

Abstract
Background and purpose:
Oxaliplatin is the first platinum-based compound effective in the treatment of colorectal cancer. Oxaliplatin combined with cetuximab for metastatic colorectal cancer is under evaluation. The preliminary results seem controversial, particularly for the use of cetuximab in K-Ras mutated patients. K-Ras mutation is known to affect redox homeostasis. Here we evaluated how the efficacy of oxaliplatin alone or combined with cetuximab varied according to the Ras mutation and redox status in a panel of colorectal tumour cell lines.
Experimental approach:
Viability was evaluated by methylthiazoletetrazolium assay, reactive oxygen species production by DCFDA and lucigenin on HT29-D4, Caco-2, SW480 and SW620 cell lines.
Key results:
Combination of oxaliplatin and cetuximab was less cytotoxic than oxaliplatin alone in colorectal cells harbouring wild-type Ras and membrane expression of receptors for epidermal growth factor receptor (EGFR), such as HT29-D4 and Caco-2 cells. In contrast, cetuximab did not affect oxaliplatin efficiency in cells harbouring K-RasV12 mutation, irrespective of membrane EGFR expression (SW620 and SW480 cells). Transfection of HT29-D4 with K-RasV12 decreased oxaliplatin IC50 and impaired cetuximab sensitivity, without affecting expression of membrane EGFR compared with HT29-D4 control. Oxaliplatin efficacy relies on endogenous production of H2O2. Cetuximab inhibits H2O2 production inhibiting the EGFR/Nox1 NADPH oxidase pathway. Oxaliplatin efficacy was impaired by short hairpin RNA for Nox1 and by catalase (H2O2 scavenger).
Conclusions and implications:
Cetuximab limited oxaliplatin efficiency by affecting the redox status of cancer cells through Nox1. Such combined therapy might be improved by controlling H2O2 elimination.
Keywords: colorectal cancer, Nox1, reactive oxygen species, oxaliplatin, cetuximab
Introduction
Colorectal cancer is one of the major types of cancer worldwide, in terms of both morbidity and mortality. Despite improvements in medical therapy, the outcomes of treatment for locally advanced and metastatic disease remains disappointing with 5 year survival rates lower than 10% in patients with metastasis. Oxaliplatin and irinotecan used in combination with 5-fluorouracil (5FU) and leucovorin (FOLFOX or FOLFIRI respectively) represent the major treatment for metastatic colorectal cancer with response rates higher than 50% (Becouarn et al., 1998). Another therapeutic approach involves the recent advances in biotherapies, which has been beneficial to the treatment of metastatic colorectal cancer through the development of a monoclonal antibody against the epidermal growth factor receptor (EGFR), cetuximab (also called C-225 or Erbitux®). Cetuximab has been proven efficient in irinotecan-resistant metastatic colorectal cancer expressing the EGFR with responses ranging between 8.8% when used in monotherapy and 22.9% when combined with irinotecan (Cunningham et al., 2004). Oxaliplatin, the other major chemotherapeutic agent used in metastatic colorectal cancer is not usually combined with cetuximab, and different trials for that combination are under evaluation (Vincenzi et al., 2006).
Oxaliplatin is a third-generation platinum analogue and is the first platinum-based compound to show efficacy in the treatment of colorectal cancer. Oxaliplatin acts by alkylating DNA, and the level of platin–DNA adducts is thought to be a main factor in platin-compound cytotoxicity. Previous studies have suggested that glutathione and glutathione related enzymes are involved in the sensitivity of cells to platin compounds (El-Akawi et al., 1996). Glutathione S-transferase (GST) catalyses the conjugation of glutathione to genotoxic compounds, preventing DNA damage and adduct formation (Watson et al., 1998). Laurent et al. showed that the glutathione system limited the cytotoxic activity of oxaliplatin through modifying the production of cellular reactive oxygen species (ROS). ROS effects are paradoxical because they can act as both disease inducers and chemotherapeutic agents (Lau et al., 2008). Indeed, ROS are usually known as cytotoxic and mutagenic and linked to tumour progression, but most anticancer drugs kill their target cells, at least in part, through the generation of elevated amounts of intracellular ROS (Benhar et al., 2001; Jackson and Loeb, 2001; Tobiume et al., 2001). Redox homeostasis of the cell is greatly dependent on pro-oxidant and antioxidant enzymes. ROS, such as superoxide anions (O2−) and hydrogen peroxide (H2O2) are produced by mitochondria, peroxisome, cytochrome P-450 and NADPH oxidase (D'Autreaux and Toledano, 2007). Superoxide anions are converted to H2O2 by the enzyme superoxide dismutase, considered to be a detoxification reaction. Catalase and glutathione peroxidase are enzymes that detoxify H2O2. Compared with mitochondria, peroxisome and cytochrome P-450, which generates ROS as normal metabolic by-products, specific enzymes such as NADPH oxidases generate ROS as a primary function (Bedard and Krause, 2007). Nox1 is a catalytic subunit of a NADPH oxidase complex initially identified in colonic adenocarcinoma cell lines (Banfi et al., 1999). Nox1 induces the production of low amount of superoxide and controls cell proliferation, apoptosis, migration and innate immune response (Morazzani et al., 2004; Rokutan et al., 2006; Sadok et al., 2008). Overexpression of Nox1 in colon seems to be related to tumour progression particularly in K-Ras mutated cells (Tominaga et al., 2007; Laurent et al., 2008). The impact of Nox1-dependent production on oxaliplatin efficiency has not yet been studied.
Colorectal cancer is frequently associated with high expression level of EGFRs (Salomon et al., 1995). The binding of a ligand to the extracellular domain of the receptor results in the phosphorylation of the tyrosine kinase domain. The activation of the receptor leads to the activation of intracellular effectors involved in mitogenic and survival pathways such as mitogen-activated protein kinases and phosphatidylinositol-3 kinase (PI3K/AKT) pathways. EGFR is also an upstream activator of Rac1-GTPase, a well-known activator of NADPH oxidase enzymes in different cell types (Sumimoto, 2008). Blockade of EGFR-mediated signalling pathways has been proposed as a potential therapeutic modality for metastatic colorectal cancer. Cetuximab (C-225, Erbitux) is a recombinant, human-murine chimeric IgG1 monoclonal antibody produced in mammalian cell culture and targeted specifically to EGFR. Although the rationale for targeting EGFR in cancer was initially oriented to directly affect signalling in tumour cells, the use of a monoclonal antibody has led to an unexpected therapeutic effect through the immune response, by antibody-dependent cell-mediated cytotoxicity (ADCC) (Iannello and Ahmad, 2005). To improve the efficiency of treatment, the combination of chemotherapy with biotherapy should present at least an additive effect through both tumour EGFR inhibition and ADCC-mediated toxicity.
The aim of this study was to evaluate the direct sensitivity of a panel of human colorectal tumour cell lines to treatment with oxaliplatin used alone and in combination with cetuximab. We found an antagonism when oxaliplatin was combined with cetuximab that was not observed in cells harbouring K-RasV12 mutation, used in this study. Our results showed that Nox1-dependent ROS production occurring through the stimulation of EGFR/Ras/Nox1 pathway is necessary for oxaliplatin cytotoxicity. Inhibition of the EGFR pathway by cetuximab leads to a decrease of oxaliplatin efficacy on tumour cells through a decreased availability of ROS.
Methods
Tumour cell lines and culture conditions
Four human colon carcinoma cell lines HT29-D4, Caco-2, SW480 and SW620 were routinely maintained in Dulbecco's modified Eagle's medium containing 10% foetal bovine serum (FBS) (GIBCO Cell Culture systems, Invitrogen, Paisley, UK), supplemented with 2 mM L-glutamine and 1% sodium pyruvate and were maintained at 37°C in a humidified atmosphere with 5% CO2. HT29-D4 cells originally derived from HT29 colon adenocarcinoma cell line (Fantini et al., 1986). None of the colorectal cell lines used were reported to present EGFR mutation, which is consistent with the absence of observed EGFR mutation in colorectal cancer (Lee et al., 2005). All experiments were performed in 1% FBS to maximize the EGFR ligand, amphiregulin, autocrine loop classically reported for colorectal cancer cells (Pichard et al., 2006).
Immunoblot
Cells were lysed in specific buffer (0.12 M Tris pH 6.8, SDS 3% and glycerol). Protein quantification was performed by the bicinchoninic acid assay (Interchim, Montluçon, France). Fifty micrograms of cellular protein lysate in Laemmli buffer was separated on 7% SDS-polyacrylamide gel and transferred onto nitrocellulose membrane. Membranes were incubated with polyclonal rabbit EGFR antibody (cell signalling technology, USA) and secondary anti-rabbit IgG peroxidase-linked antibody. Immunoblot were developed by an enhanced chemoluminescence detection system (ECL Amersham, Buckinghamshire, UK).
Flow cytometry
Epidermal growth factor receptor cell surface expression was evaluated by flow cytometry using cetuximab as primary antibody. The cells were counterstained with an Alexa Fluor 488 goat anti-human IgG (Invitrogen, France). All stainings were done on ice for 45 min followed by three washes in phosphate saline buffer. Following staining, the cell fluorescence was measured by using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). A total of 10 000 events were collected per sample. Each sample was performed in triplicate.
K-ras mutation status analysis on cell lines
DNA was extracted from cell lines pellets using the QIAamp DNA extraction kit (QIAGEN, Courtaboeuf, France) according to the manufacturer's instructions. K-ras exon 1 was PCR-amplified from tumour cells DNA using the following sense and antisense primers: 5′-AAGGCCTGCTGAAAATGACTG-3′ and 5′-CAAAGAATGGTCCTGCACCAG-3′. After purification using the QIAQuick PCR purification kit from QIAGEN, PCR-amplified K-ras exon 1 products were analysed for the presence of K-ras mutations at nucleotides nt.34, nt.35, nt.37 and nt.38, using the SNPstart Primer Extension kit (Beckman Coulter, Villepinte, France) and four primers, three of which including at their 5′ end, an additional variable poly-A chain allowing capillary electrophoresis size separation and their simultaneous detection. The sequences of the sense primers allowing the extension at nucleotides nt.34, nt.35, nt.37 and nt.38 were respectively, 5′-AACTTGTGGTAGTTGGAGCT-3′, 5′-(A)10 ACTTGTGGTAGTTGGAGCTG-3′, 5′-(A)20 TTGTGGTAGTTGGAGCTGGT-3′ and 5′-(A)30 TGTGGTAGTTGGAGCTGGTG-3′ (A indicating the additional nucleotides). The multiplex Single Base Extension reaction was performed in a final volume of 10 µL containing 100 fmol of the PCR reaction products, 4 µL of the SNPstart Master Mix and 2 µL of a mix of the four specific probes at a concentration of 1–2.5 µM. Cycling conditions were 25 cycles at 90°C for 10 s and 45°C for 20 s. Single Base Extension products were then treated for 0.5 h at 37°C with 0.25 U of shrimp alkaline phosphatase (Euromedex, Souffelweyersheim, France). After heat inactivation of the alkaline phosphatase for 15 min at 65°C, labelled products were separated by using a 16 min run on an CEQ 8000 sequencer, and data were analysed using the GenomeLab algorithm software (Beckman Coulter).
Cytotoxicity assay
Tumour cells were seeded on day 1 in 96-well plates at a density of 5 × 103 cells per well in order to be in the exponential phase of growth during the time course of experiment. Preliminary experiments has been performed to determine the linear log phase for each cell lines based on cell count after 24, 48 and 72 h with different initial cell number. The number of cells at the end of linear log phase was around 50 000 cells for Caco-2 cells and 100 000 cells for HT29-D4, SW480 and SW620 cells (data not shown). Cells were incubated on day 2 for 72 h with various concentrations of drugs. The effect of drugs alone on cell viability was evaluated at concentrations ranging from 0.1 to 100 µg·mL−1 for cetuximab and from 1 to 100 µM for oxaliplatin. A preliminary set of experiment showed that cetuximab induced only a weak effect on cell viability and proliferation, limiting the classical use of the Chou and Talalay methods for combination analysis (Chou and Talalay, 1984). Thus, combination effect was evaluated by using a fixed cetuximab concentration of 100 µg·mL−1 combined with oxaliplatin concentration ranging from 1 to 100 µM. Cetuximab was administered 15 min before oxaliplatin. Cell viability was evaluated by the reduction of methylthiazoletetrazolium to formazan (0.5 mg·mL−1). The absorbance of each well was measured at 600 nm with a multiskan spectrophotometer (Labsystems, France). Results are expressed as percentage of viable cells compared with untreated cells (which have 100% viability). The results are based on three independent experiments. Drug concentrations that inhibit 50% of cell viability (IC50) for oxaliplatin were determined by using the Chou and Talalay method (Chou and Talalay, 1981).
Transfections
The Nox1 short hairpin RNA (shRNA) corresponds to the following sequence ATATAGGCCACCAGCTTGTTGATATCCGCAAGCTGGTGGCCTATATG cloned in the pRNATH1.1/Neo expression vector (Genscript Corporation, NJ, USA) and was previously validated (Sadok et al., 2008). The same vector without insert was used as transfection control. N-terminal 3x-haemagglutinin tagged human Ras family small GTP binding protein K-Ras (G12V mutant) cloned into pcDNA3.1+ expression vector was obtained from Missouri S&T cDNA Resource Center (Rolla, USA). PcDNA3.1+ expression vector without insert was used as transfection control. HT29-D4 cells were cultured as previously described and subsequently transfected by amaxa nucleofector, according to the manufacturer's protocol. Transfection efficiency was usually over 90% as analysed by flow cytometry. Transfection was confirmed by immunoblot with goat anti-HA antibody for K-RasV12 and goat anti-Nox1 antibody, for Nox1 shRNA.
Measurement of ROS
Reactive oxygen species generation was measured by either lucigenin chemiluminescence or dichlorodihydrofluorescein diacetate (H2-DCFDA) fluorescence, detecting O2− anions and H2O2 respectively. After incubation of cells for the desired time with drugs in 96-well plates, luminescence was detected by a Fluoroskan Ascent FL fluorimeter (Labsystems, France). The detected signal was assessed each minute over the course of 60 min. Results represent the integration of the signal for 60 min. For H2-DCFDA ROS measurements, regular culture medium was replaced by measurement buffer containing 10 µM of H2-DCFDA for 30 min. Cells were then rinsed with measurement buffer without H2-DCFDA, and fluorescence was measured at 490 nm for excitation and 538 nm for emission with the Fluoroskan Ascent FL fluorimeter (Labsystems, France). All measurements were performed at 37°C. Results represent the percentage variation relative to untreated control.
Statistics
Student's t-test was used for comparison of IC50 and ROS levels. The level of significance was set at P= 0.05.
Materials
Oxaliplatin (5 mg·mL−1) was dissolved in phosphate-buffered saline to prepare a 100 µM stock solution. Cetuximab (2 mg·mL−1) was kindly provided by Merck Laboratory (Darmstadt, Germany). For in vitro experiments, stock solutions of drugs were diluted in phosphate-buffered saline. As oxaliplatin/Cetuximab is usually associated to 5FU, we evaluated the impact of a dose 100 µM of 5FU (IC30) on the dose effect of oxaliplatin plus or minus 100 µg·mL−1 of Cetuximab on HT29-D4 cell viability. IC50 for oxaliplatin in this experimental conditions equal 2,8 ± 0,4 and 0,9 ± 0,1 µM with and without Cetuximab, respectively. These data suggest that oxaliplatin/Cetuximab antagonism was still observed in presence of 5FU.
Results
EGFR expression and K-Ras mutation status of tumour cell lines
Total and surface EGFR expression was accessed by immunoblot using polyclonal rabbit EGFR antibody and flow cytometry analysis on non-permeabilized cells using cetuximab as primary antibody respectively. As shown in Figure 1A and B, over the four tested colorectal tumour cell lines total and surface expression was positive in HT29-D4 and Caco-2 cells. No EGFR expression was detected in SW620 cells while SW480 cells showed an EGFR-positive expression by immunoblot but no EGFR surface expression (Figure 1B). Because other studies reported membrane EGFR expression in SW480, one possible explanation would involve the absence of recognition of EGFR in this cell. Further studies will be needed to clarify that point. As K-Ras mutation on the codon 12 was observed for SW480 and SW620 cells but not for HT29-D4 and Caco-2 cells (Figure 1C), we checked whether K-RasV12 mutation would be responsible for the altered EGFR expression observed in SW480 and SW620 cells. K-RasV12 overexpression in HT29-D4 cells did modify neither total nor surface EGFR expression level (data not shown). These results are summarized in Table 1.
Figure 1.
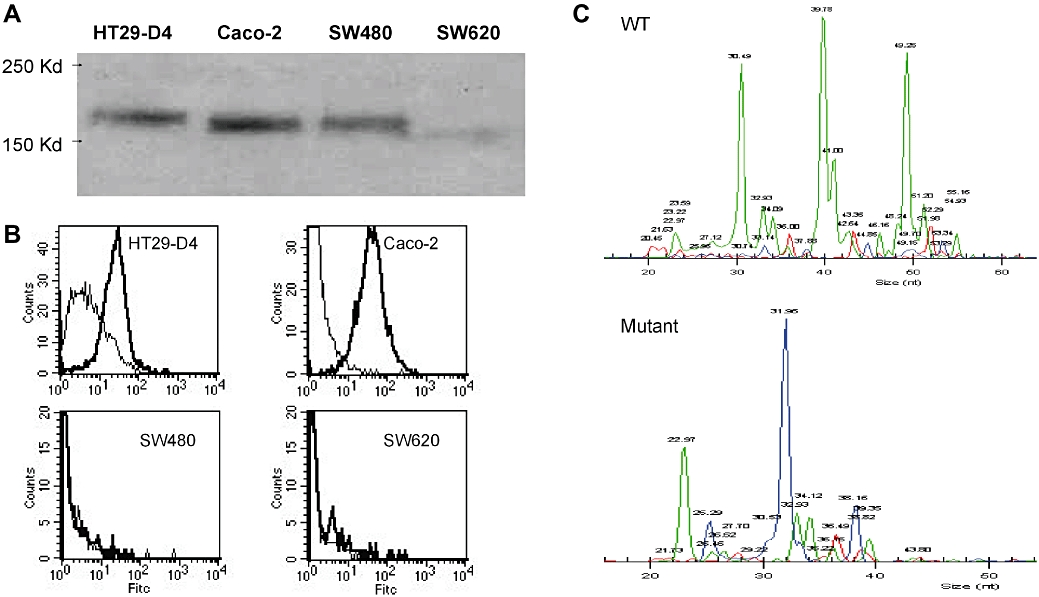
(A) Epidermal growth factor receptor protein expression was detected by immunoblotting cell lysates from four colon cancer cell lines HT29-D4, Caco-2, SW480 and SW620. (B) Epidermal growth factor receptor cell surface expression was measured by flow cytometry. Cells (5 × 105) were incubated with cetuximab as primary antibody and counterstained with an Alexa Fluor 488 goat anti-human IgG. All staining were done on ice for 45 min followed by three washes. For each cell line, a control without primary antibody was performed. (C) Detection by SNaPShot of K-Ras mutations on cell lines. Each peak corresponds to a specific extended primer. Wild type (WT) for HT29-D4 and Caco-2 (upper panel); K-Ras mutation for SW480 and SW620 (lower panel).
Table 1.
K-Ras and epidermal growth factor receptor (EGFR) status for the colorectal cell lines studied
| EGFR total expression | EGFR membrane expression | K-Ras mutation status | |
|---|---|---|---|
| HT29-D4 | + | + | − |
| Caco-2 | + | + | − |
| SW480 | + | − | + |
| SW620 | − | − | + |
Effect of cetuximab and oxaliplatin on viability of colorectal tumour cell lines
Treatment of cells with doses of cetuximab alone ranging from 0.1 to 100 µg·mL−1 showed a maximal effect on cell viability of 30–40% on cell lines with EGFR expression without K-Ras mutation (HT29-D4 and Caco-2). No inhibition of viability was observed with cetuximab treatment in K-Ras mutated cells that did not present surface EGFR (SW480 and SW620) (Figure 2A). The effective concentrations of cetuximab used in our study were similar to those previously used in vitro showing a maximal effect of cetuximab on colorectal cell line between 20 and 100 µg·mL−1 (Balin-Gauthier et al., 2006; Di Nicolantonio et al., 2008).
Figure 2.
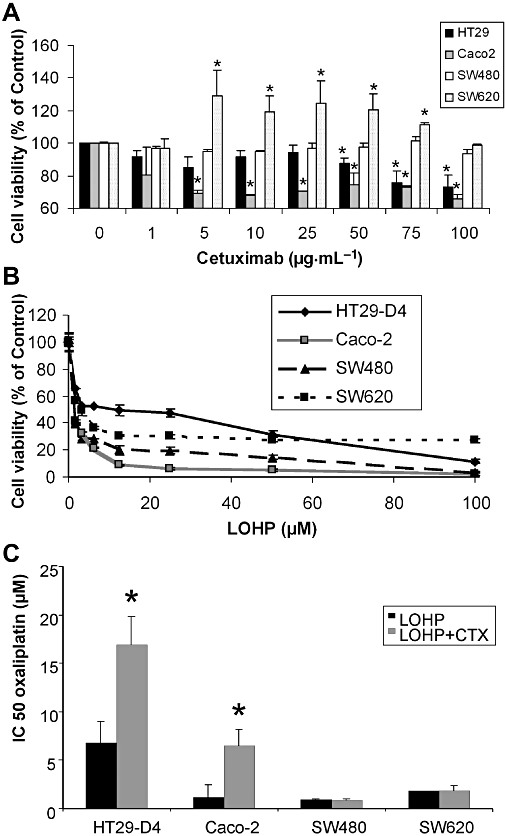
In vitro effects of a single agent, cetuximab (CTX) or oxaliplatin (LOHP), on a panel of human colorectal carcinoma cell lines. (A) Dose–response curves of cells treated with cetuximab alone at concentration ranging from 0.1 to 100 µg·mL−1 for 72 h on each cell lines using methylthiazoletetrazolium (MTT) assays. Results were presented as means ± SEM of three independent experiments. (B) Concentration–response curves of cells treated with oxaliplatin alone at concentrations ranging from 1 to 100 µM for 72 h using MTT assays. Data are expressed as mean ± SEM of three independent experiments. *P < 0.05. (C) IC50 for oxaliplatin combined with cetuximab in the panel of human colorectal carcinoma cell lines. Cells were treated with oxaliplatin at concentration ranging from 1 to 100 µM combined with a fixed cetuximab concentration of 100 µg·mL−1. Cetuximab was added 15 min before oxaliplatin. Growth inhibition was evaluated by using MTT assay. Data are expressed as mean ± SEM of three independent experiments.
Viability of cell lines treated with oxaliplatin alone (1–100 µM) is shown in Figure 2B, with the derived IC50 values in Figure 2C. All four cell lines HT29-D4, Caco-2, SW480 and SW620 were sensitive to oxaliplatin.
For the combined treatment, we choose a fixed concentration of 100 µg·mL−1 of cetuximab applied 15 min before oxaliplatin treatment. Cetuximab treatment did not significantly modify the effect of oxaliplatin on cell viability in K-RasV12 mutated cells, whether they expressed EGFR or not (SW480 and SW620) (Figure 2C). In contrast, cetuximab combined with oxaliplatin significantly increased the IC50 of oxaliplatin in HT29-D4 and in Caco-2 cells (Figure 2C; P < 0.05) compared with oxaliplatin alone. Cetuximab is a humanized monoclonal antibody directed against EGFR also named Herl or ErbBl according to the Guide to Receptors and Channels (Alexander et al., 2008).
Effect of RasV12 mutation on cetuximab/oxaliplatin antagonism
As shown in Figure 2C, cetuximab did not increase the IC50 for oxaliplatin, that is, antagonize oxaliplatin, in the K-RasV12 mutated cell lines, SW480 and SW620. Previous studies have showed that K-Ras mutation impairs responses to cetuximab (Di Fiore et al., 2007). We therefore studied the effect of K-RasV12 mutation in HT29-D4 cells on the efficacy of oxaliplatin, cetuximab and their combination. Transfection of HT29-D4 with HA-tagged K-RasV12 significantly decreased oxaliplatin IC50 compared with that in control HT29-D4 cells (P < 0.05; Figure 3B). Addition of cetuximab did not affect the IC50 for oxaliplatin in HT29-D4 transfected with K-RasV12, compared with oxaliplatin alone (Figure 3A and B).
Figure 3.
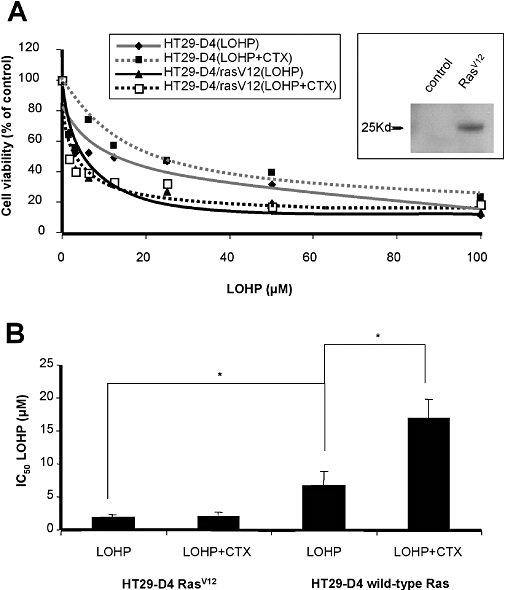
(A) In vitro effects of oxaliplatin (LOHP) combined with cetuximab (CTX) in HT29-D4 control cells compared with HT29-D4 cells transfected with HA-tagged RasV12. Insert shows immunoblot for HA in transfected HT29-D4 cells. (B) IC50 values for oxaliplatin alone or combined with cetuximab on RasV12-transfected HT29-D4 cells compared with HT29-D4 control cells. *P < 0.05.
Impact of cetuximab, oxaliplatin and combination on ROS production
The EGFR pathway is known to stimulate ROS production through NADPH oxidase activation (Juarez et al., 2008). The intracellular availability of oxaliplatin is known to be affected by glutathione metabolism, and ROS production is needed for efficient cytotoxic activity of oxaliplatin (Laurent et al., 2005). We thus assessed the contribution of redox metabolism to the cetuximab/oxaliplatin antagonism. Exposure of HT29-D4 cells to 100 µg·mL−1 of cetuximab decreased O2− production by 90% and H2O2 production by 50%, compared with control cells (Figure 4A and B). These observations were consistent with an inhibition of the production of O2− and also a limitation of the dismutation of O2− to H2O2. In HT29-D4 cells exposed to various concentration of oxaliplatin, a dose-dependent decrease of O2− production and a concomitant increase of H2O2 production were observed (Figure 4C and D). These results suggest that oxaliplatin by a yet unidentified mechanism accelerated O2− dismutation. When cetuximab was combined with oxaliplatin on HT29-D4 cells, O2− production was decreased compared with untreated cells (Figure 4E), as already observed in cells exposed to oxaliplatin or cetuximab alone. In contrast, the increase in H2O2 production induced by oxaliplatin was prevented by adding cetuximab (Figure 4F). This correlation of redox status modulation by cetuximab, oxaliplatin or their combination with effects on cell viability was further evaluated.
Figure 4.
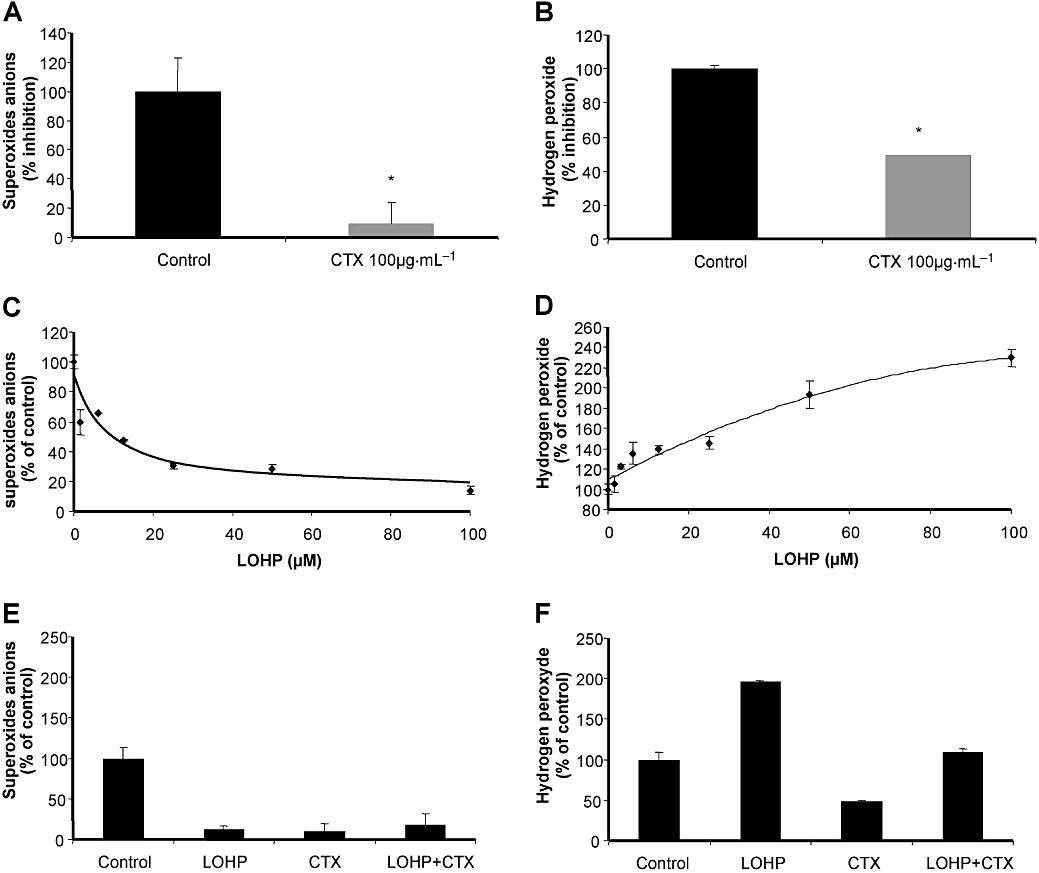
Production of O2− and H2O2 in HT29-D4 cells exposed to cetuximab (CTX) or oxaliplatin (LOHP). Production of O2− was determined by lucigenin and H2O2 production by DCFDA. Data from at least three independent experiments have been pooled. (A,B) Cetuximab significantly decreased O2− production and affected significantly the production of H2O2 compared with untreated cells (*P < 0.05). (C,D) Effects of oxaliplatin at concentrations ranging from 1 to 100 µM on O2− and H2O2 production. (E,F) Production of O2− and H2O2 in human colon carcinoma cells exposed to cetuximab, oxaliplatin or combination of the two drugs. Cells were treated with a fixed oxaliplatin concentration of 100 µM combined with a fixed cetuximab concentration of 100 µg·mL−1. Cetuximab was added 15 min before oxaliplatin. Cetuximab, oxaliplatin and the combination of both significantly decreased O2− production compared with untreated cells (P < 0.05). Oxaliplatin significantly increased H2O2 production, whereas cetuximab alone or combined with oxaliplatin decreased H2O2 production compared with untreated cells (P < 0.05).
Cetuximab/oxaliplatin combination efficiency relies on Nox1-dependent ROS production
Nox1, a homologue of the gp91phox, the catalytic moiety of the NADPH oxidase, increases O2− production and further dismutation to H2O2 in colorectal cancer cell lines. In addition, K-Ras is a known upstream modulator of Nox1 and is associated with tumourigenesis in colon (Laurent et al., 2008). Nox1 was expressed in the four cell lines tested although to a differing extent with HT29-D4 and SW480 cells expressing more Nox1 than Caco-2 and SW620 cells (Figure 5A). Nox1-dependent NADPH oxidase has been reported as the major source of superoxide in HT29 cells (Gianni et al., 2008; de Carvalho et al., 2008). We have thus evaluated Nox1 involvement in oxaliplatin-induced modulation of ROS in HT29-D4 and Caco-2 cells. We found that oxaliplatin was significantly less efficient on HT29-D4 and Caco-2 transfected with Nox1 shRNA compared with shRNA control (Figure 5B upper and lower panel respectively). Transfection of HT29-D4 with specific Nox1 shRNA markedly increased oxaliplatin IC50.
Figure 5.
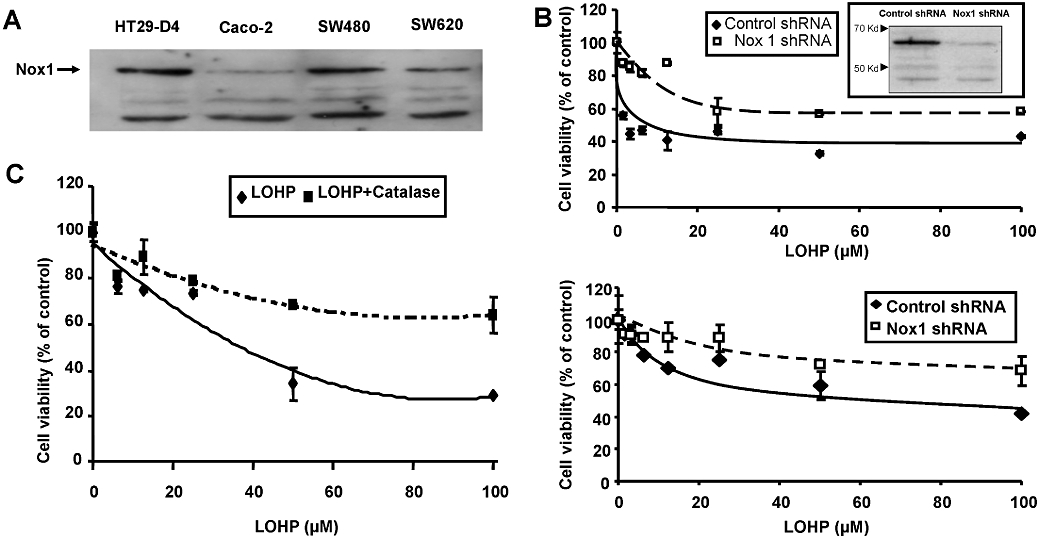
(A) Nox1 protein expression detection by immunoblot in HT29-D4, Caco-2, SW480 and SW620 cell lines. (B) In vitro effect of oxaliplatin in HT29-D4 (upper panel) and Caco-2 cells (lower panel) transfected with Nox1 short hairpin RNA (shRNA) compared with control shRNA; Immunoblot for Nox1 in HT29-D4 cells transfected with indicated shRNA; blot is representative of three independent experiments. (C) In vitro effect of oxaliplatin in HT29-D4 with or without catalase used at non-cytotoxic concentrations.
As oxaliplatin increased the level of H2O2, we used catalase to decrease the intracellular levels of H2O2 to confirm the involvement of H2O2 in oxaliplatin efficacy. Catalase was used on HT29-D4 cells at a concentration of 200 UI·mL−1, a concentration that decreased H2O2 levels without affecting cell proliferation or viability (data not shown) and was found to inhibit the cytotoxic effects of oxaliplatin. Cell viability was only decreased by 30% in presence of oxaliplatin (100 µM) when combined with catalase at 200 UI·mL−1, compared with 70% inhibition for oxaliplatin alone (Figure 5C).
Discussion
The study presented here demonstrated that combination of cetuximab with oxaliplatin produced antagonistic interactions in colonic adenocarcinoma cells expressing EGFR and wild-type K-Ras (HT29-D4 and Caco-2 cells). This antagonism was not observed in colonic adenocarcinoma cells carrying K-RasV12 mutation, whether they expressed EGFR or not (SW480 and SW620). Moreover, such antagonism was not seen in HT29-D4 cells transfected with K-RasV12 (Figures 2C and 5B). We showed that such antagonism was linked to inhibition by cetuximab, of Nox1-dependent ROS production, which impaired oxaliplatin efficiency.
The combination of targeted therapy, cetuximab, with chemotherapy, irinotecan, provides an improvement in the treatment of colorectal metastatic cancer. However, Cunningham et al. showed only 20% of objective response, suggesting there is a clear need for new and improved therapies. Oxaliplatin is the first platinum-based compound to show efficacy in the treatment of colorectal cancer. Its use in combination with cetuximab for metastatic colorectal cancer is under evaluation in numerous studies and the preliminary results seem controversial, especially for patients with K-Ras mutations in their tumours (Borner et al., 2008). Indeed, a recent trial evaluating the efficacy of cetuximab plus oxaliplatin as second-line therapy was stopped early after the interim analysis, because of the lack of response (no objective clinical response) (Vincenzi et al., 2006). More recently, the OPUS trial showed an increase of survival for the oxaliplatin/cetuximab combination in metastatic colorectal cancer patients harbouring K-Ras wild type and a significant decrease of survival in patients harbouring K-Ras mutations (Bokemeyer et al., 2009). Thus, the rationale for such a combination is still under evaluation.
At the clinical level, the effect of cetuximab is said to depend on two different mechanisms: a direct action on tumour cell signal transduction, limiting proliferation or increasing apoptosis, and a cytotoxic activity mediated by the microenvironment through ADCC. To improve the efficiency of treatment, the combination of chemotherapy with monoclonal antibody-targeted therapy should present at least an additive effect through both tumour signalling and ADCC-mediated toxicity. Our report has focused on the direct effect of the combination on signal transduction in tumour cells, considering also Ras mutations and their redox status. As previously reported (Balin-Gauthier et al., 2006), our study showed that cetuximab treatment alone has little effect on cell viability in vitro in cell lines. These results are also consistent with the fact that cetuximab should be used in combination with other drugs during therapy (Cunningham et al., 2004). Our data showed however that the effect of cetuximab on viability of the cell lines was dependent on K-Ras mutation status. Cell lines harbouring wild-type K-Ras expression were sensitive (although only moderately) to EGFR inhibition by cetuximab, while cell lines carrying K-RasV12 mutation were insensitive (Figure 2A). Recent clinical evidence showed that all patients with metastatic colorectal cancer having activating K-Ras mutations were resistant to cetuximab treatment combined with irinotecan (Lievre et al., 2006; Di Fiore et al., 2007). Because Ras is a major downstream target of EGFR, Ras activating mutations impair cell sensitivity to EGFR inhibition. Our present report showed that cetuximab antagonized the cytotoxicity of oxaliplatin, when combined with oxaliplatin in cancer cell lines expressing wild-type Ras. Only one previous study has analysed the in vitro effects of cetuximab combined with oxaliplatin (Balin-Gauthier et al., 2006). While two cell lines tested (HCT-8 and HT29) where responsive to the cetuximab/oxaliplatin combination, two other cell lines (HCT-116 and SW620) were unresponsive. They suggested that cetuximab synergized with oxaliplatin on tumour xenografts but this in vivo data probably involved an immune response. They concluded that the anti-proliferative effect of cetuximab observed in vitro cannot fully explain its anti-tumour activity. As SW620 and HCT-116 cells have activating Ras mutations, while HCT-8 and HT29 cells have wild-type Ras expression, these results are compatible with our present findings that Ras mutation impairs the direct effect of cetuximab on tumour cell signalling. As observed here for cetuximab, a combination of oxaliplatin with gefitinib, an EGFR tyrosine kinase inhibitor, led to antagonistic or synergistic activity depending on the cell lines used (Ciardiello et al., 2000; Van Schaeybroeck et al., 2005). The oxaliplatin/gefitinib combination was antagonistic in the K-Ras mutated cell line HCT-116, suggesting that mechanisms other than K-Ras mutation also led to an antagonism between oxaliplatin and EGFR inhibitors. HCT-116 cells are also mutated on PIK3CA leading to a constitutive activation of PI3K (Wang et al., 2007). Apart from K-Ras, PI3K is another major downstream EGFR signalling intermediate (see Figure 6 for details), and the PIK3CA mutation has been associated with resistance to EGFR-targeted monoclonal antibody (Sartore-Bianchi et al., 2009). However, gefitinib efficacy seems largely linked to EGFR mutation that is not the case for cetuximab and suggests a different mechanism of action. Finally, oxaliplatin and cetuximab are widely used in combination with 5FU in the treatment of metastatic colorectal cancer. Under our experimental conditions, the antagonism between oxaliplatin and cetuximab was still expressed in the presence of 100 µM of 5FU (IC30), suggesting that this mechanism might be relevant in clinical treatments for colorectal cancer, combining these three drugs. We thus further delineate the mechanism by which cetuximab can affect oxaliplatin efficacy.
Figure 6.
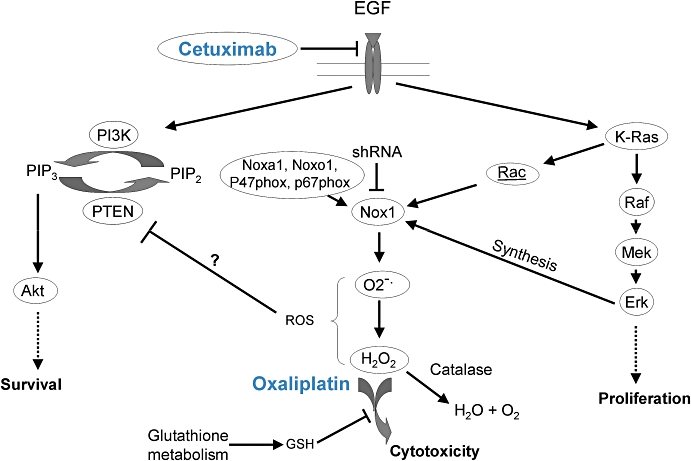
Scheme of possible mechanisms underlying the antagonism between cetuximab and oxaliplatin, involving modulation of redox status. CTX, cetuximab; EGF, epidermal growth factor; GSH, glutathione; PI3K, phosphatidylinositol-3 kinase; PIP2, phosphatidylinositol bisphosphate; PIP3, phosphatidylinositol trisphosphate; PTEN, phosphatase and TENsin homologue, ROS, reactive oxygen species; shRNA, short hairpin RNA.
The beneficial role of ROS production on chemotherapeutic effectiveness is an emerging concept (Doroshow, 2006). Limitation of ROS production or increase of ROS elimination has been identified as one of the resistance factors to chemotherapy (Alexandre et al., 2006). Oxaliplatin resistance was known to be linked to glutathione S-transferase (GST) activity and glutathione metabolism, a known regulator of redox homeostasis (Godwin et al., 1992; El-Akawi et al., 1996). In HT29-D4 cells exposed to various concentrations of oxaliplatin, a dose-dependent decrease of O2− production and a concomitant increase of H2O2 production were observed (Figure 3C and D). We showed that cetuximab significantly decreased ROS production (O2− anions and H2O2) through the blockade of the EGFR pathway (Figure 3A and B). Combination of cetuximab with oxaliplatin inhibited the increase of H2O2 induced by oxaliplatin alone (Figure 4). Our results are consistent with a previous study showing a major involvement of H2O2 production in oxaliplatin cytotoxicity. Laurent et al. (2005) showed that incubating tumour cells with oxaliplatin in association with increasing concentration of NAC resulted in decreased H2O2 production and a dose-dependent decrease in the cytotoxic action of oxaliplatin. In contrast, superoxide dismutase mimetics that increase H2O2 level by dismutation of superoxide also increased oxaliplatin cytotoxicity (Laurent et al., 2005). Consequently, ROS modulation could also explain the antagonism observed when oxaliplatin was combined with cetuximab.
Investigation of the mechanism of the observed antagonism in this study has led to several novel observations. We showed that ROS modulation involved Nox1 that is most highly expressed in colon epithelium (Bedard and Krause, 2007). Nox1 is a major source of superoxide production in many colonic epithelial cells. We previously described that Nox1 knockout with specific shRNA decreased superoxide production by 90% in HT29-D4 cells (de Carvalho et al., 2008; Sadok et al., 2008). EGFR is known to induce ROS production through Nox1 pathways (Morazzani et al., 2004; Park et al., 2006), and cetuximab inhibited O2− production in HT29-D4 cell to the same extent as the Nox1 shRNA. Concentration-dependent effects of oxaliplatin on Nox1 knockout HT29-D4 or Caco-2 cells showed a decreased cytotoxicity compared with control cells. These results are consistent with a Nox1 inhibition by cetuximab mediating the observed antagonism for the oxaliplatin/cetuximab combination. Nox1 inhibition leads to a decreased O2− production and a consequently decreased H2O2 production, limiting oxaliplatin efficacy. Finally, we showed that oxaliplatin/cetuximab antagonism is maintained in presence of 5FU. As 5FU has been reported to induce ROS production that is needed for its effects (Hwang, 2007), the mechanism of the redox-dependent antagonism reported in this report might be even more pronounced for the 5FU/oxaliplatin/cetuximab combination.
Nox1 expression level is under the control of K-Ras activation, and activating Ras mutations induce an up-regulation of Nox1 expression level in fibroblasts and participated in the transformation and tumourigenic phenotype downstream of Ras (Mitsushita et al., 2004). We found that transfection of HT29-D4 cells with K-RasV12 suppressed cetuximab/oxaliplatin antagonism (Figure 5B). This result is consistent with our observation that the K-Ras mutated cell lines (SW480, SW620) were insensitive to EGFR inhibition by cetuximab. Laurent et al. (2008) recently suggested that an increased Nox1 expression level is associated with colorectal tumour progression in patients harbouring K-Ras activating mutation. However Nox1 activity is not directly linked to Nox1 expression level but also depends on different cytosolic activators such as Noxa1, Noxo1 and Rac1. Nox1 expression level in HT29-D4 cells is not a limiting factor for Nox1-dependent O2− production, which mainly depends on limiting cytosolic activators (de Carvalho et al., 2008). The K-Ras/Raf pathway controls Nox1 at different levels, so K-Ras might stimulate Nox1 activity by increasing Rac1-GTP levels or increase Nox1 expression level through Raf stimulation (Adashi et al., 2008). The HT29 cells harbour the B-Raf V600E activating mutation. These data are consistent with low expression of Nox1 in cells harbouring wild-type B-Raf and K-Ras (Caco-2) and high Nox1 expression in cells harbouring mutated B-Raf or K-Ras (HT29-D4, SW480 or SW620). The fact that HT29-D4 cells were sensitive although only moderately to cetuximab alone suggests that stimulation of Nox1 activity is more important than the increase in Nox1 expression level by the Raf pathway. This data were supported by the fact that in Caco-2 cells expressing low Nox1 level and having comparable Nox1-dependent ROS production to HT29-D4 cells, the knock-down of Nox1 by the Nox1 shRNA induced a comparable decrease in oxaliplatin efficiency in both cell lines compared with control shRNA.
Finally, we showed that oxaliplatin had a significantly higher cytotoxic effect (lower IC50) when cells were transfected with K-RasV12. This result was consistent with an earlier study from Vekris et al. (2004) showing that oxaliplatin was more active in cell lines with a mutation in one of the Ras genes, whereas there was no correlation between Ras mutation and the activity of the other platinum compounds. This may explain why oxaliplatin is active against colon cancers, which frequently exhibit a Ras mutation. We have recently started a study on 50 patients with metastatic colorectal cancer benefiting from oxaliplatin-based treatment, seeking to correlate K-Ras mutation with benefit from oxaliplatin treatment. We found a trend (Fisher's exact test; P > 0.05) towards a better response in K-Ras mutated patients with 62.5% of controlled disease (response + stable) in K-Ras mutated patients compared with 50% in K-Ras wild-type patients. A prospective study with a larger sample, taking into account other major mutations observed in colorectal cancer (B-Raf, PIK3CA and EGFR) and studying Nox1 activity in addition to Nox1 expression level might be necessary to establish significance for this result.
To conclude, we present here an experimental study showing an antagonism between oxaliplatin and cetuximab involving modulation of the cellular redox status, through Nox1-dependent ROS production in non-mutated K-Ras colorectal cancer cell lines.
Acknowledgments
We are grateful for the following Grants: Association de la Recherche contre le Cancer (Doctoral fellowship to A Sadok and L Dahan), Institut National du Cancer R07157AA (H Kovacic), AORC Assistance Publique des Hôpitaux de Marseille (JF Seitz).
Glossary
Abbreviations:
- 5FU
5-fluorouracil
- ADCC
antibody-dependent cell-mediated cytotoxicity
- CTX
cetuximab
- EGFR
epidermal growth factor receptor
- FBS
foetal bovine serum
- GST
glutathione S-transferase
- H2-DCFDA
dichlorodihydrofluorescein diacetate
- LOHP
oxaliplatin
- MTT
methylthiazoletetrazolium
- PI3K
phosphatidylinositol-3 kinase
- PIP2
phosphatidylinositol bisphosphate
- PIP3
phosphatidylinositol trisphosphate
- PTEN
phosphatase and TENsin homologue
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
References
- Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene. 2008;27:4921–4932. doi: 10.1038/onc.2008.133. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd) 2008;153(Suppl 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre J, Nicco C, Chereau C, Laurent A, Weill B, Goldwasser F, et al. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98:236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- Balin-Gauthier D, Delord JP, Rochaix P, Mallard V, Thomas F, Hennebelle I, et al. In vivo and in vitro antitumor activity of oxaliplatin in combination with cetuximab in human colorectal tumor cell lines expressing different level of EGFR. Cancer Chemother Pharmacol. 2006;57:709–718. doi: 10.1007/s00280-005-0123-3. [DOI] [PubMed] [Google Scholar]
- Banfi B, Schrenzel J, Nusse O, Lew DP, Ligeti E, Krause KH, et al. A novel H(+) conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J Exp Med. 1999;190:183–194. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becouarn Y, Ychou M, Ducreux M, Borel C, Bertheault-Cvitkovic F, Seitz JF, et al. Phase II trial of oxaliplatin as first-line chemotherapy in metastatic colorectal cancer patients. Digestive Group of French Federation of Cancer Centers. J Clin Oncol. 1998;16:2739–2744. doi: 10.1200/JCO.1998.16.8.2739. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol. 2001;21:6913–6926. doi: 10.1128/MCB.21.20.6913-6926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- Borner M, Koeberle D, Von Moos R, Saletti P, Rauch D, Hess V, et al. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first-line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Ann Oncol. 2008;19:1288–1292. doi: 10.1093/annonc/mdn058. [DOI] [PubMed] [Google Scholar]
- de Carvalho DD, Sadok A, Bourgarel-Rey V, Gattacceca F, Penel C, Lehmann M, et al. Nox1 downstream of 12-lipoxygenase controls cell proliferation but not cell spreading of colon cancer cells. Int J Cancer. 2008;122:1757–1764. doi: 10.1002/ijc.23300. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem. 1981;115:207–216. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6:2053–2063. [PubMed] [Google Scholar]
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- Doroshow JH. Redox modulation of chemotherapy-induced tumor cell killing and normal tissue toxicity. J Natl Cancer Inst. 2006;98:223–225. doi: 10.1093/jnci/djj065. [DOI] [PubMed] [Google Scholar]
- El-Akawi Z, Abu-hadid M, Perez R, Glavy J, Zdanowicz J, Creaven PJ, et al. Altered glutathione metabolism in oxaliplatin resistant ovarian carcinoma cells. Cancer Lett. 1996;105:5–14. doi: 10.1016/0304-3835(96)04245-0. [DOI] [PubMed] [Google Scholar]
- Fantini J, Abadie B, Tirard A, Remy L, Ripert JP, el Battari A, et al. Spontaneous and induced dome formation by two clonal cell populations derived from a human adenocarcinoma cell line, HT29. J Cell Sci. 1986;83:235–249. doi: 10.1242/jcs.83.1.235. [DOI] [PubMed] [Google Scholar]
- Gianni D, Bohl B, Courtneidge SA, Bokoch GM. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol Biol Cell. 2008;19:2984–2994. doi: 10.1091/mbc.E08-02-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin AK, Meister A, O'Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IT, Chung YM, Kim JJ, Chung JS, Kim BS, Kim HJ, et al. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem Biophys Res Commun. 2007;359:304–310. doi: 10.1016/j.bbrc.2007.05.088. [DOI] [PubMed] [Google Scholar]
- Iannello A, Ahmad A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev. 2005;24:487–499. doi: 10.1007/s10555-005-6192-2. [DOI] [PubMed] [Google Scholar]
- Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Juarez JC, Manuia M, Burnett ME, Betancourt O, Boivin B, Shaw DE, et al. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc Natl Acad Sci USA. 2008;105:7147–7152. doi: 10.1073/pnas.0709451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AT, Wang Y, Chiu JF. Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J Cell Biochem. 2008;104:657–667. doi: 10.1002/jcb.21655. [DOI] [PubMed] [Google Scholar]
- Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65:948–956. [PubMed] [Google Scholar]
- Laurent E, McCoy JW, 3rd, Macina RA, Liu W, Cheng G, Robine S, et al. Nox1 is over-expressed in human colon cancers and correlates with activating mutations in K-Ras. Int J Cancer. 2008;123:100–107. doi: 10.1002/ijc.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Soung YH, Kim SY, Park WS, Nam SW, Lee JY, et al. Absence of EGFR mutation in the kinase domain in common human cancers besides non-small cell lung cancer. Int J Cancer. 2005;113:510–511. doi: 10.1002/ijc.20591. [DOI] [PubMed] [Google Scholar]
- Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64:3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- Morazzani M, de Carvalho DD, Kovacic H, Smida-Rezgui S, Briand C, Penel C. Monolayer versus aggregate balance in survival process for EGF-induced apoptosis in A431 carcinoma cells: implication of ROS-P38 MAPK-integrin alpha2beta1 pathway. Int J Cancer. 2004;110:788–799. doi: 10.1002/ijc.20198. [DOI] [PubMed] [Google Scholar]
- Park HS, Park D, Bae YS. Molecular interaction of NADPH oxidase 1 with betaPix and Nox Organizer 1. Biochem Biophys Res Commun. 2006;339:985–990. doi: 10.1016/j.bbrc.2005.11.108. [DOI] [PubMed] [Google Scholar]
- Pichard V, Berthois Y, Roccabianca M, Prevot C, Sarrazin M, Portugal H, et al. Concomitant cell growth and differentiation are dependent on erbBl and integrin activation in an autonomously surviving colon adenocarcinoma: involvement of autocrine amphiregulin secretion. Anticancer Res. 2006;26:2769–2783. [PubMed] [Google Scholar]
- Rokutan K, Kawahara T, Kuwano Y, Tominaga K, Sekiyama A, Teshima-Kondo S. NADPH oxidases in the gastrointestinal tract: a potential role of Nox1 in innate immune response and carcinogenesis. Antioxid Redox Signal. 2006;8:1573–1582. doi: 10.1089/ars.2006.8.1573. [DOI] [PubMed] [Google Scholar]
- Sadok A, Bourgarel-Rey V, Gattacceca F, Penel C, Lehmann M, Kovacic H. Nox1-dependent superoxide production controls colon adenocarcinoma cell migration. Biochim Biophys Acta. 2008;1783:23–33. doi: 10.1016/j.bbamcr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. Febs J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K, Kawahara T, Sano T, Toida K, Kuwano Y, Sasaki H, et al. Evidence for cancer-associated expression of NADPH oxidase 1 (Nox1)-based oxidase system in the human stomach. Free Radic Biol Med. 2007;43:1627–1638. doi: 10.1016/j.freeradbiomed.2007.08.029. [DOI] [PubMed] [Google Scholar]
- Van Schaeybroeck S, Karaiskou-McCaul A, Kelly D, Longley D, Galligan L, Van Cutsem, et al. Epidermal growth factor receptor activity determines response of colorectal cancer cells to gefitinib alone and in combination with chemotherapy. Clin Cancer Res. 2005;11:7480–7489. doi: 10.1158/1078-0432.CCR-05-0328. [DOI] [PubMed] [Google Scholar]
- Vekris A, Meynard D, Haaz MC, Bayssas M, Bonnet J, Robert J. Molecular determinants of the cytotoxicity of platinum compounds: the contribution of in silico research. Cancer Res. 2004;64:356–362. doi: 10.1158/0008-5472.can-03-2258. [DOI] [PubMed] [Google Scholar]
- Vincenzi B, Santini D, Tonini G. Lack of response of cetuximab plus oxaliplatin in advanced colorectal cancer patients resistant to both oxaliplatin and cetuximab plus irinotecan. Ann Oncol. 2006;17:527–528. doi: 10.1093/annonc/mdj014. [DOI] [PubMed] [Google Scholar]
- Wang J, Kuropatwinski K, Hauser J, Rossi MR, Zhou Y, Conway A, et al. Colon carcinoma cells harboring PIK3CA mutations display resistance to growth factor deprivation induced apoptosis. Mol Cancer Ther. 2007;6:1143–50. doi: 10.1158/1535-7163.MCT-06-0555. [DOI] [PubMed] [Google Scholar]
- Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]