

Abstract
The widely used cholesterol-lowering drugs, statins, were reported to reduce the incidence of stroke and the progression of Alzheimer's disease. However, little is known on how statins exert these beneficial effects. In this study, we investigated the molecular mechanisms underlying the neuroprotective actions of statins in primary cultured cortical neurons. We found that chronic treatment of neurons with a low dosage of two CNS-permeable statins (lovastatin and simvastatin) selectively reduced NMDA-induced cell death but not the caspase-mediated apoptosis. The protective effects of stains were inhibited by mevalonate, a PI3K inhibitor, and tyrphostin AG538, suggesting roles for cholesterol and insulin/IGF-1 signaling in the neurotoxic response. We further demonstrate that statins block calcium-dependent calpain activation, resulting in complete suppression of protein truncation events on multiple calpain substrates that are involved in neuronal death including CDK5 coactivator p35 cleavage to p25, GSK3 and β-catenin. This is followed by reduced and increased nuclear translocation of p25 and β-catenin, respectively. Under excitotoxic conditions, the activities of CDK5 and β-catenin are exclusively regulated by calpain-mediated cleavage while apoptosis modulates β-catenin mainly through phosphorylation. Strikingly, our data demonstrate that the calpain-blocking effect of statins is largely mediated by stimulation of α-secretase cleavage of APP, resulting in increased secretion of its soluble form, sAPP. Finally, our data suggest that statin-regulated sAPP secretion occurs via activation of the PI3K pathway and inhibition of ROCK signaling. Altogether, our study provides novel insights into statin-mediated neuronal excitoprotection through both cholesterol-dependent and -independent mechanisms and links them to calpain-mediated neuronal death.
Introduction
Excitotoxicity mediated by glutamate-gated ion channels is a well documented form of neuronal death caused by brain ischemia (Sattler and Tymianski, 2001), which has also been associated with several neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease (Choi, 1988, 1995). Elevated extracellular glutamate has long been recognized as a hallmark phenomenon during neuronal excitotoxicity (Choi, 1988, 1995). Although excitotoxicity is triggered by an exaggerated and prolonged rise in intracellular Ca2+, little is known about the subsequent events that ultimately lead to cell death. Excessive glutamate triggers massive Ca2+ influx through NMDA receptors (NMDARs), which in turn can activate the Ca2+-dependent protease calpains. Calpains are likely to be involved in processing of numerous enzymes and cytoskeletal components, thereby linking their activity to a variety of intracellular events implicated in excitotoxicity-related conditions such as hypoxia, ischemia, epilepsy, and Alzheimer's disease (Ray and Banik, 2003).
Hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, known as “statins,” have been highly effective in lowering serum cholesterol and reducing the incidence of coronary events. Epidemiological studies have also supported a beneficial effect in the human subjects taking statins through a reduced prevalence of AD and stroke (Switzer and Hess, 2006; Whitfield, 2006; Miida et al., 2007). A limited number of clinical and experimental studies have been published to elucidate the multiple mechanisms, dependent and independent of statins' anti-cholesterol effect (Delanty et al., 2001). In addition to its widely studied effect on APP processing and reducing Aβ production, which is likely mediated via its action on protein isoprenylation (Cordle et al., 2005; Cole and Vassar, 2006), statins have also been reported to be excitoprotective (Zacco et al., 2003; Bösel et al., 2005). However, the detailed mechanisms are largely undefined.
Cortical neuronal culture model of NMDA toxicity has been used extensively to investigate the mechanisms of neuronal injury and to test/screen for neuroprotective agents. Excitotoxic neuronal death induced by NMDA has been shown to occur through both necrosis and apoptosis, with apoptosis being the predominant form when the insults are relatively mild (30–300 μm NMDA) (Bonfoco et al., 1995). Using this model, we examined the excitoprotective effect of statins. Chronic treatment with statins protected cortical neurons against NMDA toxicity. Neuroprotection was substantially abolished by cotreatment with either 1 mm mevalonate or cholesterol, suggesting that the neuroprotective effect of statins is mediated by inhibition of de novo cholesterol synthesis. In addition, our data revealed a cholesterol-independent mechanism by which statin excitoprotection involves stimulation of soluble APP secretion, which is likely modulated by Rho-ROCK signaling and subsequent attenuation of calcium-dependent calpain activation.
Materials and Methods
Antibodies and chemicals.
Spectrin α II C-3 (sc-48382), CDK5 (cyclin-dependent kinase 5) C-8 (sc-173), p35 C-19 (sc-820), IGF-IR β C-20 (sc-713), and β-actin (sc-1615) antibodies were from Santa Cruz Biotechnology. Glycogen synthase kinase 3 β (GSK3β, 9315), pGSK3β (Ser-9, 9323), GSK3α (9338), pGSK3α (Ser-21, 9316), pGSK3α/β (Ser-21/9, 9327), AKT (2966), pAKT/Ser-473 (4058), phospho-β-catenin (Ser33/37/Thr41, 9561), insulin receptor β (3025) antibodies were from Cell Signaling Technology. β-Catenin (C-terminal, 610153) was from BD Transduction Laboratories. The monoclonal antibody 22C11, MAP-2 (MAB 3418), NeuN (MAB377), and pY (4G10) were from Millipore. Alexa 488-conjugated anti-mouse IgG and Alexa-594-conjugated anti-rabbit IgG were from Invitrogen.
Lovastatin (LOV), simvastatin, mevalonic acid (MVA), cholesterol, NMDA, glycine, I-OMe-tyrphostin AG 538, farnesyl pyrophosphate (FPP), geranylgeranyl pyrophosphate (GGPP), 4′,6′-diamidino-2-phenylindole (DAPI), trypan blue, poly-d-lysine, and SP600125 were obtained from Sigma. In situ cell death detection kit (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, TUNEL) was obtained from Roche. The chemical compounds including LY294002, wortmannin, APV, staurosporine (STS), BH3I-1, camptothecin (CPT), Y27632, FTI-277, FTase inhibitor II, FPT inhibitor II, TAPI-2, PD98059, U0126, SU203580, Z-VAD-FMK, calpain inhibitor I/N-acetyl-Leu-Leu-norleucinal, calpastatin/CS peptide, and PD150606 were obtained from Calbiochem. Aβ25-35 peptides were from Bachem.
Primary neuronal cell culture.
Primary cortical neurons were isolated and purified from embryos of Sprague Dawley rats at embryonic day 17 (E17) as described previously (Han et al., 2005). Isolated primary neurons were plated onto coverslips precoated with poly-d-lysine (100 μg/ml) at a density of 75,000 per well in 24-well plates for staining or plated into 6-well plates at a density of 600,000 per well or 100 mm dishes at a density of 3,000,000 per dish precoated with poly-d-lysine for Western blot analysis. The cultures were maintained in serum-free Neurobasal medium and were treated with 5 μm AraC to inhibit proliferation of non-neuronal cells. All experiments presented in this work were performed on pure neuronal cells (>95% neuronal purity assessed by staining with neuronal marker proteins: neuronal-specific nuclear protein/NeuN and microtubule-associated protein-2/MAP-2) after 14 DIV.
NMDA-induced neuronal cell death.
The cultures were maintained in serum-free Neurobasal medium for 2 weeks to allow development of NMDA receptors before being challenged with NMDA (100 μm, 15 min) in Mg2+-free Eagle's balanced salt solution (EBSS) containing 1.8 mm CaCl2 and 100 μm glycine. After NMDA exposure, cells were gently washed with EBSS/1.8 mm CaCl2 and 1.2 mm MgCl2 and returned to the original culture medium for an additional 16–24 h at 37°C with 9.6% CO2 before being assessed for cell death. Neurons were also treated with 200 nm STS (6 h), 50 μm BH3I-1 (6 h), 10 μm CPT (6 h), or 25 μm Aβ25-35 peptide (24 h). To test the effect of LOV, 500 nm LOV was added 3 d before NMDA exposure except as otherwise indicated in some experiments. LY294002, APV, Y27632, PD98059, U0126, SU203580, Z-VAD-FMK, calpain inhibitors, and SP600125 were added 1 h before NMDA exposure.
After treatment with cell death-inducing or blocking agents, cell death was assessed for necrosis and apoptosis. In most experiments, cell survival was assessed by trypan blue exclusion. After staining with trypan blue at 0.4% for 5 min, cells were then washed with PBS and fixed in 4% paraformaldehyde for 10 min. Trypan blue-positive cells were counted in 10 randomized fields. Apoptotic neurons were identified by TUNEL, followed by counterstaining with membrane-permeable DAPI (0.1 μg/ml, 5 min). In addition to TUNEL, apoptosis was also assessed by condensed morphological changes of the nuclei.
Immunofluorescence staining.
Two week-cultured neurons seeded on coverslips were fixed with 4% paraformaldehyde/PBS at room temperature for 15 min and permeabilized in 0.2% Triton X-100/PBS for 5 min. After washing in PBS, The neurons were blocked with PBS containing 10% goat serum at room temperatures for 60 min in a humid chamber. Primary antibodies, including NeuN (1:500), MAP-2(1:500), p35 (1:200), and β-catenin (1:250), diluted in the blocking buffer were applied to the specimens and incubated overnight at 4°C overnight. After extensive washes, Alexa 488-conjugated anti-mouse or Alexa-594-conjugated anti-rabbit secondary antibody (1:500) was applied and incubated for 1 h at room temperature, followed by counterstaining with DAPI. Slides were mounted in Fluoromount medium, and immunofluorescent signals were observed under confocal microscopy (LSM510; Zeiss). For MAP-2 staining, the length of neurites was quantified from 50 of the MAP-2-positive cells, and the number of processes projecting from each positive cell was also quantified. For p35/25 staining, p35/25 immunofluorescence signals in the nucleus were compared with those in the cytoplasm, and the neurons showing higher fluorescence signals in the nucleus than in the surrounding cytoplasm were counted as “nuclear p35/25-positive cells.” For β-catenin staining, immunofluorescence signals in the nucleus were quantified.
Western blot analysis.
The assays were performed as described (Han et al., 2005). Primary antibodies used were as follows: spectrin α II C-3 (1:1000), cleaved caspase-3 (1:1000), CDK5 C-8 (1:1000), p35 C-19 (1:1000), GSK3α/β (1:1000), GSK3β (1:1000), pGSK3β (1:1000, Ser-9), AKT (1:1000), pAKT (1:1000, Ser-473), β-catenin (1:1000, C-terminal), phosphor-β-catenin (1:1000, Ser33/37/Thr41), IR-β (1:1000), IGF-IR β (1:1000), pY (1:1000), and β-actin (1:5000). In some experiments, Western blots were scanned and protein bands were quantified using Scion Image software. Mouse monoclonal antibody 22C11 (1:200) was used to detect sAPP secretion from the cultured conditioned media.
Detection of cytosolic β-catenin.
Cells were collected in a lysis buffer containing 20 mm HEPES, pH 7.4, 2 mm MgCl2, 1 mm EDTA, 100 mm KCl, 0.5 mm DTT, and a cocktail of protease inhibitors. Cell mixtures were incubated on ice for 20 min and homogenized using a Dounce homogenizer for 25 strokes followed by centrifugation at 15,000 × g for 30 min. Equal amounts of lysate samples were then subjected to SDS-PAGE and Western analysis.
Preparation of dominant-negative mutant of ROCK1-expressing adenovirus.
The cDNA for human ROCK1 was purchased from Open Biosystems. The dominant-negative mutant (DN ROCK) was generated according to Itoh et al. (1999). Accordingly, the cDNA encoding the amino acids 1-477 was amplified by PCR using the following primers: (1) 5′ forward primer, ATG TCGACTGGGGACAGTTTTGAG; (2) 3′ reverse primer, TCATCTAGATTTCTTCTTTGATTTCCCTCTTC (ending at amino acid 477); (3) mutated forward primer, AAGGTATATGCTATGATG (for K-to-M mutation) CTTCTCAGC; and (4) mutated reverse primer, GCTGAGAAGCATCATAGCATATACCTT. The PCR product was then cloned into the pCR-BluntII-Topo vector (Invitrogen) and verified by sequencing. The insert was subsequently subcloned into pAdTrack-CMV at the NotI and EcoRV sites, which was further used for transient transfection and adenovirus preparation. Adenoviral genome was made by carrying out recombination of pAdTrack-CMV-ROCK1 with pAdEasy1 and transfection into HEK293 cells, as described by He et al. (1998). The virus was amplified two rounds and purified by anionic exchange cartridge (Sartorius).
Statistical analysis.
Data are presented as mean ± SD. For statistical comparison, the Student's t test was used. p values smaller than 0.05 were considered to be statistically significant.
Results
Chronic statin treatment at low dosage protects primary cortical neurons against NMDA-induced excitotoxicity
It has been reported that statins are excitoprotective in neurons insulted by NMDA. Various statins have been compared and it has been shown that the protective potency is proportional to their ability to inhibit the HMG-CoA reductase activity (Zacco et al., 2003). To study the mechanisms underlying statin's protective effect, we treated primary cultured cortical neurons (14 DIV) with two different statins that are known to be the most permeable to the CNS, lovastatin and simvastatin. We sought to investigate these statins using a chronic treatment regimen with low dosages (<1 μm) to mimic the clinical setting. All work presented herein was done in 2-week-cultured primary cortical neurons to allow expression of a full spectrum of the NMDA receptors. Similar results were obtained with lovastatin and simvastatin, we therefore only present data on lovastatin.
In a pilot study, we used a classic trypan blue assay to assess cell viability and tested two statins at various concentrations ranging from 0.1 to 1 μm. Concentrations of >2 μm resulted in acute cytotoxicity whereas the low concentrations, ∼0.5–1.0 μm, provided the optimal maximum protection when neurons were challenged with NMDA (100 μm), as assessed by trypan blue permeability. At an optimal concentration (0.5 μm), statins conferred protection in NMDA-treated neurons of >60%, comparable to that of APV, a specific NMDAR antagonist (Fig. 1A). Interestingly, the protection was only achieved with either statin after chronic treatment (3–5 d). These optimized conditions of low dosage and chronic treatment (0.5 μm and 3 d) are relevant to the clinical regimen and were used consistently throughout our studies. As previously reported (Bonfoco et al., 1995; Han et al., 2005), exposure to 100 μm NMDA for 15 min resulted in ∼70% neuronal death with the majority being apoptosis, as evidenced by the TUNEL-positive staining (Fig. 1B). The apoptosis was significantly reduced by statin pretreatment (Fig. 1B), an effect comparable to that of three calpain inhibitors, N-acetyl-Leu-Leu-norleucinal, calpastatin/CS peptide, and PD150606 (data not shown). In contrast, NMDA-induced neuronal death was not prevented by Z-VAD-fmk, a pan caspase 3 inhibitor (data not shown).
Figure 1.

Lovastatin protects NMDA-induced excitotoxicity in primary cultured cortical neurons. A, Representative micrographs of trypan blue-positive cells. Neurons were pretreated with 500 nm LOV for 3 d or 100 μm APV, a specific NMDAR antagonist, for 30 min or left untreated before a 15 min exposure to 100 μm NMDA and 100 μm glycine. Viability was measured by cell counting after trypan blue staining at 24 h after NMDA exposure. B, Representative photographs and quantitative assessment of apoptotic cell death by TUNEL assay. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA. C, Photomicrographs showing fluorescence staining of MAP-2 with DAPI. Primary cortical rat neurons were pretreated with 500 nm lovastatin or vehicle for 3 d before exposure to NMDA for 15 min. After fixation, cells were stained with the neuronal markers (MAP-2 and NeuN, green) to delineate the cell morphology, followed by counterstaining with membrane-permeable DAPI (blue). The length of neurites was quantified from 50 of the MAP-2-positive cells, and the number of processes projecting from each positive cell was also quantified. Data are presented in the bar graph as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA.
We also examined morphological alterations on apoptotic cells using a fluorescent microscope, which were characterized by condensed nuclei and cell shrinkage. Using microtubule associated protein 2 (MAP2) as a neuron-specific marker, we assessed NMDA-receptor-mediated neurotoxicity in neuron-enriched cultures. We found that, compared with LDH assay, MAP2 staining plus DAPI for nuclei morphology was more sensitive and reliable in evaluating neuronal damage and death. As shown in Figure 1C, 30 μm NMDA exposure for 15 min resulted in an almost complete loss of dendritic structures of all the neurons. Chronic pretreatment of neurons with either statin for 3 d prevented dendritic loss to a great extent (>60% protection), as quantified by both the number of dendrites per neuron and the length of the remaining dendrites (Fig. 1C).
Lovastatin selectively protects neurons against NMDA-induced excitotoxicity but not against caspase-mediated apoptosis—a specific effect of calpain inhibition
To test whether statins are universal inhibitors of apoptosis or specific to NMDA-induced neurotoxicity, we assessed their protective effects on cell death induced by different proapoptotic agents, including STS (200 nm), a potent protein kinase C inhibitor, the cell-permeable Bcl-2 homology (GH)-3 domain inhibitor BH3I-1, the DNA topomerase inhibitor CPT, or the amyloid peptide (Aβ25-35). All these agents are known to induce classic apoptotic cascades involving cytochrome c release from the mitochondria and the subsequent caspase activation. To our surprise, statins only exhibited inhibitory effect on NMDM-induced cell deaths without effect on caspase-mediated apoptosis (Fig. 2A).
Figure 2.
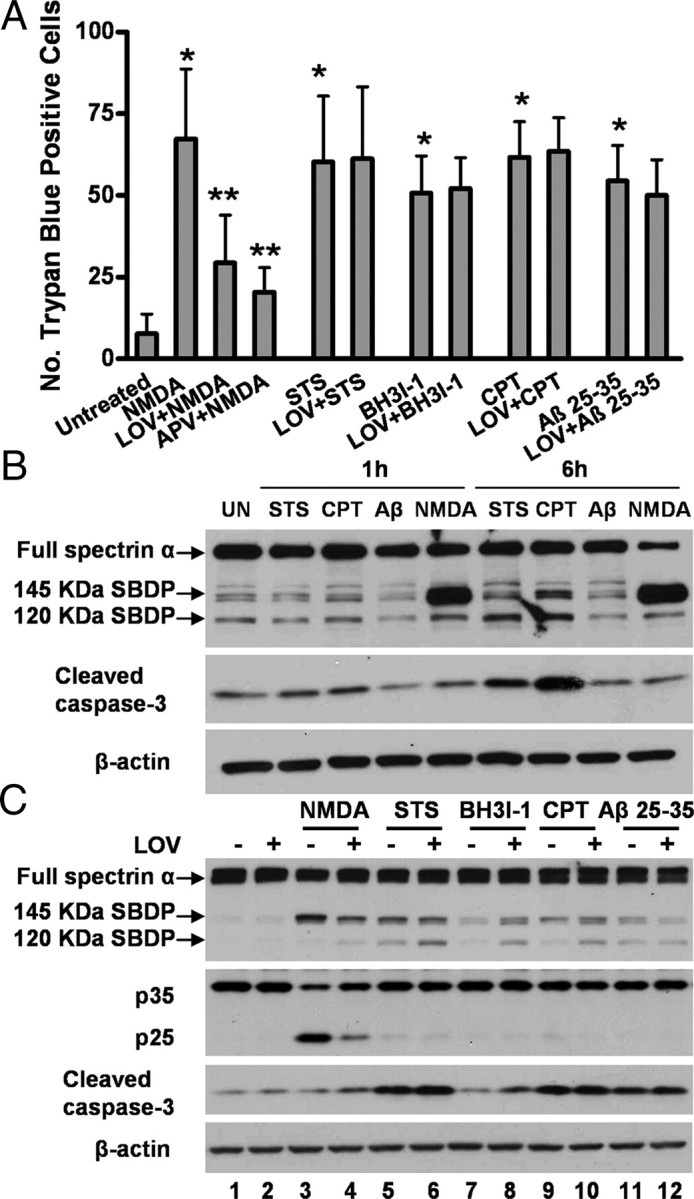
Lovastatin selectively protects neurons against NMDA-induced excitotoxicity but not against caspase-mediated apoptosis. A, Quantitative assessment of trypan blue-positive cells. Neurons were pretreated with 500 nm LOV for 3 d or left untreated before exposure to 100 μm NMDA, 200 μm STS, 50 μm BH3I-1, 10 μm CPT, or 25 μm Aβ25-35 peptide. Viability was measured by cell counting after trypan blue staining. Trypan blue-positive cells were quantitated as described in Materials and Methods. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA. B, NMDA induces significant calpain activation compared with other stresses. Neuronal lysates were prepared 1 or 6 h after exposure to STS, CPT, Aβ25-35 peptide, or NMDA. Representative Western blot autographs of spectrin and cleaved caspase-3 are shown. C, Lovastatin selectively suppresses calpain activation but not caspase-3 activation. Neurons were pretreated with or without 500 nm lovastatin for 3 d before an exposure to NMDA, STS, BH3I-1, CPT, or Aβ25-35 peptide. Representative Western blots of spectrin, p35/25, and cleaved caspase-3 are shown.
Overactivation of NMDA receptors is known to mediate excitotoxicity due to excessive entry of calcium, leading to the activation of several calcium-dependent enzymes. One of them is the family of calcium-activated proteases, calpains that appear to play a dominant role in excitotoxic neuronal death. We therefore examined the effect of statins on NMDA-induced cleavage of spectrin α, a cytoskeletal protein that is a major substrate for both calpain and caspases, albeit at differential cleavage sites. Indeed, unlike several classic proapoptotic agents tested that induced caspase-mediated pathways, NMDA induced primarily calpain activation, as evidenced by calpain-specific cleavage of spectrin and p35 to p25 conversion (Fig. 2B,C). Notably, lovastatin treatment remarkably suppressed calpain activation in response to NMDA exposure (Fig. 2C, lanes 1–4) but with almost no effect on caspase-3 activation (lanes 5–12).
Calpains are also known to alter properties of many other substrate proteins by cleavage regulation. To validate statin's effect on calpain activity, we investigated the proteolytic truncation of three additional substrates that are crucial mediators in neuronal cell death. They include p35 (CDK5 coactivator), GSK3, and β-catenin.
Lovastatin suppresses CDK5 activation induced by NMDA
We examined statin's effect on preventing the calpain-mediated cleavage from p35 to p25. Upon NMDA treatment, the cleavage took place within the first few minutes (Fig. 3A) as shown by a time course study. Increases in levels of the truncated products (mostly N terminus) appeared within 5 min of NMDAR activation and were stable for periods >30 min (Fig. 3A). Statin pretreatment completely inhibited the conversion from p35 to p25 as detected by immunoblot analysis (Fig. 3B) and also prevented the p25 translocation into the nuclei upon NMDA insult (Fig. 3C).
Figure 3.
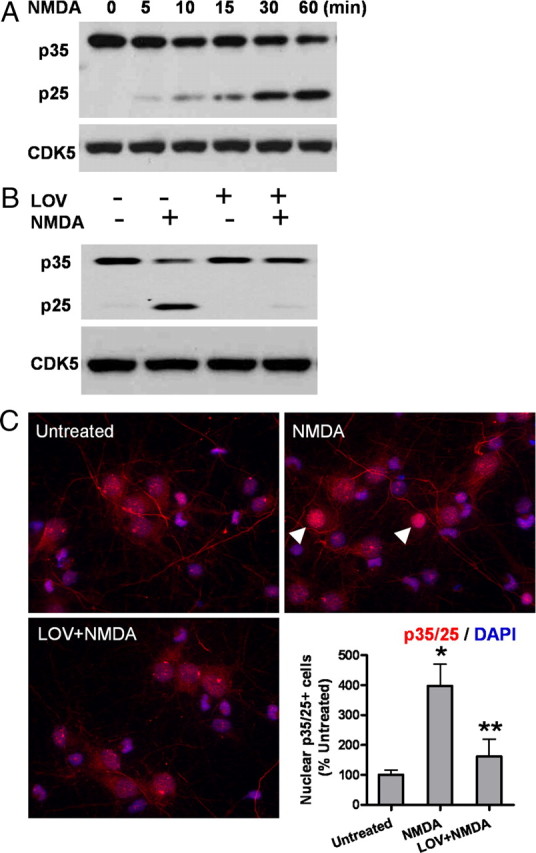
Lovastatin suppresses CDK5 activation induced by NMDA in primary neurons. A, The time course of p35 to p25 conversion after NMDA exposure. B, Lovastatin pretreatment completely inhibits the NMDA-induced conversion from p35 to p25. The protein level of CDK5 was not changed. C, Cells were fixed and immunostained for p35/p25 (red), followed by counterstaining with DAPI (blue). The p35/p25 immunofluorescent signals in the nucleus were compared with those in the cytoplasm, and the neurons showing higher fluorescence signals in the nucleus than in the surrounding cytoplasm were counted as “nuclear p35/25-positive cells.” Representative positive cells are shown by the arrowheads. Data are presented as the mean ± SD. *p < 0.001 versus untreated; **p < 0.001 versus NMDA.
Lovastatin suppresses NMDA-induced GSK3β activation and preserves β-catenin
We next examined the consequences of the other two effector molecules downstream of both PI3K/Akt and Wnt signaling pathways upon statin-mediated neuroprotective action. The biological activities of both GSK3β and β-catenin are classically recognized to be regulated by their specific phosphorylation events. However, they have recently been found to also be regulated by calpain-mediated truncation events, resulting in modulated protein activities (Abe and Takeichi, 2007; Goñi-Olover et al., 2007). We thus investigated the relationship between these two regulatory mechanisms.
Upon treatment of cells with NMDA, a detectable increase in calpain-mediated truncation of GSK3 was found (Fig. 4A). Concurrently, we observed a decrease in Ser-9 phosphorylation, an event which is associated with elevated kinase activity (Fig. 4B). All these changes were reversed by statin treatment. No effect was found from statin treatment on the pSer-21 of GSK3 α which inversely correlates with its kinase activity (data not shown). It is not clear whether this protein truncation event is related to its Ser-9 phosphorylation, though it was reported that the truncated GSK3 is associated with elevated kinase activity (Goñi-Olover et al., 2007).
Figure 4.
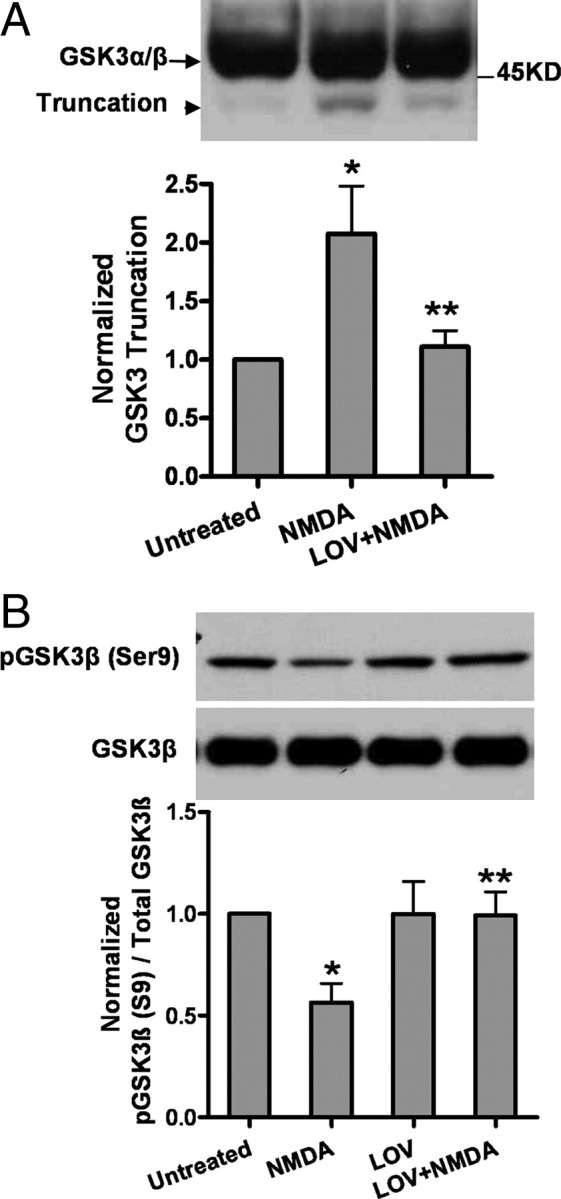
Lovastatin suppresses NMDA-induced GSK3 truncation and GSK3β activation in primary neurons. A, Lovastatin pretreatment attenuates the GSK3 truncation induced by NMDA exposure. The representative Western blots and densitometry analysis of GSK3 truncation are shown. Data are presented as the mean ± SD. *p < 0.001 versus untreated; **p < 0.001 versus NMDA. B, Lovastatin suppresses GSK3β activation upon NMDA. GSK3β activity was measured by the Ser 9 phosphorylation which inversely correlates with its enzymatic activity. Densitometry analysis of pGSK3β/GSK3β ratio is shown in the bar graph. Data are presented as the mean ± SD. *p < 0.001 versus untreated; **p < 0.001 versus NMDA.
We next examined whether changes in GSK3β concurred with alteration of the levels of β-catenin, a crucial transcriptional activator whose turnover and translocation to the nucleus is known to be mostly regulated by GSK3β phosphorylation. We first tested whether the turnover of β-catenin differed between NMDA-treated and statin-rescued neurons. Indeed, statins could rescue degradation of β-catenin induced by NMDA, resulting in elevated β-catenin levels (Fig. 5A). Moreover, as reported (Hagen et al., 2004), we found that cytosolic β-catenin correlated with its nuclear level, reflective of its transcriptional activity (Fig. 5B,C). Most strikingly, we found that β-catenin was regulated solely by the calpain-mediated truncation events (Fig. 5C) rather than by phosphorylation, which is typically induced by conventional apoptotic stimuli (Fig. 5D). Statin exerted no effect on the phosphorylated β-catenin, which was selectively induced by staurosporine but not by NMDA. The question remains of whether β-catenin is regulated exclusively by calpain-mediated truncation and whether this is dependent on the GSK3 phosphorylation and/or its truncation mechanism.
Figure 5.

Effect of lovastatin on β-catenin truncation and translocation induced by NMDA. A, Lovastatin pretreatment attenuates the β-catenin truncation induced by NMDA exposure. The representative Western blots and densitometry analysis of β-catenin truncation are shown. Data are presented as the mean ± SD. *p < 0.001 versus untreated; **p < 0.001 versus NMDA. B, The protein level of cytosolic β-catenin was analyzed by Western blotting. Densitometry analysis of cytosolic β-catenin is shown below. Data are presented as the mean ± SD. *p < 0.05 versus untreated; **p < 0.001 versus NMDA. C, Neurons were fixed and immunostained for β-catenin (red), followed by counterstaining with DAPI (blue). β-Catenin immunofluorescent signals in the nucleus was quantified. Data are presented as the mean ± SD. *p < 0.001 versus untreated; **p < 0.001 versus NMDA. D, The representative Western blots of phospho β-catenin (Ser33/37/Thr41) are shown.
Lovastatin's excitoprotective effect is mediated via insulin signaling, regulated by PI3K/Akt activation pathways
We then investigated which signaling pathway(s) underlies statin's excitoprotective mechanism. Since statins are reported to activate the phosphatidylinositol-3-kinase (PI3K) pathway, resulting in the phosphorylation of Akt (Kureishi et al., 2000) in endothelial cells, we tested whether a similar regulatory pathway exists in statin-elicited neuronal protection against excitotoxic insult.
We first tested a panel of pharmacological inhibitors against PI3K, MARK, Jun/JUNK, and tyrphostin AG538 and found that only the PI3K-specific inhibitor LY294002 and AG538 completely abolished statin-mediated excitoprotection (Fig. 6A), whereas the other inhibitors (PD98059/MEK1/2, U0126/MEK1/2, SU203580/p38MARK, and SP600125/Jun/JNK) exhibited no significant effect (data not shown). Consistently, treatment of cells with statins resulted in sustained and prolonged activation of Akt, an immediate downstream effector of PI3K (Fig. 6B), which was completely abolished by LY294002 (Fig. 6C). We further demonstrated that statins augmented insulin-induced autophosphorylation of the insulin receptor and IGF-1 receptor on tyrosine residues (Fig. 6D). Although the effect of lovastatin alone on tyrosine phosphorylation was weak, it was visible, reflecting mild, but sustained phosphorylation of Akt (Fig. 6B,D).
Figure 6.

Excitoprotection is mediated via PI3K/Akt pathway regulated by insulin signaling. A, Preincubation with the PI3K-specific inhibitor LY 294002 (10 μm) or insulin growth factor 1 (IGF-1) receptor protein tyrosine kinase inhibitor I-OMe-tyrphostin AG 538 (10 μm) completely abolishes the neuroprotective effect of lovastatin. Trypan blue-positive cells were quantified. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA; ***p < 0.001 versus LOV+NMDA. B, Lovastatin pretreatment results in sustained Akt activation upon NMDA challenge. The protein levels of the phosphorylated and total Akt were measured by Western blot analysis at various time points (0, 5, 10, 15, 30, 60, and 120 min) after NMDA exposure with or without 500 nm lovastatin pretreatment. C, The increased protein level of the phosphorylated Akt by lovastatin is abolished by LY294002. The protein levels of the phosphorylated and total Akt were measured by Western blot analysis. Densitometry analysis of pAkt(S473)/Akt ratio is shown in the bar graph. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA; ***p < 0.001 versus LOV+NMDA. D, Lovastatin enhances insulin-induced autophosphorylation of both the insulin receptor (IR) and IGF-1β receptor (IGF-1Rβ). Cells were pretreated with or without lovastatin for 48 h and were then stimulated with or without 1 μm insulin for 15 min. The IP-Western was performed using standard procedures. The representative Western blots and densitometry analysis are shown. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus insulin.
Statin's excitoprotection is likely mediated via both cholesterol-dependent and -independent mechanisms
Previous studies have shown that mevalonate, an intermediate in cholesterol biosynthesis, at low concentrations (200–250 μm), restores isoprenylation of proteins; however, at a high concentration (1 mm), it also rescues de novo cholesterol synthesis (Cole et al., 2005). To examine whether statin's excitoprotective mechanism is dependent on cholesterol levels, we added mevalonate to neurons which were pretreated with statins for 3 d. Cotreatment with 1 mm mevalonate completely abolished statin's protective effect, resulting in neuronal death comparable to the NMDA-treated control (Fig. 7A). Similar results were achieved by adding cholesterol (50 μm), indicating a cholesterol-dependent mechanism for statin's excitoprotection.
Figure 7.

Excitoprotection is likely mediated via both the cholesterol-dependent and -independent mechanisms. A, Neurons were pretreated with 500 nm LOV or coincubated with lovastatin and MVA (1 mm or 200 μm) or cholesterol (50 μm) before exposure to NMDA. Trypan blue-positive cells were quantitated. Data are presented as the mean ± SD from three independent experiments. *p < 0.001; **p < 0.01. B, Neurons were pretreated with LOV or coincubated with lovastatin and Y27632 (10 μm, Rock-specific inhibitor), FPP, or GGPP before exposure to NMDA. Trypan blue-positive cells were quantitated. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA; ***p < 0.001 versus LOV+NMDA. C, Neurons were infected with GFP or DN ROCK before exposure to NMDA. Trypan blue-positive cells were quantified. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus GFP; **p < 0.001 versus GFP+NMDA.
We then investigated whether statin's known function in modulating protein isoprenylation is involved in its excitoprotective action. We found that 200 μm mevalonate partially restored statin's effect against NMDA (Fig. 7A) while mevalonate alone, at 200 μm or 1 mm, exerted no significant effect (data not shown). This finding suggests the involvement of protein isoprenylation in statin's excitoprotective mechanism, which may be independent of its cholesterol-lowering effect.
Statin's excitoprotective mechanism likely involves Rho-ROCK signaling
Posttranslational isoprenylation is an important process regulating the activity of small G-proteins of the Ras superfamily by controlling their membrane association (Zhang and Casey, 1996; Seabra, 1998). Isoprenoids (FPP and GGPP), are essential for the farnesylation and geranylgeranylation of certain proteins such as Ras and Rho, respectively. Moreover, these two isoprenoids are the intermediate products downstream of mevalonate and they can be inhibited by statins (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To test which signaling pathway is involved in statin' s excitoprotection, we added FPP and GGPP (5 μm) to cultured neurons, with or without statin. Indeed, addition of FPP completely abolished statin's protective effect while GGPP partially interfered with statin's effect (Fig. 7B). This finding is consistent with a previous report on a reciprocal relationship between isoprenoid-mediated Rho-ROCK signaling and sAPPα secretion (Pedrini et al., 2005) based on studies using dominant-negative ROCK1 and a specific inhibitor of farnesyl transferase (FTI-1), the enzyme mediating Rho modification by FPP.
We then further tested whether a series of compounds inhibiting farnesyltransferases (FTIs) that modulate isoprenoid levels, along with a specific ROCK inhibitor Y27632 (10 μm), exert effects on excitoprotection. Interestingly, the three FTIs we tested exerted no significant effect on NMDA-induced neuronal death (date not shown), while Y27632 displayed a countereffect on NMDA (Fig. 7B), suggesting the involvement of Rho-ROCK signaling. Interestingly, statin and Y27632 did show an additive effect, further suggesting that Rho-ROCK lies downstream of statins. This was further confirmed by expression of DN-ROCK in neurons: adenovirus-mediated expression resulted in increased cell survival upon NMDA exposure, an effect comparable to Y27632 (Fig. 7C).
Lovastatin's protective effect is partially mediated by its stimulating action on APP shedding by α-secretases
Rho-ROCK has been reported to modulate APP shedding by α-secretases, resulting in increased sAPP secretion (Kojro et al., 2001; Parvathy et al., 2004). Moreover, sAPP is also known to be a potent mediator attenuating excessive calcium entry and excitotoxicity (Mattson et al., 1993; Mattson, 1997). Indeed, we found that 500 nm lovastatin increased sAPP release in a time-dependent manner with maximum effect achieved following at least 24 h treatment with lovastatin (Fig. 8A), which is in agreement with previous work (Kojro et al., 2001; Parvathy et al., 2004). This effect was further demonstrated to be due to the conventional α-secretases since it was completely abolished by TAPI-2, a potent inhibitor to the conventional enzymes (i.e., adamalysins ADAM 10 or ADAM17/TACE) (data not shown).
Figure 8.

Excitoprotection is partially mediated by stimulating APP shedding from α-secretases. A, The time course of sAPP following lovastatin treatment. Mouse monoclonal antibody 22C11 was used to detect sAPP secretion from the conditioned culture media. B, Both ROCK-specific inhibitor Y27632 and DN ROCK infection stimulate sAPP secretion similarly to lovastatin pretreatment. Soluble sAPP secretion from the conditioned media was assayed as described in A. C, Depletion of sAPP blunts the excitoprotective effect of lovastatin. The conditioned medium after incubation of neurons with lovastatin was removed, followed by replacement of fresh medium or immunoprecipitation with the 22C11 antibody before adding it back to the neurons. Depletion of sAPP was confirmed by Western blot. Trypan blue-positive cells were quantified. Data are presented as the mean ± SD from three independent experiments. *p < 0.001 versus untreated; **p < 0.001 versus NMDA; ***p < 0.001 versus LOV+NMDA. D, Depletion of sAPPα abolishes lovastatin's ability to suppress calpain activity. Neurons were treated the same way as in C and cell extracts prepared were blotted with antibodies, as indicated. E, LY294002 inhibits lovastatin-induced sAPP secretion. The representative Western blots and densitometry analysis are shown. Data are presented as the mean ± SD. *p < 0.01 versus untreated; **p < 0.01 versus LOV.
We then examined whether ROCK inhibition can also modulate sAPP secretion. Interestingly, both the Y27632 compound and the expression of the Dn-ROCK in neurons exhibited an effect in boosting sAPP secretion similar to that exhibited by statin (Fig. 8B), suggesting that ROCK signaling is likely involved in statin's excitoprotection.
We then used two experimental designs to investigate whether the stimulation of sAPP secretion is indispensable for the excitoprotective action of statins. Removal of the conditioned medium after incubation of neurons with lovastatin for 3 d, followed by replacement with fresh medium at the time of NMDA exposure, almost completely abolished the excitoprotective effect of statin. Similar results were obtained with an N-terminal monoclonal antibody 22C11 (epitope AA 68–81) used to deplete all soluble forms of APP from the culture medium during coincubation with lovastatin. At the concentration tested (0.5 μg/ml), 22C11 alone did not induce significant cell death (Fig. 8C). These results indicate that the statin-induced excitoprotection is mediated mainly by sAPP We then further demonstrated that the statin-triggered suppression of calpain proteolytic conversion of p35 to p25 was abolished by greatly reduced soluble APP molecules from the cell culture medium by removal of the statin-treated condition medium or by a specific monoclonal antibody 22C11 antibody (0.5 μg/ml) (Fig. 8D). This finding is consistent with the known effect of soluble sAPP in preventing calcium disturbances in excitotoxic conditions (Mattson et al., 1993; Mattson, 1997).
We previously showed that statins can activate the PI3K pathway, resulting in phosphorylation of Akt. We therefore investigated whether the lovastatin-induced sAPP secretion is regulated by this upstream insulin-mediated PI3K/Akt pathway by testing whether the PI3K inhibitors LY294002 could inhibit lovastatin-induced sAPP secretion. Indeed, we found that coincubation of neurons with LY294002 and lovastatin for 3 d suppressed the sAPP release induced by statin, while the basal level of sAPP shedding and secretion appeared to be unaffected (Fig. 8E). This result indicates that the insulin signaling-induced PI3K/Akt pathway is upstream of statin's stimulation of secretases. Moreover, the blocking effect of the PI3K inhibitor is likely mediated through the effectors such as sAPP. In contrast, this blocking effect was not seen by MAPK inhibitors including U0126, PD98059, and SU203580 (data not shown). This result indicates that statin stimulation of α-secretases is mediated by the PI3K pathway.
Discussion
Statins are widely used cholesterol-lowering drugs that act by inhibiting HMG-CoA reductase, the rate-limiting enzyme in cholesterol biogenesis. Recent evidence suggests that statin use may be associated with a decreased risk for Alzheimer's disease, although the mechanisms underlying this apparent risk reduction are poorly understood. One popular hypothesis for statin action is related to the drug's ability to activate α-secretase-type shedding of the Alzheimer amyloid precursor protein (APP) ectodomain (sAPPα). In the present study, we demonstrate that, like statin's effect on reducing Aβ production, which is likely through its stimulating effect on α-secretases and sAPPα secretion, this stimulating action also mediates its excitoprotective mechanism.
Statins have been documented to exhibit potent neuroprotective effects in stroke, both for prevention and for recovery. Therefore, they are expected to exert protective actions against a broad spectrum of neuronal insults. Interestingly, our results indicate a rather selective action on excitotoxicity. We report here that the two CNS-permeable statins can selectively protect cultured cortical neurons against NMDA excitotoxic challenge, and that this protection requires chronic pretreatment of neurons with statins, which is consistent with their anti-cholesterol action. Moreover, statins are found to selectively modulate the calpain-mediated truncations on a series of molecules crucial for the neuronal survival pathways. These findings greatly expand our knowledge on statin's neuroprotective mechanisms and the signaling survival pathways involved, which will be instrumental to future therapeutic design. In particular, given the fact that there is currently no success with various pharmacological calpain inhibitors, these clinically approved statins represents a significant and safe option.
Although the calpain-mediated truncation was individually identified on CDK5 activation (e.g., p35 to p25 conversion), GSK3, and β-catenin (Abe and Takeichi, 2007; Goñi-Oliver et al., 2007), our work, for the first time, suggests a strong link for these events in a unified pathway. We showed that all these molecules are truncated by NMDA and that statins can prevent these truncations and thus suppress the NMDA-induced GSK3β activation and further preserve β-catenin. These findings identify the nuclear β-catenin as the major mediator of survival in statin-treated neurons. Specific phosphorylation events triggered by an imbalance of kinases and phosphatases are known to be the major regulatory mechanisms on the biological activities of both the GSK3β and β-catenin. Our work now suggests that calpain-mediated truncation can be the additional crucial mechanism regulating the biological activity of GSK3β. This truncation of GSK3 takes place in parallel to its Ser-9 phosphorylation, which is known to inversely correlate with its kinase activity. Interestingly, statins appear to be able to modulate on both events. Moreover, the NMDA-induced calpain-mediated truncation appears to be the sole mechanism regulating the nuclear translocation of β-catenin, which determines its biological activity on transcription, whereas other apoptotic stimulus, such as staurosporine, only elicits alterations on the phosphorylation state of β-catenin. It remains to be determined whether this novel regulation of β-catenin mediated by calpain cleavage is an independent event to its upstream modulator GSK3β and how would this truncation event further regulate β-catenin translocation into the nucleus.
Our work also brings another crucial player, CDK5/p35/p25, into the paradigm. Despite its ubiquitous expression, CDK5 activity is almost exclusively restricted to postmitotic neurons because of the neuron-specific expression of its regulators p35/p39. During neuronal insult and subsequent disruption of calcium homeostasis, conversion of inactive p35 to active p25 is mediated via proteolytic cleavage by the calcium-regulated calpains. Recently, CDK5 has received considerable attention as a regulator of neuronal death (Cruz and Tsai, 2004). In particular, studies using apoptotic and excitotoxic death models demonstrate that p25/CDK5 complexes accumulate within the nucleus and that this activity is required in excitotoxic death but not apoptotic death (O'Hare et al., 2005). It is worth stressing that statins can modulate both CDK5 activation and β-catenin via a unified protein truncation mechanism, though it is not clear whether the CDK5-mediated pathway converges with the Wnt signaling pathway.
One important question remains as to how statins suppress calpain activation. As a major family of calcium-induced proteases, calpains are activated in response to calcium flux as a result of overactivated NMDA receptor channel. Given the well defined role of soluble APP in suppressing calcium influx upon excitotoxic conditions (Mattson et al., 1993; Mattson, 1997), along with statins' stimulating action on α-secretase-mediated shedding of APP (Kojro et al., 2001, Parvathy et al., 2004), we speculated that sAPP plays a key role in statin's excitoprotection. Indeed, results from a number of experimental designs all pointed to a conclusion that statin's excitoprotection is largely dependent on its stimulating effect on sAPP. In addition, we also identified the PI3K/Akt pathway as one of the crucial upstream signaling pathways activated by insulin receptor, which is preserved by statins upon NMDA challenge. Interestingly, the specific pharmacological inhibitor LY 294002 not only abolishes statin's excitoprotective effect against NMDA, but also attenuates its effect on sAPP secretion (Fig. 8E). It is not clear whether and how this insulin signaling-regulated PI3K pathway affects statin's modulation on the sAPP secretion directly or indirectly. Together, the notions that statins can also modulate on APP processing, resulting in reduced Aβ production, likely through its stimulating effect on sAPP production which later is found to be modulated on protein isoprenylation (Buxbaum et al., 2001; Puglielli et al., 2001; Ostrowski et al., 2007), it appeared that sAPP is a key executor in the pluripotent actions of statins in CNS. Therefore, we continued to investigate the upstream signaling events that mediate statins' stimulation on sAPP production upon excitotoxic conditions.
The primary action of statins is the inhibition of HMG-CoA reductase, blocking the de novo synthesis of cholesterol and resulting in lower plasma cholesterol levels. However, recent observations demonstrate that statins have pleiotropic actions that are not dependent on cholesterol reduction (Liao and Laufs, 2005; Cole and Vassar, 2006). Specifically, statins have been shown to inhibit vascular inflammation, enhance endothelial function, inhibit the proliferation of vascular smooth muscle, reduce platelet activation and aggregation, and increase atherosclerotic plaque stability. Many of these functions were postulated to arise from disruption of the actions of small G-proteins. Although blockade of HMG-CoA reductase prevents de novo synthesis of cholesterol, it also decreases the pools of intermediate metabolites, namely the two isoprenoids FPP or GGPP, in the biosynthetic pathway that have ancillary functions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Statins are known to inhibit the isoprenoid pathway, thereby modulating the activities of the Rho family of small GTPases-Rho A–C as well as the activities of Rac and cdc42. Rho proteins, in turn, exert many of their effects via Rho-associated protein kinases (ROCKs/ROCK1 and ROCK2). Since it is known that the statin-activated shedding of APP ectodomain can be modulated by ROCKs (Pedrini et al., 2005), we therefore examined whether statin's excitoprotective mechanism requires Rho-ROCK signaling. Our findings using a ROCK-specific inhibitor Y27632, as well as direct testing of the two isoprenoids (FPP and GGPP), confirmed the involvement of both the protein isoprenylation and the Rho-ROCK signaling in statin-mediated excitoprotective mechanism. However, it is still not clear how the negative regulation by the Rho-ROCK modulates sAPP production and its interplay with the insulin signaling-mediated PI3K pathway. In several reports, the insulin/IGF-mediated signaling appears to be able to activate RhoA and thus negatively regulates on Rho-ROCK signaling in cancer and in metabolic settings (Zhang et al., 2005; Tapia, 2006). It is not clear whether the insulin/IGF-mediated survival signaling is upstream to the Rho-ROCK in neurons under excitotoxic stressed conditions.
ROCK family members (ROCK1 and ROCK2) were initially identified as a Rho-binding protein with serine/threonine protein kinase activity. They have been generally implicated in cell death and survival processes in many cell types (Shi and Wei, 2007) and reported to counter-regulate insulin signaling and the subsequent PI3-K activation in adipocytes and muscle cell lines (Begum et al., 2002; Furukawa et al., 2005). Moreover, PTEN, the phosphatase and tensin homolog, is a newly identified ROCK substrate (Li et al., 2005) and ROCK phosphorylates and stimulates its phosphatase activity, which in turn antagonizes PI3K activity, resulting in reduced Akt phosphorylation/activation. Reduced PTEN is typically found to be associated with excitotoxic response in neurons, accompanied by elevated Akt activation (Liao, unpublished data), which may contribute to the protective effect of ROCK inhibition seen with either its inhibitor Y27632 or with statin-mediated inhibition on isoprenylation (Fig. 7B). Together with the notion that Rho-ROCK signaling is required for soluble APP production, the latter is being recognized to be one of the most potent neurotrophic and neuroprotective factors in CNS and the beneficial therapeutic potentials of Rho-ROCK inhibitors are further warranted.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS054880 (F.-F.L.), R01 AG021173 (H.X.), R01 NS046673 (H.X.), R01 AG030197 (H.X.), and R01 CA118918 (Z.L.), and by Alzheimer's Association Investigator-Initiated Research Grant IIRG-06-26070 (F.-F.L.). We thank Traci Fang for assisting in preparation of primary neurons.
References
- Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–397. doi: 10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho kinase (ROK-alpha) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cells. J Biol Chem. 2002;277:6214–6222. doi: 10.1074/jbc.M110508200. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösel J, Gandor F, Harms C, Synowitz M, Harms U, Djoufack PC, Megow D, Dirnagl U, Hörtnagl H, Fink KB, Endres M. Neuroprotective effects of atorvastatin against glutamate-induced excitotoxicity in primary cortical neurons. J Neurochem. 2005;92:1386–1398. doi: 10.1111/j.1471-4159.2004.02980.x. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Geoghagen NS, Friedhoff LT. Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Abeta peptide. J Alzheimers Dis. 2001;3:221–229. doi: 10.3233/jad-2001-3207. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium: still center-stage in hypoxia-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- Cole SL, Vassar R. Isoprenoids and Alzheimer's disease: a complex relationship. Neurobiol Dis. 2006;22:209–222. doi: 10.1016/j.nbd.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem. 2005;280:18755–18770. doi: 10.1074/jbc.M413895200. [DOI] [PubMed] [Google Scholar]
- Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 2005;280:34202–34209. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Delanty N, Vaughan CJ, Sheehy N. Statins and neuroprotection. Expert Opin Investig Drugs. 2001;10:1847–1853. doi: 10.1517/13543784.10.10.1847. [DOI] [PubMed] [Google Scholar]
- Furukawa N, Ongusaha P, Jahng WJ, Araki K, Choi CS, Kim HJ, Lee YH, Kaibuchi K, Kahn BB, Masuzaki H, Kim JK, Lee SW, Kim YB. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005;2:119–129. doi: 10.1016/j.cmet.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Hagen T, Sethi JK, Foxwell N, Vidal-Puig A. Signalling activity of beta-catenin targeted to different subcellular compartments. Biochem J. 2004;379:471–477. doi: 10.1042/BJ20031749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Dou F, Li F, Zhang X, Zhang YW, Zheng H, Lipton SA, Xu H, Liao FF. Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation. J Neurosci. 2005;25:11542–11552. doi: 10.1523/JNEUROSCI.3831-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med. 1999;5:221–225. doi: 10.1038/5587. [DOI] [PubMed] [Google Scholar]
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci U S A. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- Miida T, Takahashi A, Ikeuchi T. Prevention of stroke and dementia by statin therapy: experimental and clinical evidence of their pleiotropic effects. Pharmacol Ther. 2007;113:378–393. doi: 10.1016/j.pharmthera.2006.09.003. [DOI] [PubMed] [Google Scholar]
- O'Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS. Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci. 2005;25:8954–8966. doi: 10.1523/JNEUROSCI.2899-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski SM, Wilkinson BL, Golde TE, Landreth G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem. 2007;282:26832–26844. doi: 10.1074/jbc.M702640200. [DOI] [PubMed] [Google Scholar]
- Parvathy S, Ehrlich M, Pedrini S, Diaz N, Refolo L, Buxbaum JD, Bogush A, Petanceska S, Gandy S. Atorvastatin-induced activation of Alzheimer's alpha secretase is resistant to standard inhibitors of protein phosphorylation-regulated ectodomain shedding. J Neurochem. 2004;90:1005–1010. doi: 10.1111/j.1471-4159.2004.02521.x. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Carter TL, Prendergast G, Petanceska S, Ehrlich ME, Gandy S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005;2:e18. doi: 10.1371/journal.pmed.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM. Acryl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid-peptide. Nat Cell Biol. 2001;3:905–912. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24:107–129. doi: 10.1385/MN:24:1-3:107. [DOI] [PubMed] [Google Scholar]
- Seabra MC. Membrane association and targeting of prenylated Ras-like GTPases. Cell Signal. 1998;10:167–172. doi: 10.1016/s0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 2007;55:61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer JA, Hess DC. Statins in stroke: prevention, protection and recovery. Expert Rev Neurother. 2006;6:195–202. doi: 10.1586/14737175.6.2.195. [DOI] [PubMed] [Google Scholar]
- Tapia PC. RhoA, Rho kinase, JAK2, and STAT3 may be the intracellular determinants of longevity implicated in the progeric influence of obesity: insulin, IGF-1 and leptin may all conspire to promote stem cell exhaustion. Med Hypotheses. 2006;66:570–576. doi: 10.1016/j.mehy.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Whitfield JF. Can statins put the brakes on Alzheimer's disease? Expert Opin Investig Drugs. 2006;15:1479–1485. doi: 10.1517/13543784.15.12.1479. [DOI] [PubMed] [Google Scholar]
- Zacco A, Togo J, Spence K, Ellis A, Lloyd D, Furlong S, Piser T. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci. 2003;23:11104–11111. doi: 10.1523/JNEUROSCI.23-35-11104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lin M, van Golen KL, Yoshioka K, Itoh K, Yee D. Multiple signaling pathways are activated during insulin-like growth factor-I (IGF-I) stimulated breast cancer cell migration. Breast Cancer Res Treat. 2005;93:159–168. doi: 10.1007/s10549-005-4626-8. [DOI] [PubMed] [Google Scholar]