

Abstract
Background and purpose:
The chronic use of organic nitrates is limited by serious side effects including oxidative stress, nitrate tolerance and/or endothelial dysfunction. The side effects and potency of nitroglycerine depend on mitochondrial aldehyde dehydrogenase (ALDH-2). We sought to determine whether this concept can be extended to a new class of organic nitrates with amino moieties (aminoalkyl nitrates).
Experimental approach:
Vasodilator potency of the organic nitrates, in vitro tolerance and in vivo tolerance (after continuous infusion for 3 days) were assessed in wild-type and ALDH-2 knockout mice by isometric tension studies. Mitochondrial oxidative stress was analysed by L-012-dependent chemiluminescence and protein tyrosine nitration.
Key results:
Aminoethyl nitrate (AEN) showed an almost similar potency to glyceryl trinitrate (GTN), even though it is only a mononitrate. AEN-dependent vasodilatation was mediated by cGMP and nitric oxide. In contrast to triethanolamine trinitrate (TEAN) and GTN, AEN bioactivation did not depend on ALDH-2 and caused no in vitro tolerance. In vivo treatment with TEAN and GTN, but not with AEN, induced cross-tolerance to acetylcholine (ACh)-dependent and GTN-dependent relaxation. Although all nitrates tested induced tolerance to themselves, only TEAN and GTN significantly increased mitochondrial oxidative stress in vitro and in vivo.
Conclusions and implications:
The present results demonstrate that not all high potency nitrates are bioactivated by ALDH-2 and that high potency of a given nitrate is not necessarily associated with induction of oxidative stress or nitrate tolerance. Obviously, there are distinct pathways for bioactivation of organic nitrates, which for AEN may involve xanthine oxidoreductase rather than P450 enzymes.
Keywords: organic nitrates, bioactivation, mitochondrial aldehyde dehydrogenase, mitochondrial oxidative stress, vascular function
Introduction
A central constituent of the regulatory instruments of the vascular system is the synthesis of the endothelium-derived relaxing factor, nitric oxide (NO), which is a potent vasodilator and also represents an anti-aggregatory signal (Arnold et al., 1977; Palmer et al., 1987; Radomski et al., 1987). Endothelial dysfunction has been identified as a hall-mark of most cardiovascular diseases (Cai and Harrison, 2000) and is always associated with vascular oxidative stress, decreased NO bioavailability and/or impaired activity (uncoupling) of endothelial NO synthase (Munzel et al., 2005b; Forstermann and Munzel, 2006). Vice versa, it was demonstrated that patients with an impaired endothelial function have a higher risk of cardiovascular events (Schachinger et al., 2000; Heitzer et al., 2001). Endothelial dysfunction of coronary arteries is associated with an increased risk of myocardial infarction (Schachinger et al., 2000) because dysfunctional vessels are prone to atherosclerosis and thrombus formation (Wennmalm, 1994).
Therefore, NO substitution seemed to be an attractive pharmacological strategy to overcome endothelial (vascular) dysfunction in the setting of cardiovascular diseases (Anderson et al., 1994; Abrams, 1996). Organic nitrates [such as nitroglycerine/glyceryl trinitrate (GTN) and isosorbide-5-mononitrate (ISMN)] are the most prominent representatives of this pharmacological class of vasodilators (Gryglewski et al., 1992). Unfortunately, the chronic use of these drugs is limited by serious side effects such as nitrate tolerance, a phenomenon which is not only associated with the loss of vasodilator potency of these drugs but also with the induction of vascular oxidative stress resulting in endothelial and vascular dysfunction (Munzel et al., 2005a; Daiber et al., 2008).
During the last two decades, many efforts have been made to identify the mechanisms underlying clinical nitrate tolerance and endothelial dysfunction in response to chronic organic nitrate treatment (Munzel et al., 2005a; Daiber et al., 2008). The ‘oxidative stress concept in nitrate tolerance’ was established (Munzel et al., 1995) and further refined by identifying mitochondrial reactive oxygen and nitrogen species formation as a key event for the development of nitrate tolerance and endothelial dysfunction (Sydow et al., 2004; Daiber et al., 2005; Esplugues et al., 2006). The identification of the mitochondrial aldehyde dehydrogenase (ALDH-2) as an organic nitrate reductase (Chen et al., 2002; 2005; Sydow et al., 2004; Wenzel et al., 2007b) revived the Needleman ‘thiol depletion concept in nitrate tolerance’. This concept was further substantiated by the alleviation of nitrate tolerance by the dithiol dihydrolipoic acid, an essential co-factor for ALDH-2 nitrate reductase activity (Jakschik and Needleman, 1973; Wenzel et al., 2007a).
In previous studies, we have demonstrated that organic nitrate potency correlates with the number of nitrate groups and that high potency nitrates with pD2-values >6 [pentaerithrityl tetranitrate (PETN), GTN and pentaerithrityl trinitrate (PETriN)] are bioactivated by ALDH-2 (Daiber et al., 2004; Koenig et al., 2007a; Wenzel et al., 2007b). However, this concept was recently challenged when the mononitrate 2-aminoethyl nitrate (AEN) was found to be a highly potent organic nitrate with a calculated pD2-value of 7.5 in this study (for comparison the pD2-value of GTN was 7.44) (Koenig et al., 2007b). In the present study we sought to determine whether bioactivation of this new class of high potency mononitrates involves ALDH-2, whether AEN induces in vitro and in vivo tolerance or cross-tolerance to other vasodilators and, finally, whether AEN in comparison with triethanolamine trinitrate (TEAN) and methyl-3-nitrooxypropanoate (NPME) leads to increased mitochondrial oxidative stress and protein tyrosine nitration.
Methods
Animals and in vivo treatment
Young adult (8 ± 2 weeks) male ALDH-2 knockout (ALDH-2−/−) mice were age-matched with C57Bl6 mice. The generation of the ALDH-2 null mutant was as described elsewhere (Kitagawa et al., 2000). Mice were equipped with micro osmotic pumps from Alzet (Cupertino, CA, USA) containing 450 mM GTN solved in ethanol (50 µg·h−1 for 3 days) or ethanol as a control. Mice were also treated in vivo with the mononitrate AEN or the trinitrate TEAN by implanted micro osmotic pumps containing 1 M AEN (150 µg·h−1 for 3 days) or 500 mM TEAN dissolved in dimethyl sulphoxide (140 µg·h−1 for 3 days). All animal treatment was in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health and was granted by the Ethics Committee of the University Hospital Mainz.
Isometric tension studies
Vasodilator responses to acetylcholine (ACh), AEN, TEAN, NPME and nitroglycerine (GTN) were assessed with endothelium-intact isolated murine aortic rings mounted for isometric tension recordings in organ chambers and pre-constricted with prostaglandin F2α, as described previously (Wenzel et al., 2007b). To avoid development of tachyphylaxis, the potency of organic nitrates was not assessed in two sequential concentration–relaxation response curves. However, for some experiments we induced tachyphylaxis (in vitro tolerance) by performing two sequential concentration–relaxation response curves (Koenig et al., 2007b) resulting in a challenge of the isolated vessel with the EC70 concentration of the organic nitrate (100 µM for AEN and 1 mM for TEAN). AEN potency was also determined in aortic ring segments after pre-incubation with NS2028 (4H-8-bromo-1,2,4-oxadiazolo[3,4-d]benz[b][1,4]oxazin-1-one; 3 µM), 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) (10 µM), miconazol (25 µM), benomyl (10 µM), allopurinol (100 µM) and L-NAME (Nω-nitro-L-arginine methyl ester hydrochloride; 200 µM) for 30 min.
Mitochondrial reactive oxygen species formation
Isolated mitochondria were prepared from mice hearts according to a previously published protocol and reactive oxygen species (ROS) formation was detected by L-012 (8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H)dione sodium salt; 100 µM) enhanced chemiluminescence as recently described (Raha et al., 2000; Daiber et al., 2005). Briefly, hearts were glass/glass homogenized in HEPES buffer and subjected to centrifugation steps at 4°C of 1500×g for 10 min and 2000×g for 5 min (the pellets were discarded). The resulting supernatant was centrifuged at 20 000×g for 20 min, and the pellet resuspended in 1 mL of Tris buffer. The last centrifugation step was repeated and the pellet was finally resuspended in 1 mL of Tris buffer and the protein content was determined by the Lowry method. Mitochondrial suspensions were diluted to a final protein concentration of 0.1 mg·mL−1 in 0.5 mL of phosphate buffered saline (PBS) buffer containing L-012 (100 µM). ROS production was detected after stimulation with succinate (5 mM). The effect of AEN, TEAN, NPME, isopropyl nitrate (IPM) and GTN (10 and 1000 µM) on ROS formation was tested. The chemiluminescence was registered at intervals of 30 s over 5 min with a luminometer and the signal was expressed as counts min−1 at 5 min.
Detection of 3-nitrotyrosine by dot blot analysis
Cardiac mitochondria (0.2 mg·mL−1 total protein) were incubated with organic nitrates (5 mM) or GTN (50–5000 µM) for 1 h at 37°C. Soluble fractions of these samples (dissolved in Tween) were transferred to a nitrocellulose membrane as previously described (Daiber et al., 2003). Briefly, 100 µL (0.2 µg·µL−1 protein based on Bradford analysis) of the sample was transferred to a Protran BA85 (0.45 µm) nitrocellulose membrane by a Minifold I vacuum Dot-Blot system. Each slot was washed with 250 µL PBS and the membrane was dried for 15 min at 60°C. For detection of nitrated protein, a mouse monoclonal 3-NT antibody was used at a dilution of 1:1000. Positive bands were detected by enhanced chemiluminescence after incubation with a peroxidase-coupled secondary antibody (GAM-POX, 1:5000). All incubation and washing steps were performed according to the manufacturer's instructions. Densitometric quantification was performed by using a high-resolution scanner (Biometra/Epson) equipped with densitometry software Gel Pro Analyser (Media Cybernetics, Bethesda, MD, USA).
Statistical analysis
Results are expressed as mean ± SEM. One-way anova (with Bonferroni's or Dunn's correction for comparison of multiple means) was used for comparisons of vasodilator potency and efficacy and L-012-derived chemiluminescence. The EC50 value for each experiment was obtained by log-transformation. P values < 0.05 were considered significant.
Chemicals and reagents
For isometric tension studies, GTN was used from a Nitrolingual infusion solution (1 mg·mL−1) from G. Pohl-Boskamp (Hohenlockstedt, Germany). For induction of in vivo tolerance, GTN was used from a solution in ethanol (102 g·L−1) from UNIKEM (Copenhagen, Denmark). Prostaglandin F2α was obtained from Cayman Chemical (Ann Arbor, MI, USA). L-012 was purchased from Wako Pure Chemical Industries (Osaka, Japan). The organic nitrates were synthesized as previously described (Koenig et al., 2007b). 2-Nitrooxyethylammoniumnitrate (AEN) from 2-aminoethanol (Ishihara et al., 2003), TEAN from triethanolamine, NPME from methyl 3-hydroxypropanoate (McCallum and Emmons, 1956) and IPM was purchased from Sigma-Aldrich. All other chemicals were of analytical grade and obtained from Sigma-Aldrich, Fluka or Merck.
The luminometer was obtained from Berthold Techn. (Bad Wildbad, Germany); nitrocellulose membrane and Minifold I vacuum Dot-Blot system, Schleicher & Schuell (Dassel, Germany); mouse monoclonal 3-NT antibody, Upstate Biotechnology (Lake Placid, NY, USA); peroxidase-coupled secondary antibody (GAM-POX, 1:5000), Vector Laboratories (Burlingame, CA, USA).
Results
Potency of organic nitrates in ALDH-2-deficient mice and induction of tachyphylaxis (in vitro tolerance)
Figure 1 shows the chemical structure of all organic nitrates that were tested in the present study. The mononitrate AEN showed an unusually high potency (EC50= 0.83 µM, Table 1 and Figure 2A), which was superior to ISMN [EC50= 2.45 mM (Wenzel et al., 2007b)] and almost reached the potency of GTN (EC50= 0.10 µM) in murine aorta (see Table 2 and Figure 2G). Even more surprisingly, the potency of AEN was not significantly altered in ALDH-2-deficient mice indicating that bioactivation by ALDH-2 was not involved in the vasodilatation induced by this nitrate (Figure 2A). In addition, AEN only induced mild but not significant tachyphylaxis, as measured by the second of two identical concentration–relaxation response curves resulting from a cumulative pre-incubation of the vessel with the EC70 of AEN, 100 µM (Figure 2B). In contrast, the trinitrate TEAN was clearly less potent as compared with AEN or GTN (EC50= 15 µM, Table 1 and Figure 2C and G) and showed a significantly impaired potency in ALDH-2-deficient mice, suggesting the mitochondrial bioactivation pathway has a partial role in the response to this nitrate. Challenge of murine aortic segments with the EC70 of TEAN 1 mM resulted in a severe and significant shift to the right of the subsequent concentration–relaxation response curves in wild-type and ALDH-2 knockout mice (Figure 2D), compatible with the development of profound tachyphylaxis (in vitro tolerance). Interestingly, the potency of TEAN in the tachyphylactic rings was reduced to a similar degree in wild-type and ALDH-2-deficient mice, although it was different in untreated rings, indicating pretreatment with TEAN induced a complete loss of ALDH-2 activity. Finally, the mononitrate NPME showed an almost similar potency (EC50= 34 µM, Table 1 and Figure 2E and G) as compared with TEAN and to isosorbide dinitrate (ISDN) [EC50= 25 µM (Chen et al., 2005)] which again seemed to be independent of ALDH-2 because NPME potency was not significantly altered in ALDH-2-deficient mice. The mononitrate IPM, which was only used in the present study to assess mitochondrial ROS formation, was reported to have a quite low potency in rat aorta [EC50= 437 µM (Koenig et al., 2007b)]. In a subsequent second concentration–relaxation response curve the potency and efficacy of NPME was not significantly changed (Table 1 and Figure 2F) indicating the absence of any relevant tachyphylaxis. However, pretreatment of rings from ALDH-2−/− mice with NPME induced a significant shift to the right of the concentration–response curve compared with aortic sections from wild-type mice that were treated in the same way (Figure 2F).
Figure 1.
Structures of the organic nitrates used in this study. 2-Nitrooxyethylammoniumnitrate (AEN), methyl-3-nitrooxypropanoat (NPME) triethanolamine trinitrate (TEAN), glyceryl trinitrate (GTN), pentaerithrityl trinitrate (PETriN) and isopropyl nitrate (IPM).
Table 1.
Potencies and efficacies of organic nitrates in aorta from wild-type and ALDH-2−/− mice
| AEN | 2nd AEN (tachyphylaxis) | |||
|---|---|---|---|---|
| Group | Potency (pD2)# | Efficacy (%)# | Potency (pD2) | Efficacy (%) |
| Wild-type | 6.08 ± 0.10 (n= 20) | 81 ± 4 (n= 20) | 5.87 ± 0.09 (n= 16) | 77 ± 2 (n= 16) |
| ALDH-2−/− | 5.85 ± 0.11 (n= 20) | 75 ± 2 (n= 20) | 5.68 ± 0.12 (n= 16) | 71 ± 5 (n= 16) |
| TEAN | 2nd TEAN (tachyphylaxis) | |||
| Group | Potency (pD2) | Efficacy (%) | Potency (pD2) | Efficacy (%) |
| Wild-type | 4.82 ± 0.12$ (n= 20) | 71 ± 4 (n= 20) | 3.69 ± 0.12§ (n= 16) | 58 ± 8 (n= 16) |
| ALDH-2−/− | 4.22 ± 0.12& (n= 15)* | 69 ± 3 (n= 15) | 3.78 ± 0.11 (n= 16) | 48 ± 4 § (n= 16) |
| NPME | 2nd NPME (tachyphylaxis) | |||
| Group | Potency (pD2) | Efficacy (%) | Potency (pD2) | Efficacy (%) |
| Wild-type | 4.69 ± 0.13$ (n= 30) | 68 ± 3$ (n= 30) | 4.47 ± 0.17$ (n= 16) | 75 ± 2$ (n= 16) |
| ALDH-2−/− | 4.47 ± 0.09& (n= 25) | 63 ± 3& (n= 25) | 4.27 ± 0.12& (n= 14) | 56 ± 3& (n= 14) |
ALDH-2−/−, aldehyde dehydrogenase knockout; AEN, aminoethyl nitrate; TEAN, triethanolamine trinitrate; NPME, methyl-3-nitrooxypropanoate.
P < 0.05 versus wild-type
P < 0.05 versus first concentration–relaxation response curve
P < 0.05 versus AEN wild-type
P < 0.05 versus AEN knockout.
Potency is –log EC50 and efficacy is defined as maximal relaxation obtained with the highest concentration of the vasodilator used.
Figure 2.
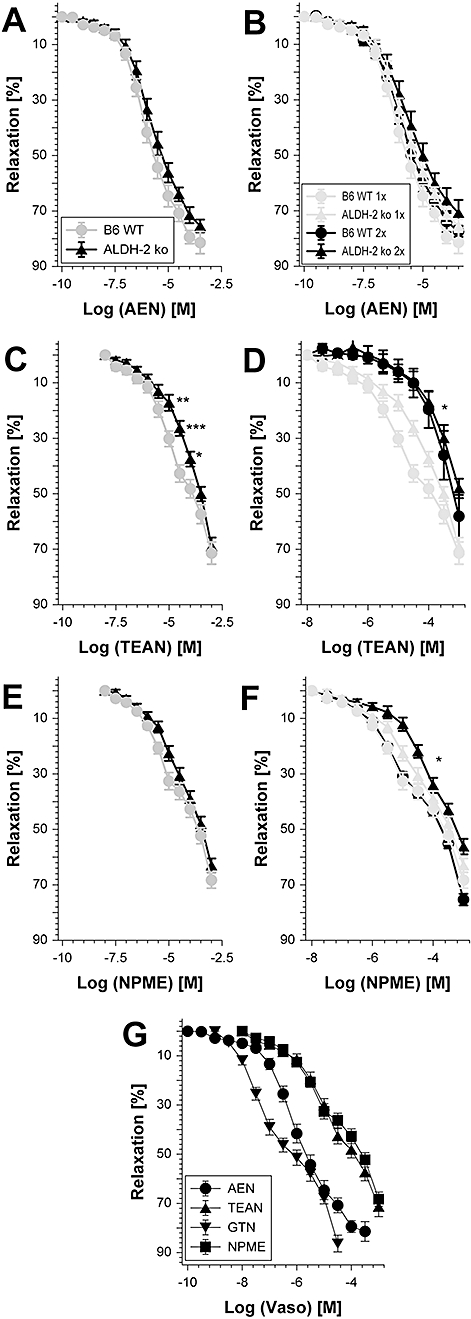
Vascular function of organic nitrates in wild-type and ALDH-2−/− aortic rings and tachyphylaxis (in vitro tolerance). Concentration–relaxation response curves for 2-nitrooxyethylammoniumnitrate (AEN) (10−10 to 10−3.5 M; A and B), triethanolamine trinitrate (TEAN) (10−7 to 10−3 M; C and D) and methyl-3-nitrooxypropanoat (NPME) (10−7 to 10−3 M; E and F) were obtained by isometric tension recordings in aortic segments from wild-type (B6 WT) and ALDH-2−/− (ALDH-2 ko) mice. (A, C and E) Potency of the nitrates was compared in aorta from wild-type and ALDH-2−/− mice. (B, D and F) Effect of repeated concentration–relaxation response curves was tested resulting in pre-incubation of the rings with the EC70 (100 µM for AEN, 1 mM for TEAN and NPME). For both strains the first concentration–relaxation response curves and the subsequent are shown. (G) Comparison of the vasodilator potency of AEN, TEAN, glyceryl trinitrate (GTN) and NPME in B6 WT mice. Data are mean ± SEM of 14–30 independent experiments with tissue from 8–17 animals per group. *P < 0.05 versus WT on the same treatment. For further statistical analysis, see Table 1.
Table 2.
Potencies and efficacies of organic nitrates in aorta from wild-type mice in response to in vivo treatment
| In vivo treatment |
ACh |
GTN |
AEN |
TEAN |
||||
|---|---|---|---|---|---|---|---|---|
| Potency (pD2)# | Efficacy (%)# | Potency (pD2) | Efficacy (%) | Potency (pD2) | Efficacy (%) | Potency (pD2) | Efficacy (%) | |
| EtOH | 6.92 ± 0.12 (n = 14) | 65 ± 4 (n = 14) | 6.72 ± 0.13 (n = 12) | 86 ± 3 (n = 12) | 6.41 ± 0.06 (n = 27) | 79 ± 3 (n = 27) | 4.11 ± 0.09 (n = 5) | 79 ± 5 (n = 5) |
| GTN | 6.63 ± 0.09 (n = 18)* | 47 ± 4 (n = 18)* | 5.95 ± 0.18 (n = 11)* | 82 ± 4 (n = 11)* | 6.29 ± 0.05 (n = 28) | 71 ± 4 (n = 28) | 3.75 ± 0.12 (n = 5) | 59 ± 4 (n = 5)* |
| DMSO | 7.04 ± 0.05 (n = 12) | 60 ± 3 (n = 12) | 6.63 ± 0.06 (n = 19) | 72 ± 2 (n = 19) | 6.13 ± 0.07 (n = 19) | 67 ± 4 (n = 19) | – | – |
| AEN | 6.78 ± 0.08 (n = 11)* | 53 ± 3 (n = 12) | 6.68 ± 0.05 (n = 17) | 76 ± 2 (n = 17) | 5.50 ± 0.06 (n = 16)* | 45 ± 3 (n = 16)* | – | – |
| DMSO | 7.13 ± 0.08 (n = 12) | 70 ± 3 (n = 12) | 6.57 ± 0.14 (n = 12) | 79 ± 4 (n = 12) | – | – | 4.81 ± 0.08 (n = 9) | 79 ± 2 (n = 9) |
| TEAN | 6.42 ± 0.07 (n = 12)* | 45 ± 4 (n = 12)* | 5.69 ± 0.05 (n = 12)* | 64 ± 7 (n = 12) | – | – | 4.48 ± 0.13 (n = 7)* | 68 ± 5 (n = 7) |
P < 0.05 versus solvent control.
Potency is –log EC50 and efficacy is defined as maximal relaxation obtained with the highest employed concentration of the vasodilator.
ACh, acetylcholine; AEN, aminoethyl nitrate; GTN, nitroglycerine; DMSO, dimethyl sulphoxide; EtOH, ethanol; TEAN, triethanolamine trinitrate.
Characterization of the AEN-dependent vasodilatation
To investigate further the mechanism involved in the relaxation evoked by AEN, isolated aortic ring segments were incubated with different inhibitors that affect distinct vasodilating signalling pathways. Pre-incubation with the inhibitor of soluble guanylyl cyclase NS2028 completely abolished AEN-dependent vasodilatation, whereas the NO scavenger PTIO only partially inhibited this process and the non-specific P450 inhibitor miconazol rather improved than impaired AEN potency (Figure 3A). We also tested the effect of the non-specific ALDH inhibitor benomyl that blocks not only the mitochondrial isoform ALDH-2, but also the cytosolic isoforms ALDH-1 and ALDH-4. As benomyl did not significantly affect the AEN-induced relaxation, the involvement of ALDH enzymes in its bioactivation may be excluded. In addition, L-NAME, an inhibitor of all NOS isoforms, did not significantly alter the AEN potency (Figure 3B). In contrast, allopurinol, the inhibitor of xanthine oxidoreductase (after conversion to its hydroxylated product oxypurinol) caused a significant impairment of the relaxation to higher concentrations of AEN (Figure 3B).
Figure 3.
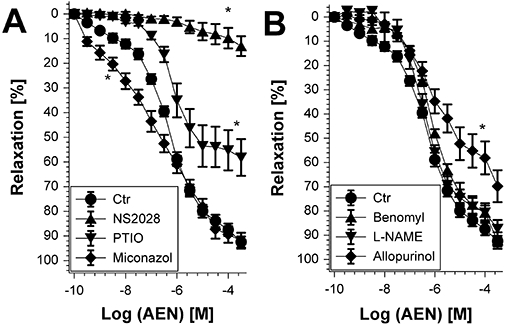
Effect of different inhibitors on vasodilatation evoked by 2-nitrooxyethylammoniumnitrate (AEN) in wild-type mice. AEN potency was determined after pre-incubation of isolated aortic ring segments for 30 min with (A) NS2028 (3 µM), PTIO (10 µM) or miconazol (25 µM) and (B) benomyl (10 µM), L-NAME (200 µM) or allopurinol (100 µM). Data are mean ± SEM of 6–12 independent experiments with tissue from different mice. *P < 0.05 versus control without treatment.
Mitochondrial ROS formation and protein tyrosine nitration after in vitro treatment with organic nitrates
In isolated heart mitochondria from ALDH-2−/− mice, the L-012-derived chemiluminescence signal increased in response to challenge with 1 mM TEAN but not with AEN, NPME or IPM (Figure 4A). A similar pattern was obtained in isolated heart mitochondria from wild-type mice (Figure 4B) and the trinitrate was found to be even more potent than GTN at inducing acute mitochondrial oxidative stress (Daiber et al., 2005; Mollnau et al., 2006). GTN was also identified as a powerful inducer of mitochondrial oxidative stress, whereas another nitrate, PETriN, did not induce mitochondrial ROS formation (Figure 4C). PETriN is the trinitrate metabolite of PETN, which was previously shown to possess antioxidative properties (Daiber et al., 2004). As expected, neither NPME nor AEN induced mitochondrial ROS formation at a concentration of 500 µM. When mitochondria from wild-type and ALDH-2−/− mice were directly compared, the pattern of organic nitrate-induced ROS formation was very similar in both groups, indicating that the presence of ALDH-2 does not have an essential role in nitrate-triggered mitochondrial oxidative stress (Figure 4D). Uric acid (50 µM), a scavenger of peroxynitrite-derived free radicals, completely abolished the chemiluminescence signals induced by GTN and TEAN (not shown).
Figure 4.
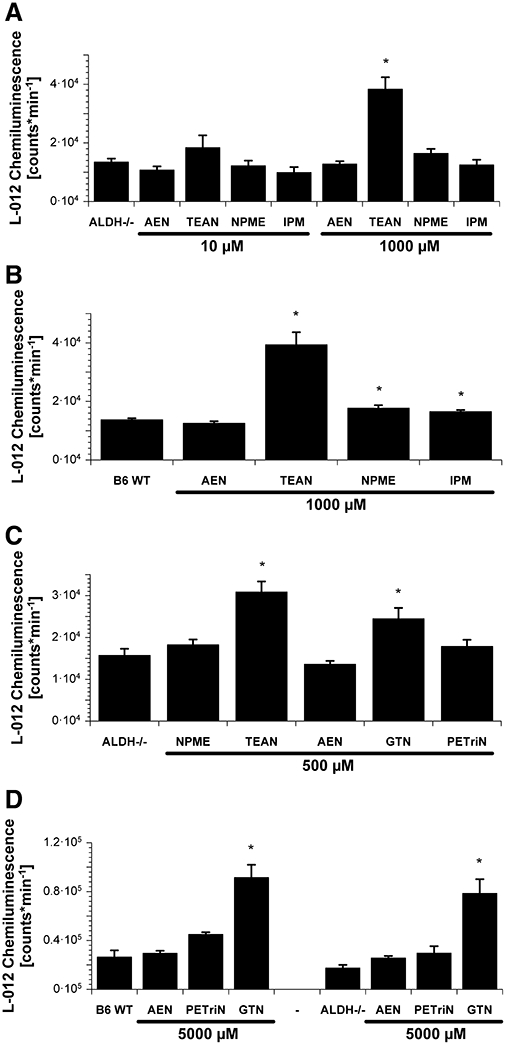
Mitochondrial reactive oxygen species (ROS) formation after acute organic nitrate treatment. Isolated cardiac mitochondria (0.1 mg protein mL−1) were stimulated with succinate (5 mM), the chemiluminescence signal was detected using a single photon counter in the presence of the dye L-012 (100 µM). The organic nitrates 2-nitrooxyethylammoniumnitrate (AEN), triethanolamine trinitrate (TEAN), methyl-3-nitrooxypropanoat (NPME) and isopropyl nitrate (IPM) were tested at 10 or 1000 µM in mitochondria from ALDH-2−/− (A) or wild-type (WT) (B) mice. ROS formation was also determined for NPME, TEAN, AEN, glyceryl trinitrate (GTN) and pentaerithrityl trinitrate (PETriN) at 500 µM in mitochondria from ALDH-2−/− mice (C). Mitochondrial ROS formation in WT versus ALDH-2−/− mice was also measured for AEN, PETriN and GTN at 5 mM (D). All nitrates were used from stocks in dimethyl sulphoxide and the vehicle was added to controls. Data are mean ± SEM of 8–24 (A), 6–24 (B), 8 (C) and 4–6 (D) experiments with mitochondria from three to six animals per group. *P < 0.05 versus untreated control.
Protein tyrosine nitration is an oxidative amino acid modification resulting from peroxynitrite-derived free radicals or peroxidase-catalysed nitration by nitrite and hydrogen peroxide. When mitochondria were incubated with GTN, the level of nitrated proteins significantly increased in a dose-dependent manner (Figure 5A). Among the nitrates tested, TEAN was the most effective inducer of nitration, while AEN and NPME did not significantly affect 3-nitrotyrosine levels, which was in accordance with the observations made with L-012-enhanced chemiluminescence (Figure 5B).
Figure 5.
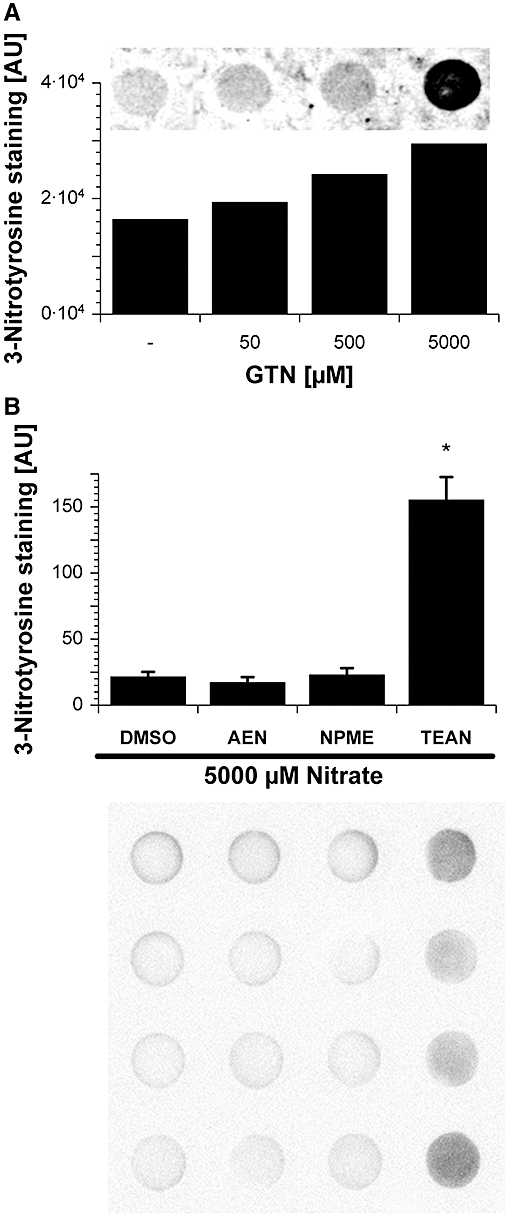
Nitration of mitochondrial proteins in response to acute organic nitrate treatment. Protein tyrosine nitration was tested in isolated cardiac mitochondria (0.2 mg protein mL−1) from control mice for glyceryl trinitrate (GTN) (5–5000 µM) (A) and 2-nitrooxyethylammoniumnitrate (AEN), methyl-3-nitrooxypropanoat (NPME), triethanolamine trinitrate (TEAN) (5 mM) (B). 3-Nitrotyrosine formation was detected by dot blot analysis using a specific antibody. Dimethyl sulphoxide (DMSO) was used as a solvent control for AEN, NPME and TEAN whereas ethanol (EtOH) was used for GTN respectively; 20 µg of protein was transferred to each well. Below the densitometric quantification the original blot is shown. Data shown in (B) are mean ± SEM of eight independent experiments. *P < 0.05 versus DMSO solvent control.
Induction of in vivo tolerance and cross-tolerance
Chronic in vivo treatment with the mononitrate AEN had no significant effect on the ACh-dependent relaxation, thus excluding the possibility that this compound causes endothelial dysfunction (Table 2 and Figure 6A). In contrast, in vivo administration of the trinitrate TEAN induced severe endothelial dysfunction, demonstrated by a significantly impaired efficacy of the endothelium-dependent vasodilator ACh (Table 2 and Figure 6B). Similar observations were made for the GTN concentration–relaxation response curves which were not significantly impaired after treatment with AEN in vivo but were inhibited by TEAN in vivo therapy, indicating a marked degree of cross-tolerance towards GTN (Table 2, Figure 6C and D). Remarkably, AEN induced a marked tolerance to itself (Table 2 and Figure 6E), which is inconsistent with the observation showing that AEN was devoid of tachyphylaxis and oxidative stress (Figures 2, 4 and 5). The TEAN concentration–relaxation response curve revealed the development of tolerance, because the curve was significantly shifted to the right in aortic ring segments from mice treated with TEAN in vivo for 3 days (Figure 6F).
Figure 6.
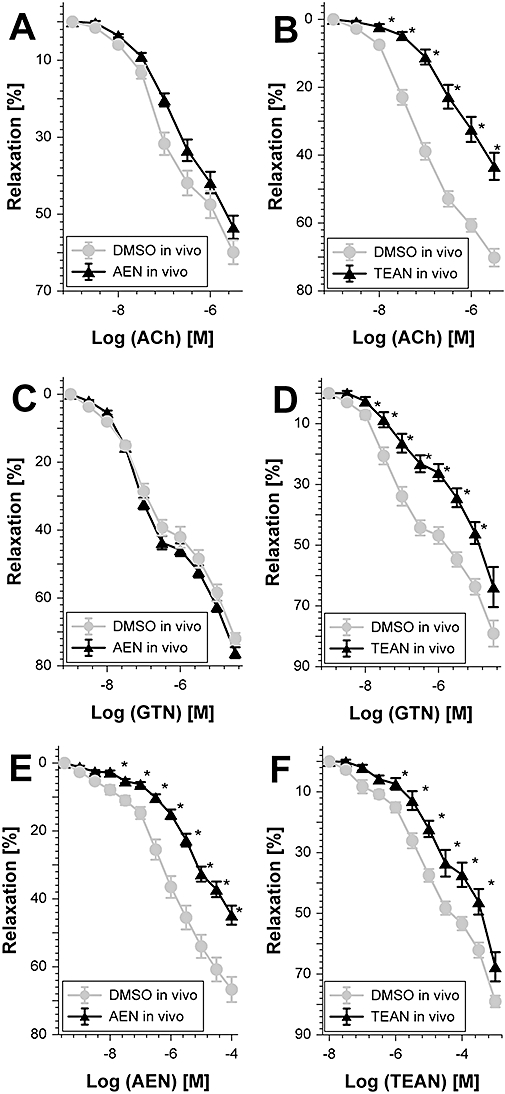
Potency of different vasodilators after chronic 2-nitrooxyethylammoniumnitrate (AEN) or triethanolamine trinitrate (TEAN) treatment. Relaxation was assessed by recording isometric tension in aortic segments from wild-type mice. The effect of AEN treatment in vivo (150 µg·h−1 per 3 days) on relaxation to acetylcholine (ACh) (A), glyceryl trinitrate (GTN) (C) and AEN (E) was tested. Similarly, the effect of TEAN (140 µg·h−1 per 3 days) on relaxation to ACh (B), glyceryl trinitrate (GTN) (D) or TEAN (F) was assessed. For both nitrates dimethyl sulphoxide (DMSO) was used as a solvent control. Data show mean ± SEM of 12–19 (AEN) and 7–12 (TEAN) independent experiments with tissue from at least five animals per group. *P < 0.05 versus DMSO solvent control.
In vivo treatment with GTN resulted in severe impairment of endothelium-dependent relaxation (ACh response) and of course tolerance to itself (Table 2, Figure 7A and B). AEN in vivo therapy did not induce cross-tolerance towards GTN (Figure 6C) and vice versa GTN in vivo treatment did not trigger cross-tolerance towards AEN (Table 2 and Figure 7C). In accordance with the observation that in vivo administration of TEAN caused cross-tolerance towards GTN (Figure 6D), the present results demonstrate the induction of cross-tolerance towards TEAN by in vivo treatment with GTN (Table 2 and Figure 7D).
Figure 7.
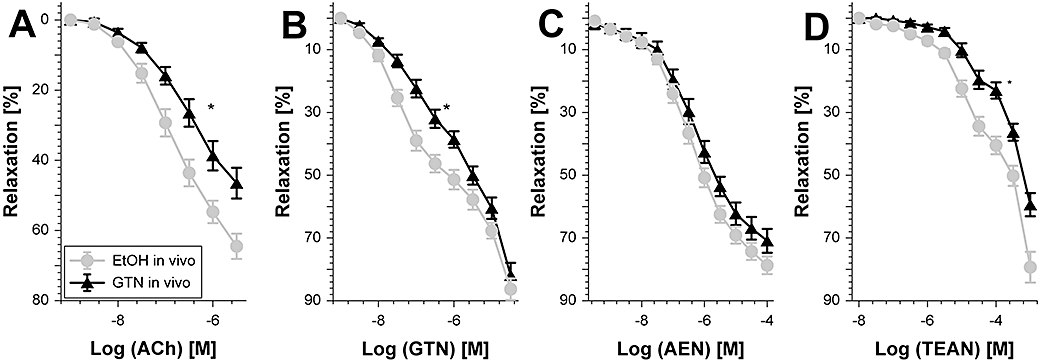
Potency of different vasodilators after chronic glyceryl trinitrate (GTN) treatment. Relaxation was assessed by recording isometric tension in aortic segments from wild-type mice. The effect of GTN treatment in vivo (50 µg·h−1 per 3 days) on relaxation to acetylcholine (ACh) (A), GTN (B), 2-nitrooxyethylammoniumnitrate (AEN) (C) and triethanolamine trinitrate (TEAN) (D) was assessed. Data are mean ± SEM of 12–18 (ACh, GTN, TEAN) or 27–28 (AEN) independent experiments with tissue from at least seven animals per group. *P < 0.05 versus ethanol (EtOH) solvent control.
Mitochondrial ROS formation upon in vivo treatment with organic nitrates
In isolated heart mitochondria from wild-type mice, the L-012-derived chemiluminescence signal increased in response to in vivo therapy with either TEAN or GTN whereas in vivo treatment with AEN had no effect on mitochondrial ROS formation (Figure 8).
Figure 8.
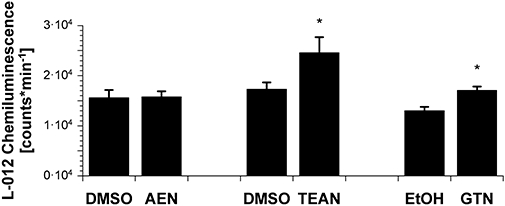
Mitochondrial reactive oxygen species (ROS) formation after chronic organic nitrate treatment. Isolated cardiac mitochondria (0.1 mg protein mL−1) where stimulated with succinate (5 mM), the chemiluminescence signal was detected using a single photon counter in the presence of the dye L-012 (100 µM). The organic nitrates 2-nitrooxyethylammoniumnitrate (AEN), triethanolamine trinitrate (TEAN) and glyceryl trinitrate (GTN) were infused at a dose of 150, 140 or 50 µg·h−1 for 3 days respectively. Data are mean ± SEM of 24 (AEN), 20–21 (TEAN) and 13–17 (GTN) independent experiments with mitochondria from at least eight animals per group. *P < 0.05 versus solvent (sham-treated) control.
Discussion
The present study extends our previous results on organic nitrate bioactivation and tolerance mechanisms (Munzel et al., 2005a; Daiber et al., 2008). We have shown that a clear correlation between the number of nitrate (-ONO2) groups in the molecule and the vasodilator potency exists (Koenig et al., 2007a; Wenzel et al., 2007b). Moreover, we demonstrated that potency correlates with the induction of the formation of mitochondrial ROS (Daiber et al., 2004). This postulate was challenged by the new mononitrate 2-aminoethyl nitrate (AEN), which was shown to have a potency similar to that of GTN in a previous study (Koenig et al., 2007b).
An impressive potency for AEN (pD2= 6.09 ± 0.06) was also found using murine aortic vessel segments. This high potency was far above the pD2-values normally reported for mononitrates: 3.24 ± 0.03 for pentaerithrityl mononitrate (PEMN), 2.61 ± 0.19 for ISMN in murine aorta (Wenzel et al., 2007b) and 4.14 ± 0.02 for PEMN, 4.48 ± 0.03 for ISMN in porcine pulmonary arteries (Koenig et al., 2007a). Consistent with previous findings, the AEN potency was quite similar to that of GTN (pD2= 6.72 ± 0.13) in the present study.
Even more surprisingly, AEN did not induce either tolerance (tachyphylaxis) in vitro or mitochondrial ROS formation and is not dependent on ALDH-2 for its activity either. In contrast, the new less potent trinitrate TEAN (pD2= 4.82 ± 0.12) showed impaired vasorelaxant activity in ALDH-2−/− mice, induced severe tolerance (tachyphylaxis) in vitro and evoked mitochondrial ROS formation. Another organic mononitrate (NPME, pD2= 4.47 ± 0.17) did not rely on ALDH-2 for its bioactivation and induced almost no mitochondrial ROS formation. NPME caused no tolerance (tachyphylaxis) in vitro either, although repeated challenge of the vessel with high concentrations caused a significant loss of potency in ALDH-2-deficient aorta. Whether this is related to the recently reported additive oxidative stress caused by ALDH-2 insufficiency and organic nitrate overload (Wenzel et al., 2008a) remains to be established. In addition, there was no significant difference between ROS formation in wild-type versus ALDH-2−/− mitochondria. Finally, it should be noted that all organic nitrates tested caused an increase in mitochondrial nitrated proteins. This might be best explained by the release of NO and simultaneous superoxide formation by organic nitrates, resulting (at least in compartments where the nitrate accumulates or by over-dosing) in nitrosative/oxidative stress, which may also cause protein tyrosine nitration (e.g. by nitrite/hydrogen peroxide and peroxidases or peroxynitrite formation) (Halliwell, 1997). However, the degree of tyrosine nitration might reflect the balance between undesired oxidative stress and vasorelaxing potency, because the nitrate with the least favourable profile (TAEN) was the most potent at evoking tyrosine nitration.
We also studied the development of in vivo tolerance in response to chronic treatment with these organic nitrates. As expected from the in vitro data, AEN did not induce cross-tolerance to the endothelium-dependent vasodilator ACh (no endothelial dysfunction) or to GTN. Surprisingly, in vivo treatment with AEN showed significant development of tolerance to itself but failed to increase mitochondrial ROS formation. In contrast, in vivo treatment with the trinitrate TEAN caused cross-tolerance to ACh (endothelial dysfunction) and GTN as well as to itself. TEAN also significantly and most efficiently increased ROS formation and protein tyrosine nitration in isolated mitochondria. Therefore, the observation that TEAN caused the most pronounced tolerance effects in vitro and in vivo is compatible with its ability to induce severe oxidative stress, supporting the concept that oxidative stress is involved in the development of nitrate and cross-tolerance.
In a last set of experiments we tested the development of cross-tolerance to AEN and TEAN induced by chronic treatment with GTN in vivo. In accordance with previous data, chronic GTN infusion caused cross-tolerance to ACh (endothelial dysfunction) and tolerance to itself whereas AEN potency was not impaired in response to in vivo GTN treatment. The latter result was expected because in vivo AEN treatment also did not cause cross-tolerance to GTN. In contrast, TEAN potency was impaired in GTN-tolerant vessels indicating cross-tolerance to TEAN, which was in accordance with the observation that in vivo TEAN treatment resulted in cross-tolerance to GTN. Although TEAN was less potent than GTN, in vivo treatment with the latter resulted in a less pronounced increase in mitochondrial ROS formation and nitrated protein as compared with TEAN.
An important finding of the present study is the observation that AEN potency is not linked to increased mitochondrial ROS formation in response to bolus challenges, which was previously observed for other nitrates (Daiber et al., 2004). Also, in vitro tolerance was absent with AEN, which was considered a feature of the low-potency nitrates so far (ISMN and PEMN). With respect to induction of in vitro tachyphylaxis and mitochondrial ROS formation, the trinitrate TEAN behaved in the same manner like a high potency organic nitrate without sharing its high potency. Even more important is the finding of an independent bioactivation pathway for AEN, which was not affected by chronic GTN treatment but obviously was affected by chronic AEN infusion. The lack of an effect of ALDH-2 deficiency and pharmacological inhibition of ALDH isozymes by benomyl on AEN potency together suggest that AEN uses a bioactivation pathway different from that used by GTN. Moreover, the superior potency of AEN compared with other mononitrates leads us to speculate that AEN uses a different bioactivation pathway from those previously described for low-potency nitrates (e.g. P450 proteins) (Munzel et al., 2005a); otherwise this high potency of AEN would be hard to achieve.
Of note, important differences exist between in vitro and in vivo nitrate tolerance: the former is a consequence of the overload of nitrate bioactivating mechanisms that follows exposure to high concentrations of organic nitrates in a time frame that is too short to allow regeneration of these mechanisms. In contrast, in vivo tolerance is determined by the activation of counter-regulatory mechanisms at the humoral, genomic and proteomic level. The observation that AEN does not cause tachyphylaxis or cross-tolerance to GTN but, in contrast, induces severe tolerance to itself, along with the lack of increased mitochondrial oxidative stress, suggests that oxidative alterations play if at all a less important role in the effects of AEN. It is more likely that AEN causes alterations in the protein and gene expression profiles that lead to the observed in vivo tolerance. Another possibility is that AEN inactivates its bioactivating system by direct interaction (e.g. by covalent binding). While chronic treatment with ISDN, ISMN and GTN result in clinical tolerance (Schulz et al., 2002; Sekiya et al., 2005; Thomas et al., 2007), PETN is devoid of tolerance development and even more importantly, preserves endothelial function (Jurt et al., 2001; Gori et al., 2003).
Previous in vitro observations clearly indicate that there is a correlation between the potency, mitochondrial ROS formation and inhibition of mitochondrial ALDH elicited by clinically used organic nitrates and which decrease according to the following order (Daiber et al., 2004): GTN >> PETN > PETriN > ISDN > PEDN = ISMN = PEMN. This sequence indicates that the protective pathways of PETN described above are not operative in response to acute challenges because they occur at the level of gene and protein expression, which requires treatment times of more than 6 h. Obviously, overcharge of isolated tissue with PETN causes very similar tolerance phenomena as observed for GTN and results in impaired vasodilator potency regardless of whether isolated aortae were pre-incubated with PETN at EC100 (1 µM) concentrations or far higher (300 µM) (Koenig et al., 2007a).
In conclusion, our results further establish that organic nitrates are a heterogeneous group of drugs, which differ significantly with respect to their vasodilator potency, induction of tolerance and mitochondrial (as well as vascular) oxidative stress. AEN may represent a new class of organic nitrates, which does not induce tachyphylaxis in vitro or cause oxidative stress, and showed an impressively high potency compared with all the mononitrates tested so far but also to di- and trinitrates. According to our data, AEN-mediated vasodilatation is based on the NO/cGMP signalling pathway (NS2028 and PTIO effects) and its bioactivation involves xanthine oxidoreductase (allopurinol effect) rather than P450 or ALDH enzymes (miconazol and benomyl incubations), but further mechanistic studies are required to reveal the bioactivation pathway as well as the precise mechanism of action of AEN. Despite these beneficial and pharmacologically relevant properties, AEN induced severe tolerance in vivo to itself. This may be of clinical interest because AEN is part of the structure of the potassium channel blocker nicorandil which consists of a fused organic nitrate moiety. Nicorandil is devoid of clinical tolerance (Sekiya et al., 2005). There is also not much known about the denitration pathway of nicorandil but earlier work has suggested that it is mainly metabolized in the kidney and the denitrated product 2-nicotinamidoethanol is the major metabolite excreted in urine (Frydman, 1992). Therefore, future strategies aimed at developing organic nitrate-based vasodilators and hybride molecules may include the insertion of AEN-like structures. According to a recent publication in Science activation of ALDH-2 protects the heart from ischaemic damage and vice versa inhibition of ALDH-2 contributes to cardiotoxic events (Chen et al., 2008). This accords with our previous observations that show ALDH-2 deficiency increases cardiotoxicity of doxorubicin and cardiovascular damage by nitroglycerine (Wenzel et al., 2008a) and aggravates age-dependent vascular dysfunction (Wenzel et al., 2008b). In view of these data the ability of an organic nitrate to prevent oxidative inhibition or down-regulation of ALDH-2 might not only be a beneficial side effect, but might even be essential in order to confer cardiac and vascular protection.
Acknowledgments
The expert technical assistance of Jörg Schreiner, Nicole Schramm and Merle Götz is gratefully appreciated. The financial support by the German Research Foundation (SFB 553 to A.D., T.M. and SCHU 1486/2–1 to E.S.), by MAIFOR and Förderfonds grants from the University Hospital Mainz (A.D.) and by the Robert-Müller-Foundation (T.M. and A.D.) is gratefully acknowledged. This study contains parts of the thesis work of Luise Sydow.
Glossary
Abbreviations:
- ACh
acetylcholine
- AEN
aminoethyl nitrate
- ALDH-2
mitochondrial aldehyde dehydrogenase
- ALDH-2−/−
ALDH-2 knockout
- DMSO
dimethyl sulphoxide
- ECL
enhanced chemiluminescence
- GTN
glycerol trinitrate (nitroglycerin)
- NPME
methyl-3-nitrooxypropanoate
- IPM
iso-propyl nitrate
- ISDN
isosorbide dinitrate
- ISMN
isosorbide-5-mononitrate
- L-012
8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazine-1,4-(2H,3H)dione sodium salt
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- NO
nitric oxide
- NS2028
4H-8-bromo-1,2,4-oxadiazolo[3,4-d]benz[b][1,4]oxazin-1-one
- PBS
phosphate buffered saline
- PETN
pentaerithrityl tetranitrate
- PETriN
pentaerithrityl trinitrate
- PTIO
2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide
- ROS
reactive oxygen species
- TEAN
triethanolamine trinitrate
Conflict of interest
The authors state no conflict of interest.
References
- Abrams J. Beneficial actions of nitrates in cardiovascular disease. Am J Cardiol. 1996;77(13):31C–37C. doi: 10.1016/s0002-9149(96)00186-5. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Meredith IT, Ganz P, Selwyn AP, Yeung AC. Nitric oxide and nitrovasodilators: similarities, differences and potential interactions. J Am Coll Cardiol. 1994;24(2):555–566. doi: 10.1016/0735-1097(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74(8):3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321(5895):1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99(12):8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, et al. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102(34):12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Bachschmid M, Kavakli C, Frein D, Wendt M, Ullrich V, et al. A new pitfall in detecting biological end products of nitric oxide-nitration, nitros(yl)ation and nitrite/nitrate artefacts during freezing. Nitric Oxide. 2003;9(1):44–52. doi: 10.1016/j.niox.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Coldewey M, Bachschmid M, Wenzel P, Sydow K, et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: a comparison of pentaerythritol tetranitrate with other organic nitrates. Mol Pharmacol. 2004;66(6):1372–1382. doi: 10.1124/mol.104.002600. [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Sulyok S, Coldewey M, Schulz E, Treiber N, et al. Heterozygous deficiency of manganese superoxide dismutase in mice (Mn-SOD+/−): a novel approach to assess the role of oxidative stress for the development of nitrate tolerance. Mol Pharmacol. 2005;68(3):579–588. doi: 10.1124/mol.105.011585. [DOI] [PubMed] [Google Scholar]
- Daiber A, Wenzel P, Oelze M, Munzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin Res Cardiol. 2008;97(1):12–20. doi: 10.1007/s00392-007-0588-7. [DOI] [PubMed] [Google Scholar]
- Esplugues JV, Rocha M, Nunez C, Bosca I, Ibiza S, Herance JR, et al. Complex I dysfunction and tolerance to nitroglycerin: an approach based on mitochondrial-targeted antioxidants. Circ Res. 2006;99(10):1067–1075. doi: 10.1161/01.RES.0000250430.62775.99. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- Frydman A. Pharmacokinetic profile of nicorandil in humans: an overview. J Cardiovasc Pharmacol. 1992;20(Suppl.)(3):S34–44. doi: 10.1097/00005344-199206203-00008. [DOI] [PubMed] [Google Scholar]
- Gori T, Al-Hesayen A, Jolliffe C, Parker JD. Comparison of the effects of pentaerythritol tetranitrate and nitroglycerin on endothelium-dependent vasorelaxation in male volunteers. Am J Cardiol. 2003;91(11):1392–1394. doi: 10.1016/s0002-9149(03)00342-4. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Zembowicz A, Salvemini D, Taylor GW, Vane JR. Modulation of the pharmacological actions of nitrovasodilators by methylene blue and pyocyanin. Br J Pharmacol. 1992;106(4):838–845. doi: 10.1111/j.1476-5381.1992.tb14422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett. 1997;411(2–3):157–160. doi: 10.1016/s0014-5793(97)00469-9. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104(22):2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- Ishihara S, Saito F, Ohhata Y, Kanai M, Mizuno H, Fujisawa M, et al. Synthesis and collateral dilator activity of nitroxyalkylamides having direct or latent sulfhydryl moieties. Bioorg Med Chem Lett. 2003;13(9):1527–1530. doi: 10.1016/s0960-894x(03)00199-9. [DOI] [PubMed] [Google Scholar]
- Jakschik B, Needleman P. Sulfhydryl reactivity of organic nitrates: biochemical basis for inhibition of glyceraldehyde-P dehydrogenase and monoamine oxidase. Biochem Biophys Res Commun. 1973;53(2):539–544. doi: 10.1016/0006-291x(73)90695-5. [DOI] [PubMed] [Google Scholar]
- Jurt U, Gori T, Ravandi A, Babaei S, Zeman P, Parker JD. Differential effects of pentaerythritol tetranitrate and nitroglycerin on the development of tolerance and evidence of lipid peroxidation: a human in vivo study. J Am Coll Cardiol. 2001;38(3):854–859. doi: 10.1016/s0735-1097(01)01414-0. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, et al. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476(3):306–311. doi: 10.1016/s0014-5793(00)01710-5. [DOI] [PubMed] [Google Scholar]
- Koenig A, Lange K, Konter J, Daiber A, Stalleicken D, Glusa E, et al. Potency and in vitro tolerance of organic nitrates: partially denitrated metabolites contribute to the tolerance-devoid activity of pentaerythrityl tetranitrate. J Cardiovasc Pharmacol. 2007a;50(1):68–74. doi: 10.1097/FJC.0b013e31805881ee. [DOI] [PubMed] [Google Scholar]
- Koenig A, Roegler C, Lange K, Daiber A, Glusa E, Lehmann J. NO donors. Part 16: investigations on structure-activity relationships of organic mononitrates reveal 2-nitrooxyethylammoniumnitrate as a high potent vasodilator. Bioorg Med Chem Lett. 2007b;17(21):5881–5885. doi: 10.1016/j.bmcl.2007.08.046. [DOI] [PubMed] [Google Scholar]
- McCallum KS, Emmons WD. Notes – the dissociation constants and infrared spectra of some nitratoacids. J Org Chem. 1956;21(3):367–368. [Google Scholar]
- Mollnau H, Wenzel P, Oelze M, Treiber N, Pautz A, Schulz E, et al. Mitochondrial oxidative stress and nitrate tolerance–comparison of nitroglycerin and pentaerithrityl tetranitrate in Mn-SOD+/− mice. BMC Cardiovasc Disord. 2006;6:44. doi: 10.1186/1471-2261-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005a;97(7):618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005b;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- Munzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95(1):187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92(3):639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha S, McEachern GE, Myint AT, Robinson BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med. 2000;29(2):170–180. doi: 10.1016/s0891-5849(00)00338-5. [DOI] [PubMed] [Google Scholar]
- Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- Schulz E, Tsilimingas N, Rinze R, Reiter B, Wendt M, Oelze M, et al. Functional and biochemical analysis of endothelial (dys)function and NO/cGMP signaling in human blood vessels with and without nitroglycerin pretreatment. Circulation. 2002;105(10):1170–1175. doi: 10.1161/hc1002.105186. [DOI] [PubMed] [Google Scholar]
- Sekiya M, Sato M, Funada J, Ohtani T, Akutsu H, Watanabe K. Effects of the long-term administration of nicorandil on vascular endothelial function and the progression of arteriosclerosis. J Cardiovasc Pharmacol. 2005;46(1):63–67. doi: 10.1097/01.fjc.0000162771.00174.a8. [DOI] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113(3):482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GR, DiFabio JM, Gori T, Parker JD. Once daily therapy with isosorbide-5-mononitrate causes endothelial dysfunction in humans: evidence of a free-radical-mediated mechanism. J Am Coll Cardiol. 2007;49(12):1289–1295. doi: 10.1016/j.jacc.2006.10.074. [DOI] [PubMed] [Google Scholar]
- Wennmalm A. Endothelial nitric oxide and cardiovascular disease. J Intern Med. 1994;235(4):317–327. doi: 10.1111/j.1365-2796.1994.tb01081.x. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Hink U, Oelze M, Schuppan S, Schaeuble K, Schildknecht S, et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH-2) activity: IMPLICATIONS FOR MITOCHONDRIAL OXIDATIVE STRESS AND NITRATE TOLERANCE. J Biol Chem. 2007a;282(1):792–799. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Hink U, Oelze M, Seeling A, Isse T, Bruns K, et al. Number of nitrate groups determines reactivity and potency of organic nitrates: a proof of concept study in ALDH-2−/− mice. Brit J Pharmacol. 2007b;150:526–533. doi: 10.1038/sj.bjp.0707116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem Biophys Res Commun. 2008a;367(1):137–143. doi: 10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008b;80(2):280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]