

Abstract
Exposure of the neonatal lung to chronic hypoxia produces significant pulmonary vascular remodeling, right ventricular hypertrophy (RVH), and decreased lung alveolarization. Given recent data suggesting that stem cells could contribute to pulmonary vascular remodeling and RVH, we tested the hypothesis that blockade of stromal derived factor-1 (SDF-1), a key stem cell mobilizer or its receptor, chemokine receptor 4 (CXCR4), would attenuate and reverse hypoxia-induced cardiopulmonary remodeling in newborn mice. Neonatal mice exposed to normoxia or hypoxia were randomly assigned to receive daily intra-peritoneal injections of normal saline (PL), AMD3100, or anti-SDF-1 antibody from postnatal day 1–7 (preventative strategy) or postnatal day 7–14 (therapeutic strategy). As compared to PL, inhibition of the SDF-1/CXCR4 axis significantly improved lung alveolarization, as well as decreased pulmonary hypertension, RVH, vascular remodeling, vascular cell proliferation and lung or RV stem cell expressions to near baseline values. We therefore conclude that the SDF-1/CXCR4 axis both prevents and reverses hypoxia-induced cardiopulmonary remodeling in neonatal mice, by decreasing progenitor cell recruitment to the pulmonary vasculature as well as by decreasing pulmonary vascular cell proliferation. These data offer novel insights into the role of the SDF-1/CXCR4 axis in the pathogenesis of neonatal hypoxia-induced cardiopulmonary remodeling and have important therapeutic implications.
Keywords: Pulmonary Hypertension, Hypoxia, Progenitor Cells, SDF-1, Vascular Remodeling
Introduction
Despite marked improvements in medical care, approximately 2 in 1000 neonates are perinatally exposed to intermittent or chronic periods of hypoxia1. Unlike that of the adult, the neonatal pulmonary vascular response to chronic hypoxic exposure is much more rapid and severe 2 and results in failure of the fetal circulation to adapt to one that supports postnatal life. This in turn contributes to the pathogenesis of persistent pulmonary hypertension (PH) of the newborn, chronic lung disease of prematurity and congenital heart disease. It is typically characterized by profound proliferation of smooth muscle and adventitial cells in the pulmonary vasculature, and abnormal extension of smooth muscle into peripheral arteries along with impairment in alveolar development in preterm neonates3, 4.
While the mechanisms underlying neonatal hypoxia-induced cardiopulmonary remodeling remain unclear, recent studies have suggested that stem cells may contribute to systemic as well as pulmonary vascular remodeling. We therefore sought to examine the role of a key stem cell mobilizer, the chemokine stromal derived factor-1 (SDF-1) and its receptor chemokine receptor 4 (CXCR4) in neonatal chronic hypoxia-induced cardiopulmonary remodeling.
SDF-1 or CXCL12 is a chemokine which is secreted by several tissues following exposure to hypoxia5, 6, in turn leading to the release of progenitor cells along a chemical gradient to the zone of tissue injury7–9. Its receptor CXCR4 is a G-protein coupled receptor that is widely expressed on several tissues, including endothelial cells, smooth muscle cells, monocytes, hematopoeitic and tissue committed stem cells 10–14. Binding of SDF-1 to CXCR4 induces several signal transduction pathways which regulate cell survival and proliferation15.
We therefore first tested the hypothesis that increased release of SDF-1 in chronically hypoxic neonatal mice would facilitate an increased number of progenitor cells in the pulmonary vasculature and right ventricle. Secondly, in order to establish the functional relevance of SDF-1 in this process, we tested the hypothesis that inhibition of SDF-1 or its cognate receptor CXCR4 would attenuate cardiopulmonary remodeling, by decreasing the recruitment of progenitor cells to the pulmonary vasculature and right ventricle as well as by decreasing vascular cell proliferation and apoptosis.
Methods and Materials
Animal Care and Treatment
Preventative Strategy
Sixty Eight FVB/NJ neonatal mice exposed to normobaric hypoxia (10% O2) or normoxia (20.9% O2) for one week were randomly assigned to receive daily intraperitoneal injections of normal saline (PL; n=19), or AMD3100 (7.5 mg/kg; n=10) or anti-SDF-1 antibody (25µg/kg; n=19) for seven days, from postnatal day 1–7.
Therapeutic Strategy
Twenty one FVB/NJ neonatal mice (1–2 d old) were exposed to normobaric hypoxia (10% O2) or normoxia (20.9% O2) for two weeks. After one week of this exposure, the mice were randomly assigned to receive daily intra-peritoneal injections of normal saline (PL; n=6), or AMD3100 (7.5 mg/kg; n=7) for seven days, from postnatal day 7–14.
The experimental protocol was performed according to guidelines set forth by the University of Miami Animal Care and Use Committee.
Hemodynamic Measurements
After set duration of hypoxic or normoxic exposure, mice were weighed and anesthetized with Avertin (tribromoethanol) 0.375 mg/g body weight injected intraperitoneally. A tracheostomy was performed with a 22 gauge angiocatheter and secured in place with a 4.0 silk suture. Mice were ventilated with a Harvard Mini-Vent with a stroke volume of 325 µl and rate of 150 stroke/min. Anesthesia was maintained throughout with 1% isoflurane mixed with room air or 10% O2. After thoracotomy, a 25 gauge needle fitted to a pressure transducer was inserted into the right ventricle. Right ventricular systolic pressure (RVSP) was measured and continuously recorded on a Gould polygraph (model TA-400, Gould instruments, Cleveland, Ohio). Immediately after RVSP measurements were obtained, the mice were sacrificed.
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org and provides details of all materials, bone marrow transplantation, pulmonary vascular morphometry, Western blot, immunohistochemistry, immunofluorescence and statistical analyses.
Results
SDF-1 Expression in Neonatal Pulmonary Hypertension
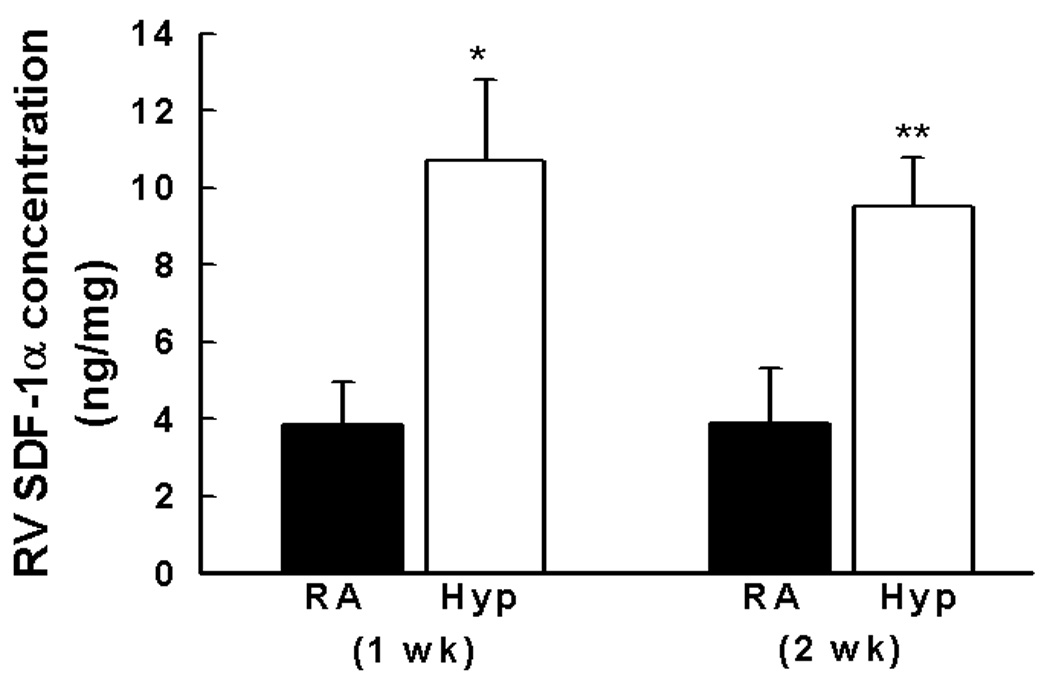
We first tested the prediction that neonatal hypoxia up regulates pulmonary SDF-1. Indeed, after hypoxia for one or two weeks, lung protein expression of SDF-1 increased 2-fold (Figure 1a). Similarly, within the right ventricle, SDF-1 was also markedly elevated (Figure 1b). In contrast, left ventricular SDF-1 was not elevated in response to hypoxia.
Figure 1. SDF-1 is up regulated in the lungs and right ventricles of neonatal mice with pulmonary hypertension.
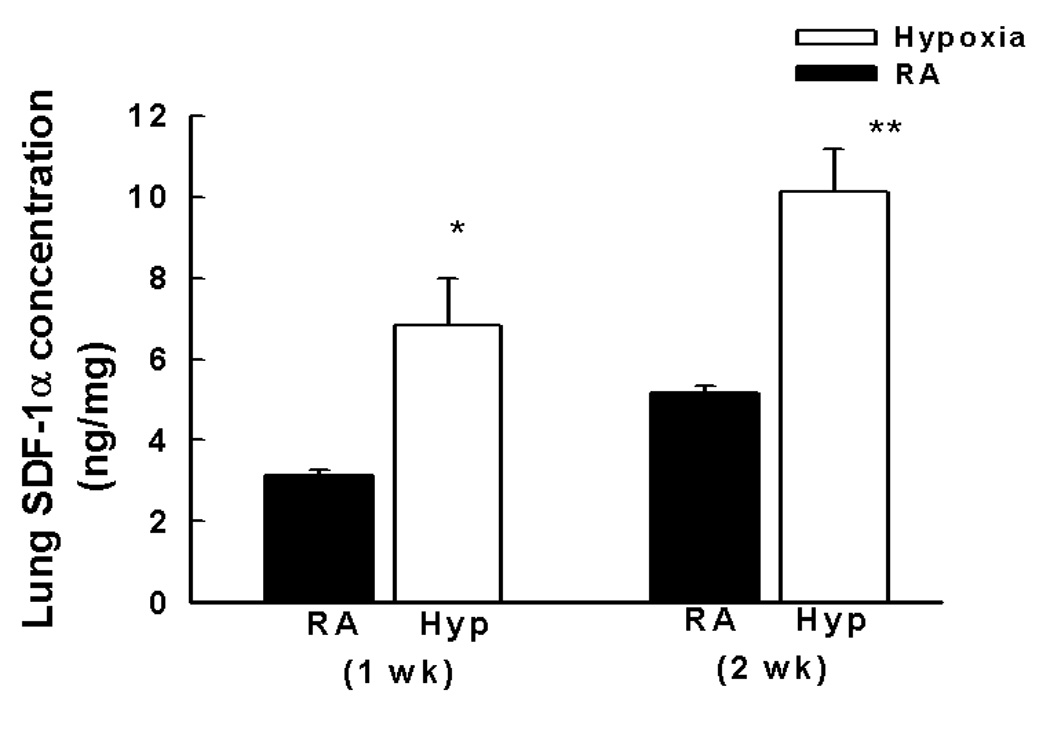
a. Neonatal mice with pulmonary hypertension following exposure to hypoxia for 1 or 2 wk (n = 5–7 mice/group) had a 2 fold increase in SDF-1 protein expression as compared to normoxia (n= 5–7 mice/group). * p<0.005 RA vs Hyp 1 wk; ** p<0.005 RA vs Hyp 2 wk.
b. Neonatal mice with pressure overload induced RVH following exposure to hypoxia for 1 or 2 wk ((n = 5–7 mice/group) had a 2 fold increase in SDF-1 protein expression as compared to normoxia. * p<0.0001, RA vs Hyp 1 wk; ** p<0.0001, RA vs Hyp 2 wk.
Bone Marrow -Derived Cells Migrate to the Pulmonary Vasculature during Hypoxia
We next examined whether bone marrow- derived cells were recruited to the pulmonary vasculature during chronic hypoxia. Six week old mice whose bone marrow had been reconstituted with GFP+ cells were exposed to hypoxia for 8 weeks. Following this exposure, GFP+ cells were visualized in the smooth muscle and adventitial layers of the hypoxic pulmonary arteries (Figures 2a and 2b).
Figure 2. BM-derived cells migrate to the pulmonary vasculature during hypoxia-induced pulmonary hypertension.


a. Confocal Immunofluorescence images of pulmonary arterioles in control and pulmonary hypertensive mice demonstrating more EGFP+ BM-derived cells (green) in the pulmonary hypertensive mice as compared to control in which these are sparsely evident. Original Magnification × 400.
b. Immunohistochemistry demonstrating more EFP+ BM-derived cells (brown) predominantly in the adventitia and media of pulmonary arterioles of mice with pulmonary hypertension as compared to control. Original Magnification × 400.
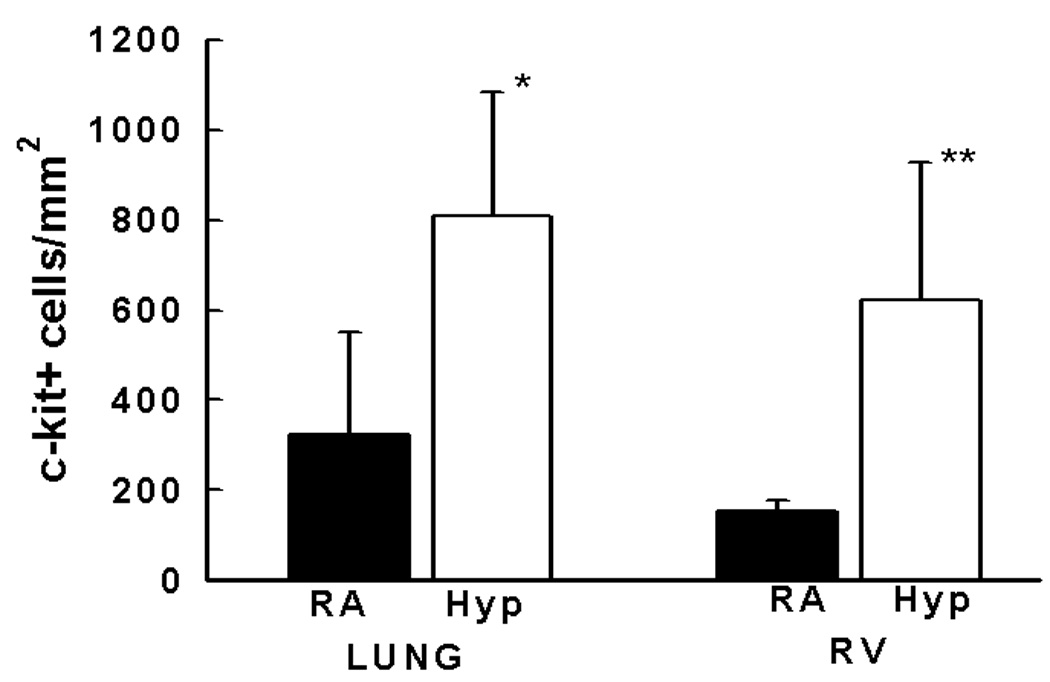
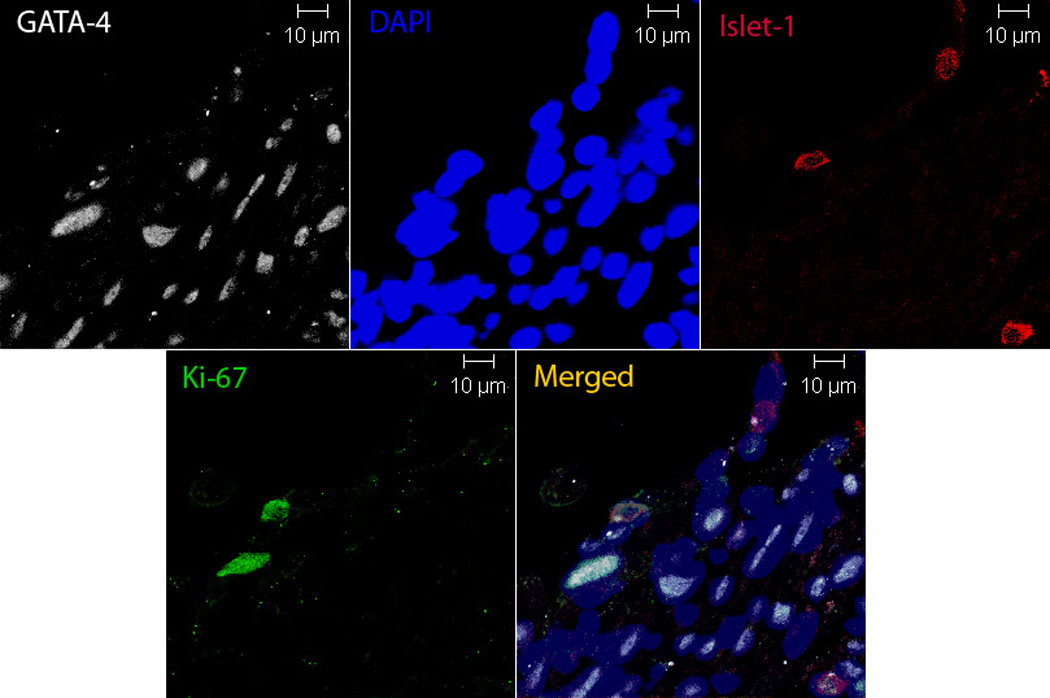
Given this, we sought to determine whether neonatal mice (1–2 d) exposed to hypoxia had increased numbers of c-kit+ cells in their lungs and right ventricles. Indeed, we demonstrated a 2.5 and 4 fold increase in the number of c-kit+ cells in the lungs and right ventricles respectively of neonatal mice with PH and right ventricular hypertrophy (RVH), (Figures 3a–3b). These c-kit+ cells were localized mainly to the adventitia of the hypoxic pulmonary arterioles. Additionally, within the hypertrophied right ventricles, double Immunofluorescence study also demonstrated c-kit+, Sca-1+ and Isl-1+ cells co-localized with GATA-4 and Ki-67 suggesting that they were proliferating and committed to a cardiac fate (Figures 3c and 3d).
Figure 3. Progenitor Cells are present in the lungs and right ventricles of neonatal mice with pulmonary hypertension.


a. Representative confocal Immunofluorescence images of lungs and right ventricles obtained from control and pulmonary hypertensive neonatal mice following staining with anti-c-kit (red) antibody and DAPI (blue). Magnification × 400.
b. Bar Graph demonstrating significantly increased c-kit+ cells/mm2 in the lungs and right ventricles of neonatal mice with pulmonary hypertension. The cells were quantified by confocal microscopy in 6 random × 400 fields in a total of 5 mice/group ( *Lung: RA vs Hyp, p< 0.03 and **RV: RA vs Hyp, p<0.05)
c. Representative confocal Immunofluorescence images of sections obtained from hypertrophied right ventricles which were double-stained with anti-c-kit (green), anti-Sca-1 (red), DAPI (blue), anti- GATA-4 or anti-Ki67 (white) demonstrating co-localization of c-kit and Sca-1 cells with GATA-4 and Ki-67. Magnification × 400.
d. Isl-1 cells (red) present in the hypertrophied right ventricles of neonatal mice with pulmonary hypertension. These cells co-localized with GATA-4 and Ki-67 (white). Magnification ×400.
Inhibition of SDF-1/CXCR4 axis Decreases Progenitor Cells in the Lungs and Right Ventricles of Mice with Pulmonary Hypertension
Given that SDF-1 is an integral component of stem cell mobilization during hypoxia, and that c-kit+ as well as Sca-1+ cells were increased in the lungs and right ventricles of neonatal mice with hypoxia-induced PH, we next evaluated whether inhibition of the SDF-1/CXCR4 axis would decrease the expression of c-kit and Sca-1 in the lungs and right ventricles of mice with PH. Administration of AMD3100 or anti-SDF-1 antibody to mice exposed to 1 wk hypoxia significantly decreased the protein expression of these stem cell markers to baseline or near baseline values (Figures 4 a–e).
Figure 4. Inhibition of the SDF-1/CXCR4 axis decreased Stem Cell Marker Expression in the Lungs and Right Ventricles of Neonatal Mice with Pulmonary Hypertension.

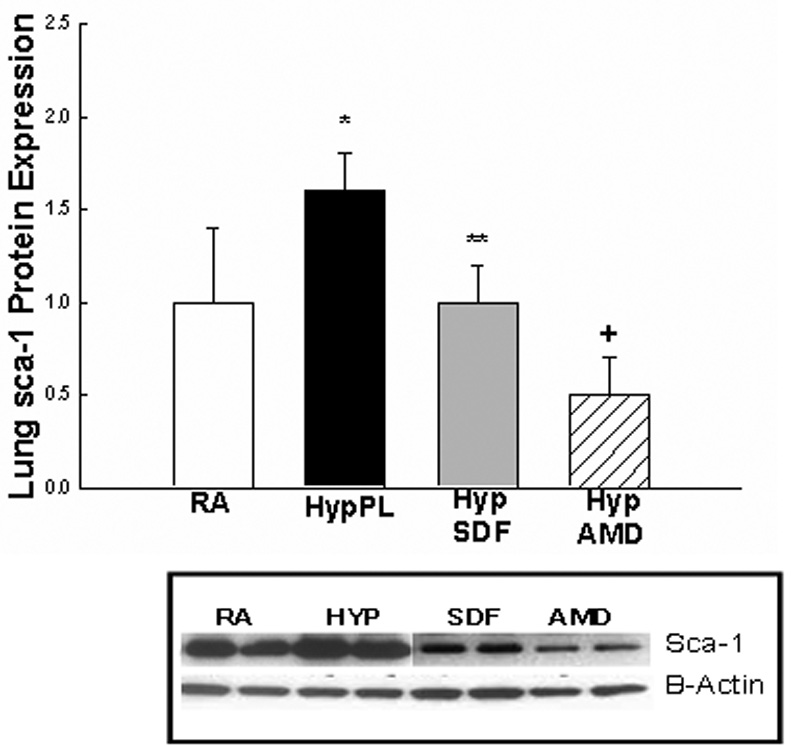
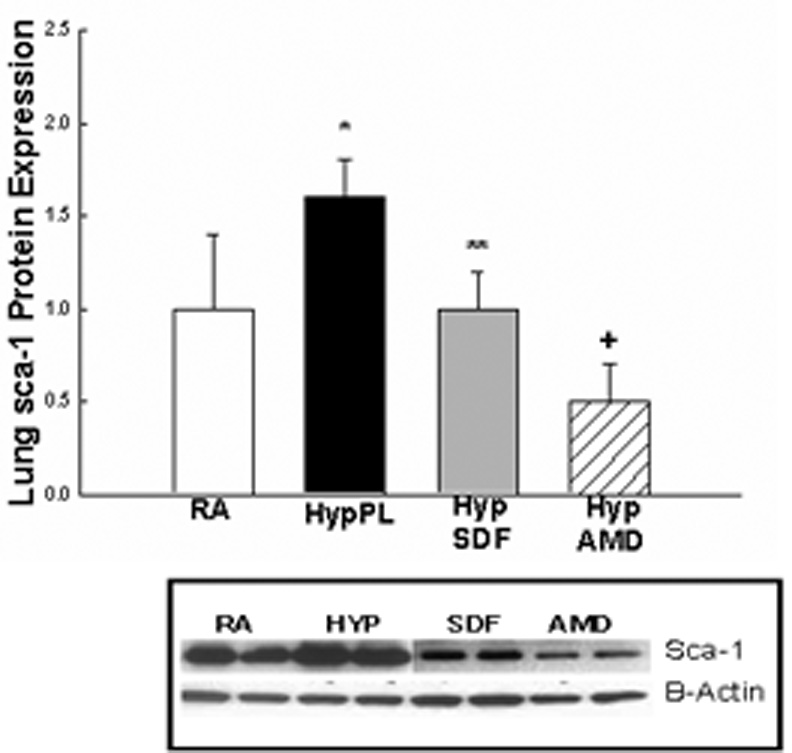
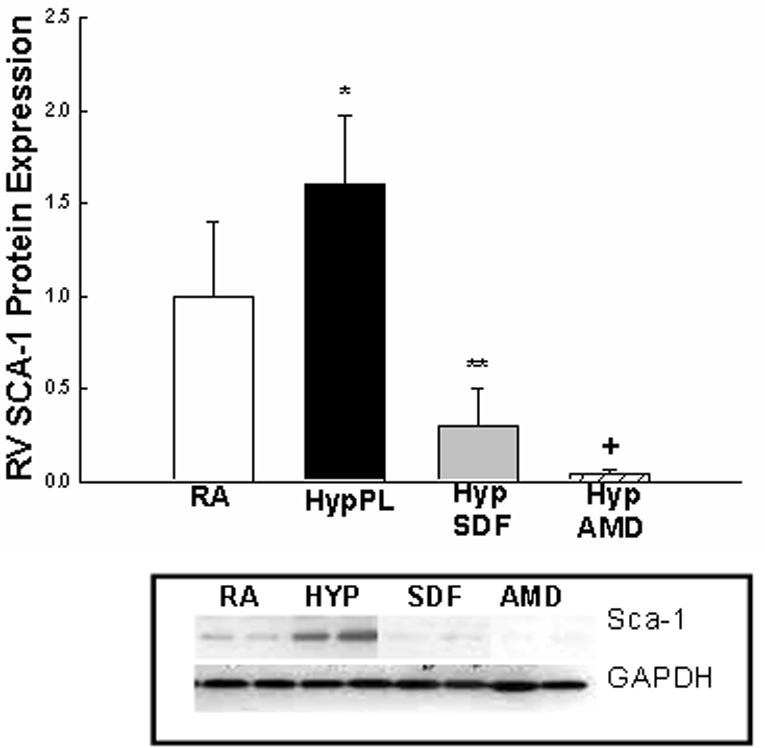
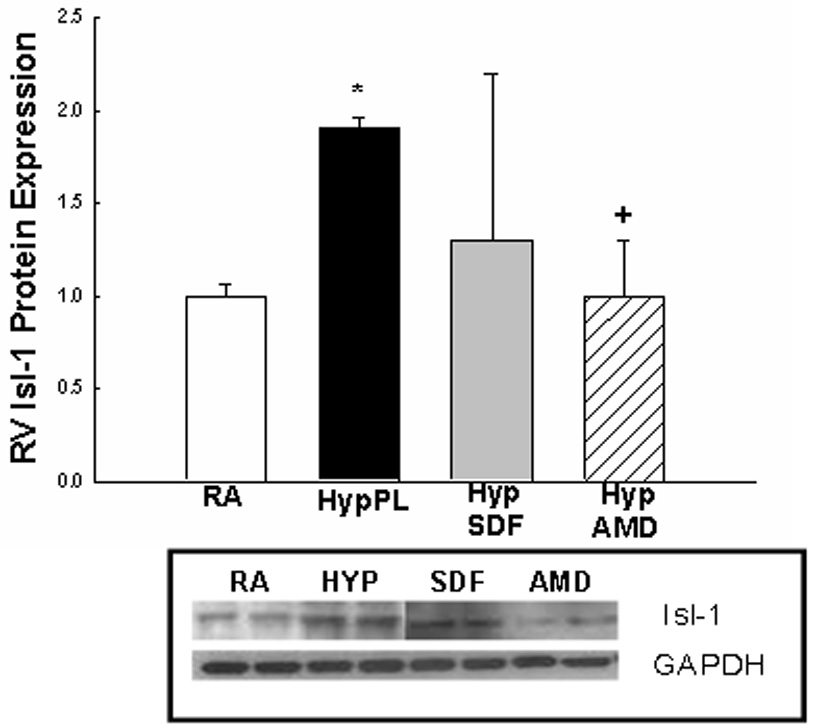
Neonatal mice with pulmonary hypertension had a marked increase in lung: a)c-kit (2 fold, *p< 0.0001) and b) sca-1 (1.9 fold, *p<0.02)); c) RV: c-kit (5 fold; *p<0.0001), d) RV sca-1(1.6 fold; *p<0.001) and e) RV Isl-1 (2 fold; *p<0.02)) expressions as compared to RA (n=5–7/group). As compared to Hyp PL mice, administration of anti-SDF-1 significantly decreased a) lung c-kit (** p<0.0008), b) lung Sca-1 (** p<0.02) and d) RV Sca-1 (** p<0.0005) expressions to near baseline values. Similarly, administration of AMD3100 significantly decreased a) lung c-kit (+ p<0.0001), b) lung Sca-1 (+ p<0.006), c) RV c-kit (+ p< 0.01), d) RV Sca-1 (+p< 0.0001) and e) RV Isl-1 (+ p< 0.05) expressions to near baseline values as compared to Hyp PL (n=5–7/group).
Inhibition of the SDF-1/CXCR4 axis Prevents and Reverses Pulmonary Vascular Remodeling Preventative Strategy
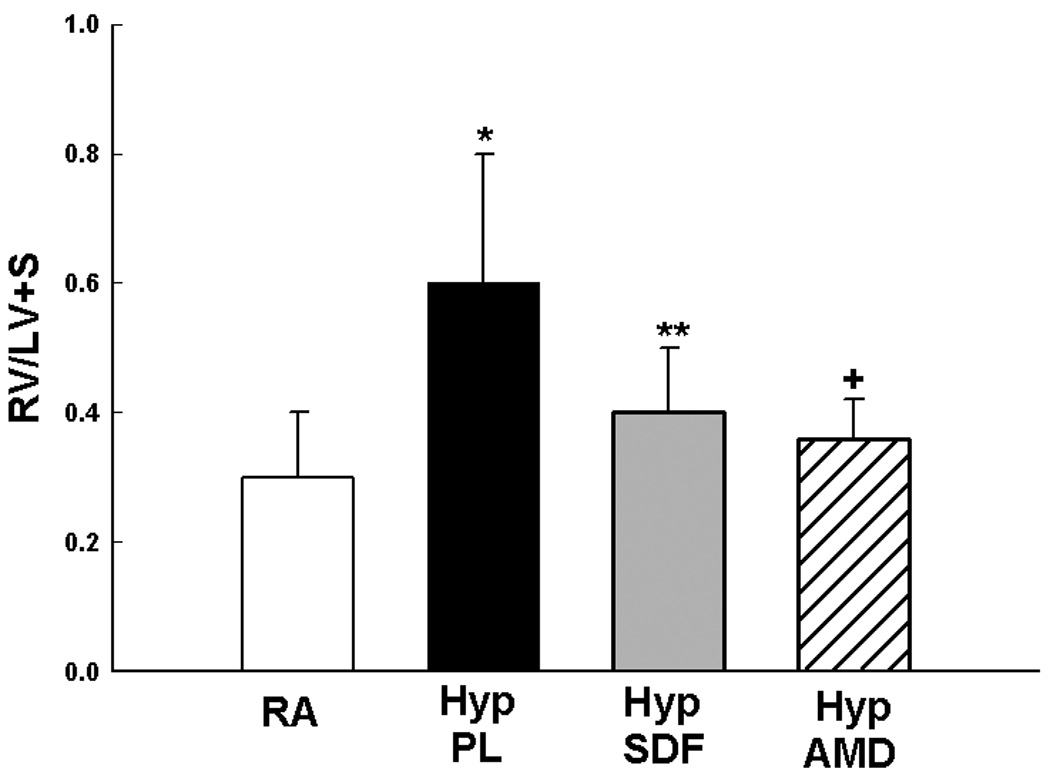
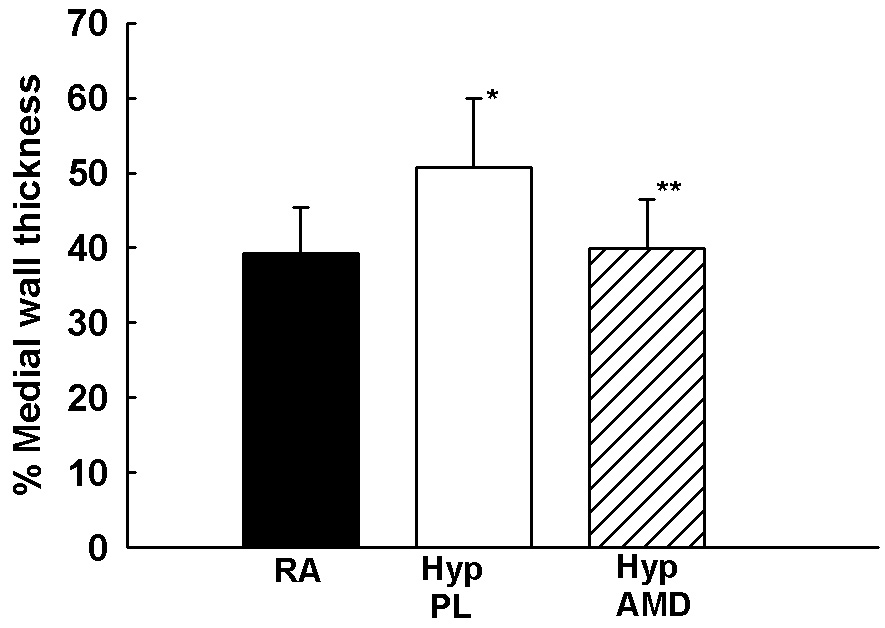
We next sought to elucidate whether inhibition of the SDF-1/CXCR4 axis would prevent or reverse pulmonary vascular remodeling. Exposure of neonatal mice to 1 wk hypoxia resulted in a significant increase in RVSP (11±2 vs 24±6 mmHg; RA vs Hyp PL, p<0.001) and RV/LV+S (0.2±0.1 vs 0.5±0.2; RA vs Hyp PL, p<0.01). To test the prediction that the increased release of SDF-1 in the lungs of hypoxic neonatal mice exacerbates PH, we administered monoclonal anti-SDF-1 antibody or AMD3100 (a CXCR4 antagonist) daily to neonatal mice exposed to 1 wk hypoxia. Importantly, both strategies to inhibit the SDF-1/CXCR4 axis restored RVSP and RV/LV+S close to baseline values (Figures 5a and 5b). In addition, as compared to hypoxic placebo mice, hypoxic AMD3100 treated mice had a marked decrease in the medial wall thickness (Figures 5c–d), and a con-comitant increased percentage of non-muscular vessels (Online Figure I).
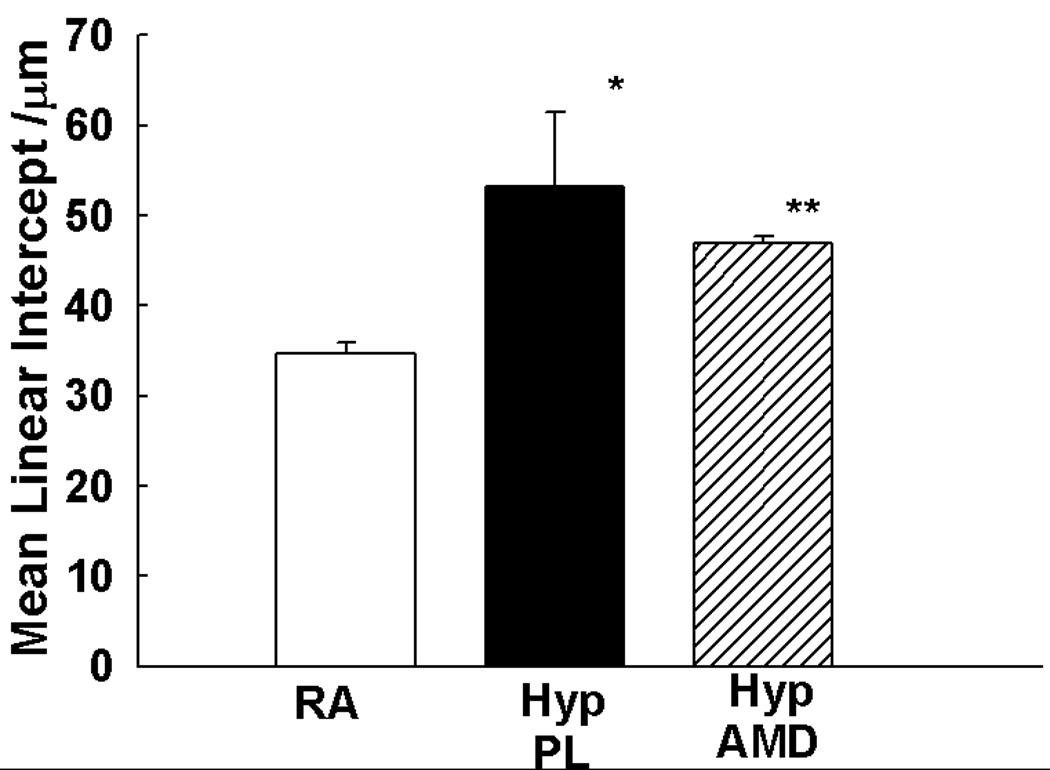
Figure 5. Inhibition of the SDF-1/CXCR4 axis Prevents and Reverses Pulmonary Hypertension in Neonatal Hypoxic Mice.
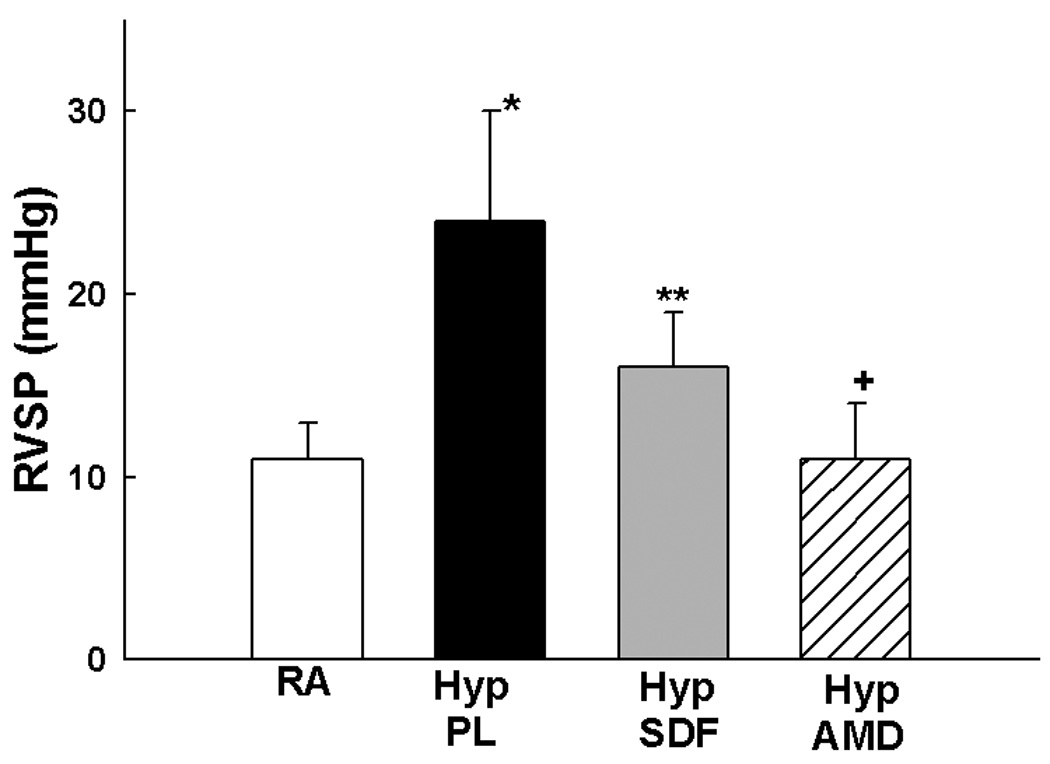

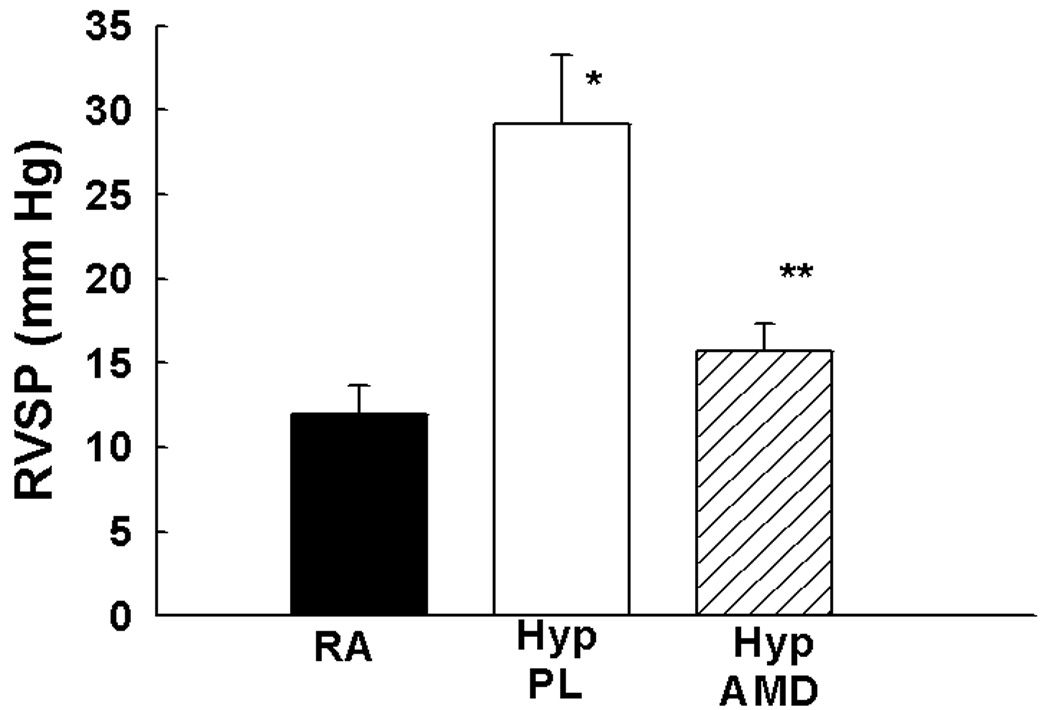
a. Administration of anti-SDF-1 antibody (n=19) or the CXCR4 antagonist AMD3100 (n=10) to neonatal mice exposed to hypoxia significantly attenuated RVSP as compared to placebo (n=19), (* p<0.0001 RA vs Hyp PL, ** p<0.0002 Hyp PL vs Hyp SDF, + p< 0.00001 Hyp PL vs Hyp AMD).
b. Administration of anti-SDF-1 antibody (n=14) or the CXCR4 antagonist AMD3100 (n=6) significantly attenuated RV Hypertrophy as compared to placebo (n=14), (* p<0.001 RA vs Hyp PL,** p<0.005 Hyp PL vs Hyp SDF, + p< 0.008 Hyp PL vs Hyp AMD).
c. Representative bright-field photomicrographs of lung sections stained with α- smooth muscle actin demonstrating decreased number of muscularized pulmonary arterioles in AMD3100 treated mice as compared to placebo. Magnification × 200.
d. Morphometric Analysis performed on 35–40 (20–50 µm diameter) pulmonary arterioles (n=5/group) demonstrated that AMD3100 markedly attenuated vascular remodeling (* p< 0.04 RA vs Hyp PL; ** p<0.05 Hyp PL vs Hyp AMD).
e. The Mean Linear Intercept was significantly decreased in the hypoxia treated mice as compared to placebo (* p< 0.005 RA vs Hyp PL; ** p<0.05 Hyp PL vs Hyp AMD; n=5/group).
f. Administration of the CXCR4 antagonist AMD3100 (n=7) to mice with established pulmonary hypertension, significantly attenuated RVSP as compared to placebo, (* p<0.0003 RA vs Hyp PL, ** p <0.007 Hyp PL vs Hyp AMD).
Moreover, since chronic hypoxia is also known to significantly alter lung alveolarization in the neonate, we measured the mean linear intercept (MLI) to evaluate the effect of the SDF-1/CXCR4 axis on lung alveolarization. Whilst exposure to chronic hypoxia was associated with a significant increase in the MLI, blockade of the SDF-1/CXCR4 axis significantly decreased the MLI (Figure 5e).
Therapeutic Strategy
Exposure of neonatal mice to 2 wk hypoxia also resulted in a marked increase in RVSP (12±2 vs 29±4 mmHg; RA vs Hyp PL, p<0.001) and RV hypertrophy ((0.2 ±0.0 vs 0.5±0.1; RA vs Hyp PL, p<0.01). In contrast, administration of AMD3100 in mice with established PH resulted in a significant decrease in RVSP (Figure 5f) but no significant difference in RV hypertrophy (Online Figure II). There was however a significant increase in the percentage of non-muscularized pulmonary arterioles in the hypoxia treated mice as compared to placebo (Online Figure III).
Inhibition of the SDF-1/CXCR4 Axis Decreases Pulmonary Vascular Cell Proliferation and Apoptosis
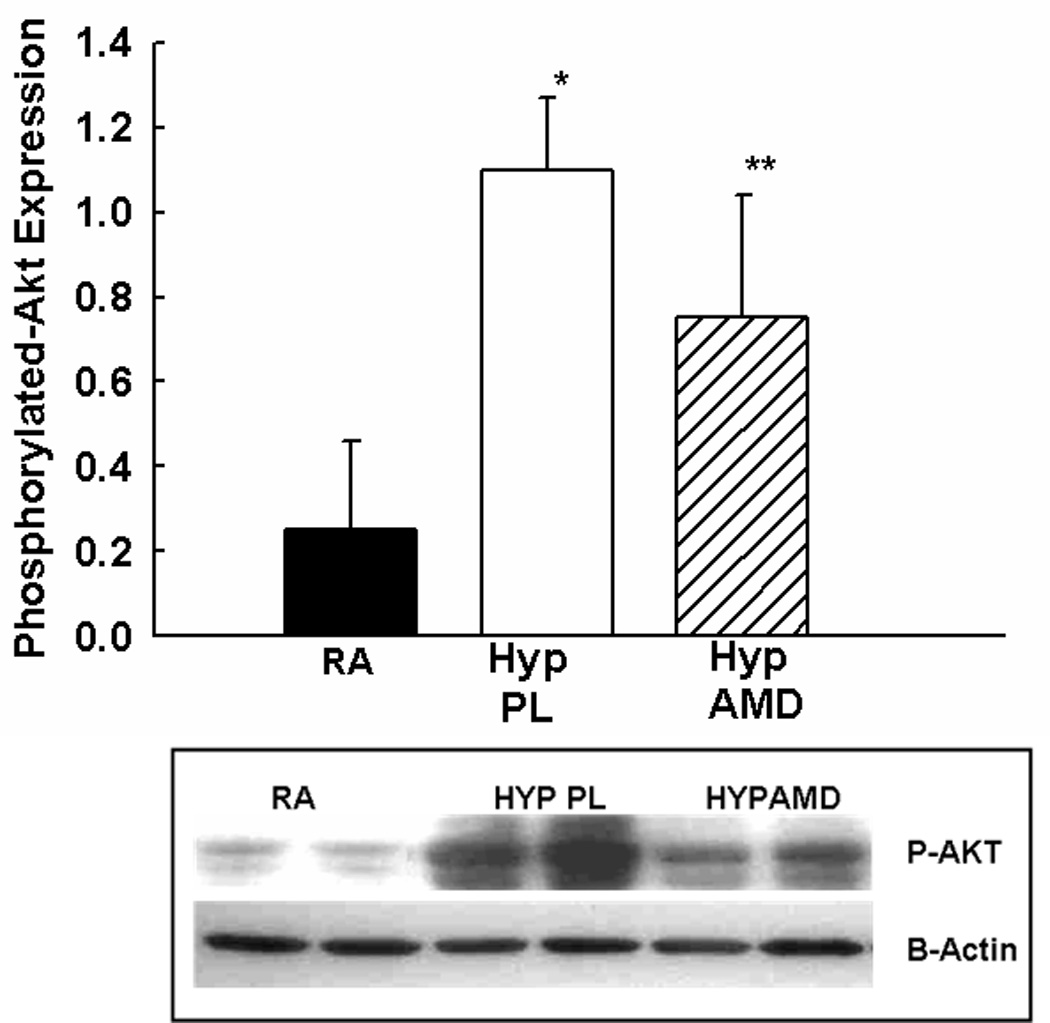
In order to ascertain other mechanisms by which inhibition of the SDF-1/CXCR4 axis attenuates hypoxia-induced pulmonary vascular remodeling, proliferating cell nuclear antigen (PCNA) immunostaining and TUNEL assay were utilized to determine cell proliferation and survival respectively. Whilst exposure to hypoxia resulted in a marked increase in the number of PCNA+ and TUNEL+ cells in the pulmonary vasculature, this was significantly attenuated in the AMD3100 hypoxic mouse pups (Figures 6a–e). Moreover, on further questioning of the down-stream signaling mechanisms that were responsible for this effect, we demonstrated a marked decrease in phosphorylated -Akt expression in the treated mouse pups as compared to placebo (Figure 6c).
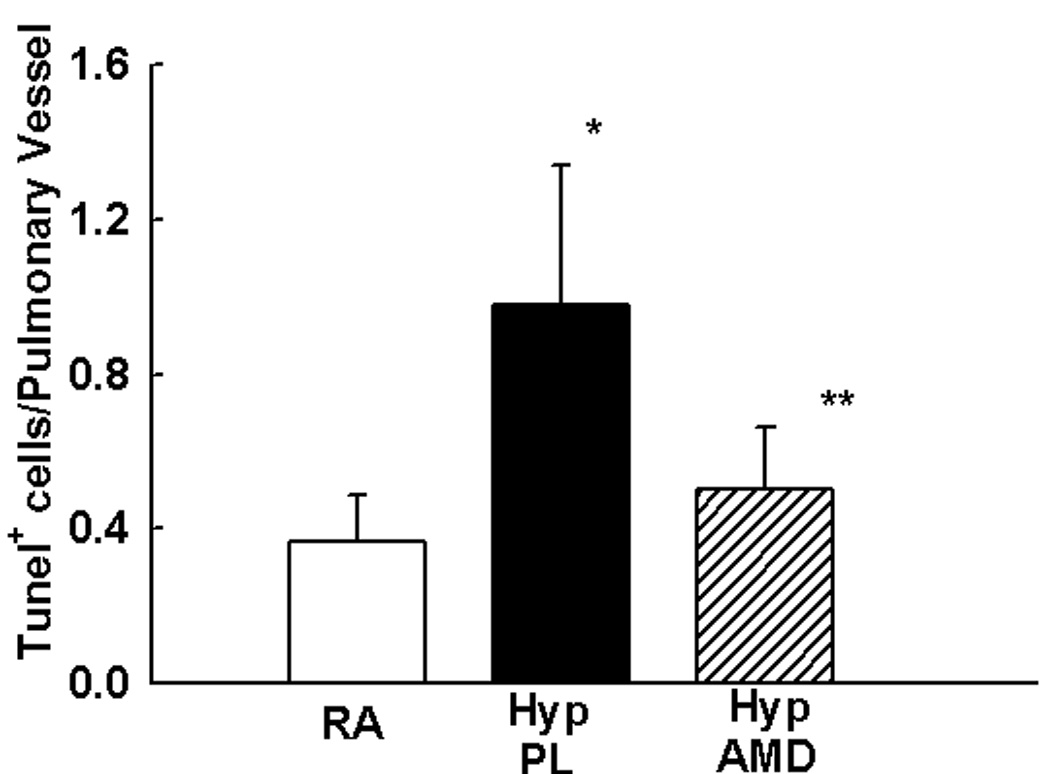
Figure 6. Inhibition of SDF-1/CXCR4 axis Decreases Pulmonary Vascular Proliferation and Apoptosis.

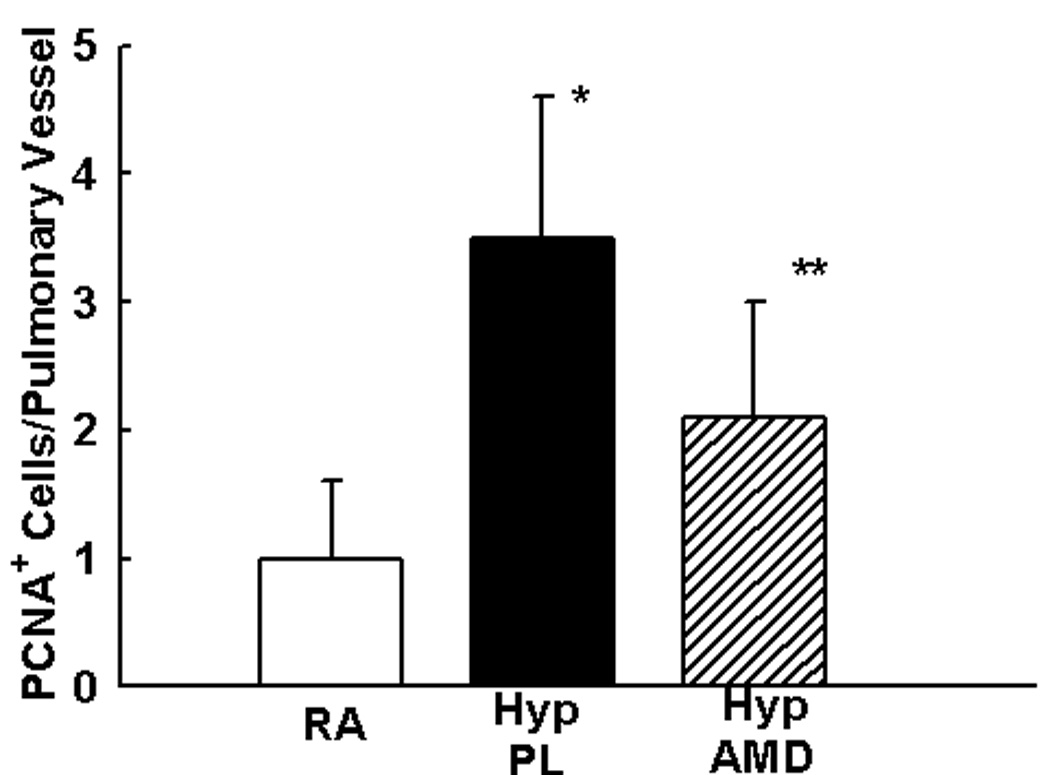

a. Representative immunofluorescence images of lungs obtained from RA, HYP PL and HYP AMD neonatal mice following staining with anti-PCNA antibody (green) and DAPI (blue). PCNA+ cells (green) were more abundantly present in HYP PL mice as compared to HYP AMD. Magnification × 400.
b. Bar Graph demonstrating significantly decreased number of PCNA+ cells/pulmonary vessel in the HYP AMD mice as compared to HYP PL, (* p< 0.005 RA vs Hyp PL; ** p<0.04 Hyp PL vs Hyp AMD; n=4/group).
c. Phosphorylated- Akt expression was markedly decreased in the lungs of neonatal mice with pulmonary hypertension following inhibition of the SDF-1/CXCR4 axis, (* p< 0.0005 RA vs Hyp PL; ** p<0.03 Hyp PL vs Hyp AMD; n=4/group)
d. Tunel Assay revealed significantly less apoptotic (pink) pulmonary vascular cells following inhibition of the SDF-1/CXCR4 axis. Magnification × 400. Scale Bar is 50 µm.
e. Bar Graph demonstrating significantly decreased number of Tunel+ cells/pulmonary vessel in the HYP AMD mice as compared to HYP PL, ((* p< 0.02 RA vs Hyp PL; ** p<0.05 Hyp PL vs Hyp AMD; n=4/group).
Discussion
In this study we provide direct systematic evidence of the participation and the mechanism of action of SDF-1 and its receptor CXCR4 in the pathogenesis of neonatal chronic hypoxia-induced cardiopulmonary remodeling. We demonstrate that inhibition of the SDF-1/CXCR4 axis improves alveolarization, prevents the development of hypoxia-induced pulmonary vascular remodeling in neonatal mice, and significantly decreases pulmonary artery pressure in neonatal mice with established disease. These findings were associated with decreased expression of progenitor cells in the lungs and right ventricles of treated mice as well as decreased pulmonary vascular cell proliferation and apoptosis. This study therefore offers important pathophysiologic insights into the role of the SDF-1/CXCR4 axis in neonatal chronic hypoxia-induced cardiopulmonary remodeling and has potential therapeutic implications for neonatal hypoxia-induced PH.
One of the major roles of SDF-1 is the mobilization of stem cells from the bone marrow to injured sites16–19. In proof of this concept, we demonstrated that inhibition of the SDF-1/CXCR4 axis decreased the pulmonary expression of c-kit and sca-1, known stem cell markers. The role of c-kit+ or sca-1+ cells in neonatal hypoxia-induced cardiopulmonary remodeling is however unclear. Davie and colleagues showed an increased number of c-kit cells in the pulmonary artery adventitia of neonatal animals with hypoxic PH 20, whilst BM-derived cells were previously shown to express smooth muscle actin in hypoxia remodeled pulmonary arteries and selective depletion of circulating mesenchymal precursors prevented pulmonary adventitial remodeling 21, suggesting that these cells may have functional relevance. Our present study adds further credence to this theory as inhibition of the SDF-1/CXCR4 axis both inhibited and reversed pulmonary vascular remodeling, and this was associated with decreased stem cell expression.
This is plausible as SDF-1 has been suggested to play a role in adult systemic vascular remodeling22–25. Whilst the remodeled systemic vasculature clearly does not mirror a remodeled neonatal pulmonary arteriole, this study does suggest that SDF-1 may be a crucial factor in both processes. In agreement with this, Satoh et al demonstrated increased expression of SDF-1 in the plasma of hypoxic adult rats and, following administration of a statin, demonstrated decreased pulmonary vascular remodeling associated with decrease SDF-1 and progenitor cell expression26. Our study extends this finding and more importantly provides direct evidence that inhibition of the SDF-1/CXCR4 axis actually does decrease neonatal pulmonary vascular remodeling.
Another possible mechanism by which the SDF-1/CXC4 axis could participate in hypoxia-induced pulmonary vascular remodeling is through its role in cell proliferation and apoptosis. Vascular cell proliferation is known to be an important component of pulmonary vascular remodeling, whilst early PH is known to be associated with increased endothelial cell apoptosis and loss of small capillaries. We demonstrated in this present study that inhibition of the SDF-1/CXCR4 axis decreased pulmonary vascular cell proliferation as well as apoptosis, and this was associated with decreased expression of phosphorylated Akt. Binding of SDF-1 to its receptor CXCR4 is known to induce several signal transcription pathways including activation of phosphoinositol 3-kinase (PI3K). The PI3K/Akt axis has been shown to affect the calcium currents that govern smooth muscle cell contraction through coupling membrane receptors to calcium channels27, and most recently, investigators demonstrated that SDF-1 may be a major regulator of smooth muscle cell proliferation through its involvement in PI3K/PTEN signaling pathway 25. Our data support these findings and further suggests that blockade of the SDF-1/CXCR4 pathway is not only therapeutically beneficial in PH through its effects on cell migration but also via its role in cell proliferation and survival.
Another important aspect of this study is the finding that inhibition of the SDF-1/CXCR4 axis also decreased right ventricular hypertrophy and this was associated with increased right ventricular expression of c-kit, Isl-1 and Sca-1. Moreover, cells which expressed these stem cell markers co-localized with GATA-4 and Ki67 suggesting that they were committed to a cardiac fate and also proliferating. Whilst we did not evaluate directly whether these stem cells were bone marrow derived or resident stem cells, we speculate that a significant fraction may be indeed derived from the bone marrow, as following inhibition of the SDF-1/CXCR4 axis we demonstrated a significant decrease in the expression of these stem cell markers in the right ventricle. Moreover, bone marrow-derived cells have been suggested by several investigators to be involved in the ventricular hypertrophic response28, 29. Fukuda and colleagues found that bone marrow-derived cells were involved in the pathogenesis of cardiac hypertrophy through cell fusion as well as transdifferentiation, while Spees et al demonstrated that some of the bone marrow derived cells in the hypertrophied right ventricles of rats with monocrotaline-induced PH were vascular cells as well cardiomyocytes28, 30. Nonetheless, the possible role of resident cardiac stem cells cannot be understated as several investigators including our group have documented several resident stem niches within the myocardium which differentiate and proliferate in response to injury31, 32.
It is unclear whether these stem cells in the hypertrophied right ventricle have adaptive or maladaptive roles. We speculate that these cells do contribute significantly to the compensatory increase in myocardial mass that is necessary to adapt to the increase overload, and our findings extend those of other investigators who have implicated stem cells in pressure-overload induced ventricular hypertrophy 33–35. Urbanek and colleagues previously demonstrated that in a model of left ventricular overload induced by aortic stenosis there was intense new formation of myocytes resulting from the differentiation of stem-like cells, whilst Lee and colleagues demonstrated that stem cells or precursor cells could regenerate cardiomyocytes in hearts subjected to pressure overload36, 37. It should however be noted that the roles of these stem cells may not be equivalent. Whilst c-kit+ cells have been shown by several investigators to have significant reparative potential, only one other published report has suggested that transplantation of Sca-1+ cells into the peri-infarct zone could attenuate left ventricular structural and functional remodeling post myocardial infarction, and no other studies have suggested that Isl-1 committed cardiomyoblasts could participate in repair38–41. This is the first study to demonstrate that Isl-1+ cells, could also contribute to RVH. This was a surprising finding as these cells were previously shown to be extremely sparse in the native heart after postnatal day 542, 43. We postulate that this response could be developmentally determined as our pilot studies evaluating the role of stem cells in pressure overload-induced RVH showed a progressive decline in the role of these cells with increasing age. It should also be noted that whilst these cells were significantly increased in the hypertrophied right ventricle, they were not changed in the left ventricle, suggesting that the right ventricular stem cell response was not only due to global hypoxia of the animal, but rather a direct reflection of the increase in the SDF-1 expression noted in the right but not the left ventricle.
This finding is clearly surprising as hypoxia is known to upregulate SDF-1 expression in several tissues in a manner proportional to the degree of hypoxia6, 44. Additionally, the promoter region of the SDF-1 gene contains at least two binding sites for the master regulator of hypoxic cell signaling, HIF-1α, and the latter is known to be a major participant in hypoxia-induced cardiopulmonary remodeling45, 46. Nonetheless, this present study does suggest that factors that are uniquely released in the pressure overloaded right ventricle may also regulate SDF-1 production. Indeed, chronic hypobaric hypoxia has been shown to induce a differential transcriptional profile in the right and left ventricle47, including genes involved in apoptosis, and SDF-1 secretion was triggered in medial smooth muscle cells following exposure to apoptotic bodies23.
In conclusion, this study clearly demonstrates that SDF-1 participates in the pathogenesis of neonatal hypoxia-induced cardiopulmonary remodeling through several mechanisms. We propose that during neonatal hypoxia, there is increased release of SDF-1 which results in the mobilization of progenitor cells to the pulmonary vasculature and right ventricle, and that these mobilized cells participate directly in pulmonary vascular remodeling and RVH. Moreover, following binding of the locally released SDF-1 to its receptor CXCR4, downstream signaling pathways regulate pulmonary vascular cell proliferation and apoptosis resulting in the findings evidenced in the lungs and hearts of hypoxic neonates. Finally, but most importantly, given the tremendous need for new therapies, the demonstration that inhibition of SDF-1/CXCR4 axis significantly improves alveolarization, attenuates neonatal PH, vascular remodeling and RVH suggest a novel and potentially highly effective therapeutic strategy for this disease.
Supplementary Material
Acknowledgments
Sources of Funding: Supported in part by Project New Born, Bank of America Charity Foundation and American Heart Association Scientist Development Grant to KY. Dr. Hare is supported by RO1 grants from the NHLBI and the NIA (HL-65455. HL-084275, HL-094849 and AG-025017).
Footnotes
Disclosures: None.
References
- 1.Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, Verter J, Stoll BJ, Lemons JA, Papile LA, Shankaran S, Donovan EF, Oh W, Ehrenkranz RA, Fanaroff AA. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics. 2000;105:14–20. doi: 10.1542/peds.105.1.14. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol Heart Circ Physiol. 1981;240:H62–H72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol. 1979;236:H818–H827. doi: 10.1152/ajpheart.1979.236.6.H818. [DOI] [PubMed] [Google Scholar]
- 4.Haworth SG, Hislop AA. Lung development--the effects of chronic hypoxia. Seminars in Neonatology. 2003;8:1–8. doi: 10.1016/s1084-2756(02)00195-1. [DOI] [PubMed] [Google Scholar]
- 5.Lima e Silva R, Shen J, Hackett SF, Kachi S, Akiyama H, Kiuchi K, Yokoi K, Hatara MC, Lauer T, Aslam S, Gong YY, Xiao W-H, Khu NH, Thut C, Campochiaro PA. The SDF-1/CXCR4 ligand/receptor pair is an important contributor to several types of ocular neovascularization. FASEB J. 2007;21:3219–3230. doi: 10.1096/fj.06-7359com. [DOI] [PubMed] [Google Scholar]
- 6.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 7.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends in Immunology. 2007;28:299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MAS, Werb Z, Rafii S. Recruitment of Stem and Progenitor Cells from the Bone Marrow Niche Requires MMP-9 Mediated Release of Kit-Ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ratajczak MZ, Majka M, Kucia M, Drukala J, Pietrzkowski Z, Peiper S, Janowska-Wieczorek A. Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells. 2003;21:363–371. doi: 10.1634/stemcells.21-3-363. [DOI] [PubMed] [Google Scholar]
- 11.Ara T, Nakamura Y, Egawa T, Sugiyama T, Abe K, Kishimoto T, Matsui Y, Nagasawa T. Impaired colonization of the gonads by primordial germ cells in mice lacking a chemokine, stromal cell-derived factor-1 (SDF-1) Proc Natl Acad Sci U S A. 2003;100:5319–5323. doi: 10.1073/pnas.0730719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiss K, Mentlein R, Sievers J, Hartmann D. Stromal cell-derived factor 1 is secreted by meningeal cells and acts as chemotactic factor on neuronal stem cells of the cerebellar external granular layer. Neuroscience. 2002;115:295–305. doi: 10.1016/s0306-4522(02)00307-x. [DOI] [PubMed] [Google Scholar]
- 13.Crane IJ, Wallace CA, McKillop-Smith S, Forrester JV. CXCR4 receptor expression on human retinal pigment epithelial cells from the blood-retina barrier leads to chemokine secretion and migration in response to stromal cell-derived factor 1 alpha. J Immunol. 2000;165:4372–4378. doi: 10.4049/jimmunol.165.8.4372. [DOI] [PubMed] [Google Scholar]
- 14.Hatch HM, Zheng D, Jorgensen ML, Petersen BE. SDF-1alpha/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells. 2002;4:339–351. doi: 10.1089/153623002321025014. [DOI] [PubMed] [Google Scholar]
- 15.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol. 2004;35:233–245. doi: 10.1023/b:hijo.0000032355.66152.b8. [DOI] [PubMed] [Google Scholar]
- 16.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ, Penn MS. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. The Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi J-i, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner JM, Asahara T. Stromal Cell-Derived Factor-1 Effects on Ex Vivo Expanded Endothelial Progenitor Cell Recruitment for Ischemic Neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 18.De Falco E, Porcelli D, Torella AR, Straino S, Iachininoto MG, Orlandi A, Truffa S, Biglioli P, Napolitano M, Capogrossi MC, Pesce M. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 2004;104:3472–3482. doi: 10.1182/blood-2003-12-4423. [DOI] [PubMed] [Google Scholar]
- 19.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal Cell-Derived Factor-1{alpha} Plays a Critical Role in Stem Cell Recruitment to the Heart After Myocardial Infarction but Is Not Sufficient to Induce Homing in the Absence of Injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 20.Davie NJ, Crossno JT, Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L668–L678. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 21.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiba Y, Takahashi M, Yoshioka T, Yajima N, Morimoto H, Izawa A, Ise H, Hatake K, Motoyoshi K, Ikeda U. M-CSF Accelerates Neointimal Formation in the Early Phase After Vascular Injury in Mice: The Critical Role of the SDF-1-CXCR4 System. Arterioscler Thromb Vasc Biol. 2007;27:283–289. doi: 10.1161/01.ATV.0000250606.70669.14. [DOI] [PubMed] [Google Scholar]
- 23.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF-1{alpha}/CXCR4 Axis Is Instrumental in Neointimal Hyperplasia and Recruitment of Smooth Muscle Progenitor Cells. Circ Res. 2005;96:784–791. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 24.Schober A, Knarren S, Lietz M, Lin EA, Weber C. Crucial Role of Stromal Cell-Derived Factor-1{alpha} in Neointima Formation After Vascular Injury in Apolipoprotein E-Deficient Mice. Circulation. 2003;108:2491–2497. doi: 10.1161/01.CIR.0000099508.76665.9A. [DOI] [PubMed] [Google Scholar]
- 25.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MCM. Targeted Deletion of PTEN in Smooth Muscle Cells Results in Vascular Remodeling and Recruitment of Progenitor Cells Through Induction of Stromal Cell-Derived Factor-1{alpha} Circ Res. 2008;102:1036–1045. doi: 10.1161/CIRCRESAHA.107.169896. [DOI] [PubMed] [Google Scholar]
- 26.Satoh K, Fukumoto Y, Nakano M, Sugimura K, Nawata J, Demachi J, Karibe A, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. Statin ameliorates hypoxia-induced pulmonary hypertension associated with down-regulated stromal cell-derived factor-1. Cardiovasc Res. 2009;81:226–234. doi: 10.1093/cvr/cvn244. [DOI] [PubMed] [Google Scholar]
- 27.Macrez N, Mironneau C, Carricaburu V, Quignard J-F, Babich A, Czupalla C, Nurnberg B, Mironneau J. Phosphoinositide 3-Kinase Isoforms Selectively Couple Receptors to Vascular L-Type Ca2+ Channels. Circ Res. 2001;89:692–699. doi: 10.1161/hh2001.097864. [DOI] [PubMed] [Google Scholar]
- 28.Endo J, Sano M, Fujita J, Hayashida K, Yuasa S, Aoyama N, Takehara Y, Kato O, Makino S, Ogawa S, Fukuda K. Bone Marrow-Derived Cells Are Involved in the Pathogenesis of Cardiac Hypertrophy in Response to Pressure Overload. Circulation. 2007;116:1176–1184. doi: 10.1161/CIRCULATIONAHA.106.650903. [DOI] [PubMed] [Google Scholar]
- 29.Muller P, Kazakov A, Semenov A, Bohm M, Laufs U. Pressure-induced cardiac overload induces upregulation of endothelial and myocardial progenitor cells. Cardiovasc Res. 2008;77:151–159. doi: 10.1093/cvr/cvm037. [DOI] [PubMed] [Google Scholar]
- 30.Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J, Prockop DJ. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 2008;22:1226–1236. doi: 10.1096/fj.07-8076com. [DOI] [PubMed] [Google Scholar]
- 31.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative Potential of Cardiosphere-Derived Cells Expanded From Percutaneous Endomyocardial Biopsy Specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 32.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 33.Angelini A, Castellani C, Della Barbera M, Toffoli S, Padalino MA, Milanesi O, Stellin G, Thiene G. Stem cells recruitment in human right ventricular remodeling. Journal of Molecular and Cellular Cardiology. 2007;42:S101–S101. [Google Scholar]
- 34.Spees JL, Whitney MJ, Sullivan DE, Lasky JA, Laboy M, Ylostalo J, Prockop DJ. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 2008;22:1226–1236. doi: 10.1096/fj.07-8076com. [DOI] [PubMed] [Google Scholar]
- 35.Endo J, Sano M, Fujita J, Hayashida K, Yuasa S, Aoyama N, Takehara Y, Kato O, Makino S, Ogawa S, Fukuda K. Bone Marrow Derived Cells Are Involved in the Pathogenesis of Cardiac Hypertrophy in Response to Pressure Overload. Circulation. 2007;116:1176–1184. doi: 10.1161/CIRCULATIONAHA.106.650903. [DOI] [PubMed] [Google Scholar]
- 36.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh PCH, Segers VFM, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fazel SS, Chen L, Angoulvant D, Li SH, Weisel RD, Keating A, Li RK. Activation of c-kit is necessary for mobilization of reparative bone marrow progenitor cells in response to cardiac injury. FASEB J. 2008;22:930–940. doi: 10.1096/fj.07-8636com. [DOI] [PubMed] [Google Scholar]
- 39.Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li RK. c-kit dysfunction impairs myocardial healing after infarction. Circulation. 2007;116:I77–I82. doi: 10.1161/CIRCULATIONAHA.107.708107. [DOI] [PubMed] [Google Scholar]
- 40.Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Hu Q, Nakamura Y, Lee J, Zhang G, From AHL, Zhang J. The Role of the Sca-1+/CD31- Cardiac Progenitor Cell Population in Postinfarction Left Ventricular Remodeling. Stem Cells. 2006;24:1779–1788. doi: 10.1634/stemcells.2005-0386. [DOI] [PubMed] [Google Scholar]
- 42.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, Sun Y, Evans SM, Laugwitz KL, Chien KR. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 43.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 46.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adrogue J, Sharma S, Ngumbela K, Essop M, Taegtmeyer H. Acclimatization to chronic hypobaric hypoxia is associated with a differential transcriptional profile between the right and left ventricle. Molecular and Cellular Biochemistry. 2005;278:71–78. doi: 10.1007/s11010-005-6629-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.