aspects. Growth Horm IGF Res. 2006;16:1–28. doi: 10.1016/j.ghir.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Franks PW, Brage S, Luan J, Ekelund U, Rahman M, Farooqi IS, et al. Leptin predicts a worsening of the features of the metabolic syndrome independently of obesity. Obes Res. 2005;13:1476–1484. doi: 10.1038/oby.2005.178. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Gavrilova O, Barr V, Marcus-Samuels B, Reitman M. Hyperleptinemia of pregnancy associated with the appearance of a circulating form of the leptin receptor. J Biol Chem. 1997;272:30546–30551. doi: 10.1074/jbc.272.48.30546. [DOI] [PubMed] [Google Scholar]
- Ge H, Huang L, Pourbahrami T, Li C. Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. J Biol Chem. 2002;277:45898–45903. doi: 10.1074/jbc.M205825200. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Heaney ML, Golde DW. Soluble hormone receptors. Blood. 1993;82:1945–1948. [PubMed] [Google Scholar]
- Hileman SM, Pierroz DD, Masuzaki H, Bjorbaek C, El-Haschimi K, Banks WA, et al. Characterizaton of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology. 2002;143:775–783. doi: 10.1210/endo.143.3.8669. [DOI] [PubMed] [Google Scholar]
- Huang L, Wang Z, Li C. Modulation of circulating leptin levels by its soluble receptor. J Biol Chem. 2001;276:6343–6349. doi: 10.1074/jbc.M009795200. [DOI] [PubMed] [Google Scholar]
- Judge MK, Zhang J, Tumer N, Carter C, Daniels MJ, Scarpace PJ. Prolonged hyperphagia with high-fat feeding contributes to exacerbated weight gain in rats with adult-onset obesity. Am J Physiol Regul Integr Comp Physiol. 2008;295:R773–R780. doi: 10.1152/ajpregu.00727.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- Li C, Ioffe E, Fidahusein N, Connolly E, Friedman JM. Absence of soluble leptin receptor in plasma from dbPas/dbPas and other db/db mice. J Biol Chem. 1998;273:10078–10082. doi: 10.1074/jbc.273.16.10078. [DOI] [PubMed] [Google Scholar]
- Li G, Klein RL, Matheny M, King MA, Meyer EM, Scarpace PJ. Induction of uncoupling protein 1 by central interleukin-6 gene delivery is dependent on sympathetic innervation of brown adipose tissue and underlies one mechanism of body weight reduction in rats. Neuroscience. 2002;115:879–889. doi: 10.1016/s0306-4522(02)00447-5. [DOI] [PubMed] [Google Scholar]
- Li G, Zhang Y, Wilsey JT, Scarpace PJ. Hypothalamic pro-opiomelanocortin gene delivery ameliorates obesity and glucose intolerance in aged rats. Diabetologia. 2005;48:2376–2385. doi: 10.1007/s00125-005-1943-8. [DOI] [PubMed] [Google Scholar]
- Li H, Matheny M, Nicolson M, Tumer N, Scarpace PJ. ). Leptin gene expression increases with age independent of increasing adiposity in rats. Diabetes. 1997;46:2035–2039. doi: 10.2337/diab.46.12.2035. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu XJ, Barry G, Ling N, Maki RA, De Souza EB. Expression and characterization of a putative high affinity human soluble leptin receptor. Endocrinology. 1997;138:3548–3554. doi: 10.1210/endo.138.8.5343. [DOI] [PubMed] [Google Scholar]
- Maamra M, Bidlingmaier M, Postel-Vinay MC, Wu Z, Strasburger CJ, Ross RJ. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology. 2001;142:4389–4393. doi: 10.1210/endo.142.10.8442. [DOI] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- Oka K, Yamamoto M, Nonaka T, Tomonaga M. The significance of artificial cerebrospinal fluid as perfusate and endoneurosurgery. Neurosurgery. 1996;38:733–736. [PubMed] [Google Scholar]
- Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Zhang Y. Elevated leptin: consequence or cause of obesity? Front Biosci. 2007;12:3531–3544. doi: 10.2741/2332. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Pollock BH, Tumer N. Leptin increases uncoupling protein expression and energy expenditure. Am J Physiol. 1997;273:E226–E230. doi: 10.1152/ajpendo.1997.273.1.E226. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Nicolson M, Matheny M. UCP2, UCP3 and leptin gene expression: modulation by food restriction and leptin. J Endocrinol. 1998;159:349–357. doi: 10.1677/joe.0.1590349. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Shek EW. Impaired leptin signal transduction with age-related obesity. Neuropharmacology. 2000;39:1872–1879. doi: 10.1016/s0028-3908(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104:1111–1117. doi: 10.1016/s0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N, Cheng KY, Zhang Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia. 2005;48:1075–1083. doi: 10.1007/s00125-005-1763-x. [DOI] [PubMed] [Google Scholar]
- Shek EW, Scarpace PJ. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regul Pept. 2000;92:65–71. doi: 10.1016/s0167-0115(00)00151-8. [DOI] [PubMed] [Google Scholar]
- Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res. 2004;12:150–160. doi: 10.1038/oby.2004.20. [DOI] [PubMed] [Google Scholar]
- Tu H, Kastin AJ, Hsuchou H, Pan W. Soluble receptor inhibits leptin transport. J Cell Physiol. 2008;214:301–305. doi: 10.1002/jcp.21195. [DOI] [PubMed] [Google Scholar]
- Wu Z, Bidlingmaier M, Liu C, De Souza EB, Tschop M, Morrison KM, et al. Quantification of the soluble leptin receptor in human blood by ligand-mediated immunofunctional assay. J Clin Endocrinol Metab. 2002;87:2931–2939. doi: 10.1210/jcem.87.6.8610. [DOI] [PubMed] [Google Scholar]
- Yang G, Ge H, Boucher A, Yu X, Li C. Modulation of direct leptin signaling by soluble leptin receptor. Mol Endocrinol. 2004;18:1354–1362. doi: 10.1210/me.2004-0027. [DOI] [PubMed] [Google Scholar]
- Zhang J, Matheny MK, Tumer N, Mitchell MK, Scarpace PJ. Leptin antagonist reveals that the normalization of caloric intake and the thermic effect of food after high-fat feeding are leptin dependent. Am J Physiol Regul Integr Comp Physiol. 2007;292:R868–R874. doi: 10.1152/ajpregu.00213.2006. [DOI] [PubMed] [Google Scholar]