

Abstract
Although exosites 1 and 2 regulate thrombin activity by binding substrates and cofactors and by allosterically modulating the active site, it is unclear whether there is direct allosteric linkage between the two exosites. To begin to address this, we first titrated a thrombin variant fluorescently labeled at exosite 1 with exosite 2 ligands, HD22 (a DNA aptamer), γ′-peptide (an analog of the COOH terminus of the γ′-chain of fibrinogen) or heparin. Concentration-dependent and saturable changes in fluorescence were elicited, supporting inter-exosite linkage. To explore the functional consequences of this phenomenon, we evaluated the capacity of exosite 2 ligands to inhibit thrombin binding to γA/γA-fibrin, an interaction mediated solely by exosite 1. When γA/γA-fibrinogen was clotted with thrombin in the presence of HD22, γ′-peptide, or prothrombin fragment 2 there was a dose-dependent and saturable decrease in thrombin binding to the resultant fibrin clots. Furthermore, HD22 reduced the affinity of thrombin for γA/γA-fibrin 6-fold and accelerated the dissociation of thrombin from preformed γA/γA-fibrin clots. Similar responses were obtained when surface plasmon resonance was used to monitor the interaction of thrombin with γA/γA-fibrinogen or fibrin. There is bidirectional communication between the exosites, because exosite 1 ligands, HD1 (a DNA aptamer) or hirudin-(54–65) (an analog of the COOH terminus of hirudin), inhibited the exosite 2-mediated interaction of thrombin with immobilized γ′-peptide. These findings provide evidence for long range allosteric linkage between exosites 1 and 2 on thrombin, revealing further complexity to the mechanisms of thrombin regulation.
As the central effector of hemostasis, thrombin is engaged in procoagulant, anticoagulant, and fibrinolytic processes. These seemingly contrasting roles are regulated, at least in part, by thrombin's interactions with other factors in the blood and vasculature. The binding of ligands to thrombin is promoted by exosites 1 and 2, which are positively charged domains that flank the active site. These exosites facilitate the binding of substrates or cofactors and align them for optimal interaction with the active site (1).
Exosite 1 is predominantly used to gain access to the active site by substrates such as fibrinogen (2), factors V (3) and VIII (4), and the protease-activated receptors (PARs)2 on platelets (5). Effectors that modulate thrombin activity, including thrombomodulin (6), hirudin (7), and heparin cofactor II (8), also utilize exosite 1. Thrombomodulin alters the specificity of thrombin by hindering access of other substrates to exosite 1 (9) and by providing new binding sites for protein C and thrombin-activable fibrinolysis inhibitor, thereby promoting anticoagulant and antifibrinolytic pathways, respectively (10, 11). Fewer processes are mediated by exosite 2, which serves largely as a tether that anchors thrombin for participation in other reactions. Thus, heparin binds exosite 2 (12) and catalyzes thrombin inhibition by antithrombin and heparin cofactor II (13, 14). Exosite 2 also is used by glycoprotein 1bα on platelets to localize thrombin for activation of PARs (15–17).
Although the prevailing role of the exosites is to bring substrates and cofactors into proximity with thrombin, there is evidence that the exosites also serve as allosteric regulators of thrombin activity. Crystallographic studies reveal that, when peptides derived from PAR1 or PAR3 are bound to exosite 1 on thrombin, an obstructing surface loop moves out of the active site pocket, thereby providing access to substrates (18). The binding of a thrombomodulin fragment to exosite 1 was shown to alter the environment of an active site fluorescent probe (19), which accelerates the rate of protein C and thrombin-activable fibrinolysis inhibitor activation in an allosteric fashion. In contrast, exosite 1-binding peptides from heparin cofactor II or fibrinogen decrease the rate of protein C activation (20). Additionally, the binding of ligands to exosite 1 alters the rates of chromogenic substrate hydrolysis (21, 22). Allosteric effects are not limited to exosite 1, because prothrombin fragment 2 (F2), a cleavage product of prothrombin, binds exosite 2 and decreases the rate at which thrombin converts fibrinogen to fibrin (23, 24) and is inhibited by antithrombin (25, 26). In support of the concept that these alterations are allosteric in origin, fluorescent probes bound to the active site of thrombin undergo a change in fluorescence intensity when exosite 2 is occupied (24, 27).
Although there is good evidence for allosteric regulation of the active site by the exosites, it remains unclear whether there is direct allosteric connection between the exosites. Reciprocal effects between exosites 1 and 2 have been observed by some investigators (28–30), but not by others (25, 31). The aim of the current study was to use different techniques and additional ligands to resolve this controversy. First, we examined the effect of exosite 2-directed ligands on the fluorescence intensity of a thrombin variant that was labeled in exosite 1. Next, we examined the effect of these ligands on thrombin binding to fibrin. To exploit the observation that thrombin binds γA/γA-fibrinogen exclusively via exosite 1 (2, 32), leaving exosite 2 accessible, this subpopulation was isolated (32). We used intact fibrin clots and surface plasmon resonance (SPR) to examine the influence of exosite 2-directed ligands on thrombin binding to γA/γA-fibrin. In addition, diffusion studies were performed to examine the effect of exosite-directed ligands on the rate of thrombin dissociation from preformed fibrin clots. Finally, we explored whether exosite 1-directed ligands modulate the binding of thrombin to an exosite 2-directed ligand.
EXPERIMENTAL PROCEDURES
Materials
Reagents
Human α-thrombin, γ-thrombin, plasminogen-free fibrinogen, and factor XIII were obtained from Enzyme Research Laboratories, Inc. (South Bend, IN), whereas Quick I-thrombin (R67C) was a generous gift from Ruth Ann Henrickson. Batroxobin from Bothrops atrox moojeni snake venom was from Pentapharm (Switzerland). Oligonucleotides HD1 (5′-GGTTGGTGTGGTTGG-3′) and HD22 (5′-AGTCCGTGGTAGGGCAGGTTGGGGTGACT-3′), the exosite 1- and 2-directed aptamers, respectively, and HD23 (5′-AGTCCGTAAAGCAGGTTAAAATGACT-3′), a scrambled variant of HD22, were synthesized by the Molecular Biology and Biotechnology Institute at McMaster University (Hamilton, Canada). Before use, the aptamers were heated to 95 °C for 10 min and then cooled on ice for an additional 10 min. A 20-amino acid analog of the COOH-terminal portion of the γ′-chain of fibrinogen, the γ′-peptide (H-Val-Arg-Pro-Glu-His-Pro-Ala-Glu-Thr-Glu-Tyr(PO3H2)-Asp-Ser-Leu-Tyr(PO3H2)-Pro-Glu-Asp-Asp-Leu-OH), was from Bachem AG (San Diego, CA). To enhance stability, Tyr residues of the γ′-peptide were modified with phosphate groups in place of sulfate groups (33). d-Phe-Pro-Arg-chloromethyl ketone (FPRck), d-Tyr-Pro-Arg-chloromethyl ketone (YPRck) and dansyl-Glu-Gly-Arg-chloromethyl ketone were obtained from Calbiochem. A peptide analog of residues 54–65 of leech protein hirudin (hirudin-(54–65)) was purchased from Bachem AG. Chromogenic substrates H-d-Phe-Pip-Arg-p-nitroanilide (S2238) and pyro-Glu-Pro-Arg-pNA-HCl (S2366) were from Chromogenix (Milano, Italy), whereas tosyl-Gly-Pro-Arg-p-nitroaniline acetate was from Aniara (Mason, OH). Unfractionated heparin, streptavidin, and biotinaminohexanoic acid-3-sulfoester were from Sigma-Aldrich. 5-Iodoacetamidofluorescein (5-IAF) was purchased from Invitrogen.
Isolation of γA/γA-Fibrinogen
γA/γA-Fibrinogen was isolated from fibrinogen by fractionation on DEAE-Sepharose, and its integrity was assessed by SDS-PAGE as previously described (32, 34).
Purification of F2
F2 was isolated from human prothrombin activation byproduct fraction (Enzyme Research Laboratories, Inc.) as previously described (29) but with the following modifications. The material was treated with FPRck and dansyl-Glu-Gly-Arg-chloromethyl ketone to inhibit residual α-thrombin and factor Xa, respectively, dialyzed overnight against 1 mm sodium phosphate (NaPi) buffer, pH 6.9 (buffer A), and then applied to a 5-ml hydroxyapatite (CHT-II) Econo-Pac Cartridge column (Bio-Rad) on a Bio-Rad Biologic Duoflow system at a flow rate of 3 ml/min. After washing the column with buffer A, a stepwise elution protocol was performed using a mixture of buffer A and 500 mm NaPi buffer, pH 6.9 (buffer B), to separate prothrombin fragment 1 (F1) from F2. F2 eluted from the column at 16% buffer B, whereas F1 eluted at 100% buffer B. The integrity of F2 and F1 preparations was assessed by SDS-PAGE on 4–15% polyacrylamide gels (Ready-Gel, Bio-Rad) under reducing conditions. Fractions containing F1 or F2 were pooled, concentrated by speed vacuum (Allegra 6R Centrifuge, Beckman Coulter), dialyzed against 10 mm Hepes-NaOH, 150 mm NaCl at pH 7.4 (HBS), and stored at −80 °C. The concentrations of F1 and F2 were determined spectrophotometrically at 280 nm using extinction coefficients (ml/mg/cm) of 1.01 (35) and 1.1 (36), respectively.
Fluorescent Labeling of Quick I-thrombin
Quick I-thrombin at 0.40 mg/ml (150 μl) in 0.2 m phosphate buffer, pH 7.5, was added to a 15-fold molar excess of 5-IAF and incubated in the dark for 45 min at 23 °C. After passage over a PD-10 column equilibrated with 20 mm Tris-HCl, pH 7.4, 150 mm NaCl (Tris-buffered saline) to remove unreacted 5-IAF, 0.5-ml fractions were collected. 5-IAF-Quick I-thrombin concentration was determined using a molecular weight of 37,000 and an extinction coefficient of 1.8 ml/mg/cm at 280 nm.
Radiolabeling of Thrombin
α-Thrombin was radiolabeled by reaction with 125I-labeled YPRck as previously described (34).
Biotinylation of γ′-Peptide
One milligram of biotinaminohexanoic acid-3-sulfoester was added to 0.5 mg γ′-peptide and dissolved in 0.5 ml of 0.1 m NaPi, pH 8, and the solution was incubated for 1 h at 23 °C. A PD-10 column (Amersham Biosciences) was used to separate the biotinylated γ′-peptide from unincorporated biotin. 0.5-ml fractions were collected, and samples containing the biotinylated γ′-peptide were identified spectrophotometrically at 214 nm.
Methods
Fluorescent Binding Studies
The binding of exosite 1- and 2-directed ligands to 5-IAF-Quick I-thrombin was assessed by fluorescence. An 800-μl aliquot of 50 nm 5-IAF-Quick I-thrombin in Tris-buffered saline containing 0.2% polyethylene glycol 8000 was maintained at 23 °C with a circulating water bath in a 10 × 4 mm quartz cuvette and stirred using a micro stir bar. Fluorescence was monitored at an excitation wavelength of 492 nm and emission wavelength of 532 nm (slit widths of 10 nm) and an emission filter at 515 nm in an LS 50B luminescence spectrometer (PerkinElmer Life Sciences). After the baseline fluorescence (Io) stabilized, 5-IAF-Quick I-thrombin was titrated with 1- to 10-μl aliquots of either unfractionated heparin (1 mg/ml), γ′-peptide (200 μm), HD1 (1 mm), HD22 (1 mm), or hirudin-(54–65) (1.27 mm), allowing the fluorescence signal to stabilize between additions. Once the fluorescence signal reached a plateau, intensity values (I) were obtained from time drive profiles and corrected for dilution. Plots of I/Io versus the titrant concentration were analyzed by nonlinear regression using Table Curve (Jandel Scientific, San Rafael, CA) to the binding equation (Equation 1), where L is the concentration of the titrated ligand, Qo is the concentration of 5-IAF-Quick I-thrombin, α is the maximal fluorescence change, and Kd is the dissociation constant.
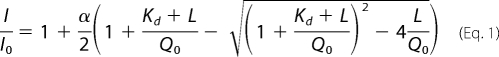 |
Effect of Exosite 2-Directed Ligands on Thrombin Binding to Fibrin
Varying concentrations of HD22 (0–20 μm), γ′-peptide (0–50 μm), or F2 (0–75 μm) were added to a series of 1.5-ml microcentrifuge tubes containing 125I-YPR-α-thrombin and α-thrombin in HBS at final concentrations of 20 nm and 5 nm, respectively. A stock solution was added to each tube to achieve a final concentration of 2 μm γA/γA-fibrinogen and 2 mm CaCl2 in a volume of 100 μl, and samples were allowed to clot for 45 min at 23 °C. Under these conditions clotting was complete within 10 min, as determined by turbidometric analysis. As a control, 4 μm bovine albumin was used in place of γA/γA-fibrinogen. After vortexing, fibrin clots were pelleted by centrifugation at 14,000 × g for 10 min. Duplicate 20-μl aliquots of supernatant were removed and counted for radioactivity using a gamma counter (1272 Clinigamma, LKB Wallace) to determine the concentration of unbound 125I-YPR-α-thrombin. The amount of bound 125I-YPR-α-thrombin was calculated by subtraction from the total amount of 125I-YPR-α-thrombin in the supernatant of the albumin controls. The fraction of 125I-YPR-α-thrombin bound relative to total 125I-YPR-α-thrombin in the supernatants of tubes without fibrinogen was calculated. Plots of bound 125I-YPR-α-thrombin versus concentration of exosite competitor were then analyzed by non-linear regression of a rectangular hyperbola using Table Curve (Jandel Scientific, Systat Software Inc., San Jose CA), and the Kiobs (concentration at half-maximal response) was determined.
To exclude the possibility that active site-blocked thrombin has properties distinct from those of active thrombin, the effect of exosite 2-directed ligands on binding of active thrombin to γA/γA-fibrin also was examined. γA/γA-Fibrinogen (5 μm) containing 2 mm CaCl2 was clotted with 25 nm α-thrombin in the presence of 1 unit/ml Batroxobin to ensure rapid and consistent clotting in the presence of varying concentrations of HD22 (0–5 μm). After incubation for 45 min at 23 °C, the fibrin was pelleted by centrifugation, and 20-μl aliquots of supernatant were removed and subjected to chromogenic assay by the addition of 80 μl of 200 μm S2238. Chromogenic activity was read in a SpectraMAX platereader (Molecular Devices, Sunnyvale, CA) at 405 nm. Based on the specific activity of thrombin, the concentration of free thrombin was determined and used to calculate the concentration of the bound fraction. The effect of HD22 on thrombin binding was then analyzed as described above.
Effect of HD22 on the Affinity of Thrombin for Fibrin
A series of microcentrifuge tubes containing varying concentrations of γA/γA-fibrinogen (0–12.5 μm), 20 nm 125I-YPR-α-thrombin, and 2 mm CaCl2 was prepared. Clotting was initiated by addition of 5 nm α-thrombin in the absence or presence of 20 μm HD22. The fraction of 125I-YPR-α-thrombin bound was determined as described above. Plots of bound 125I-YPR-α-thrombin versus concentration of γA/γA-fibrinogen were then analyzed by non-linear regression of a rectangular hyperbola to determine Kd.
SPR
The interaction of FPR-α-thrombin with γA/γA-fibrinogen or γA/γA-fibrin in the absence or presence of various exosite ligands was quantified by SPR using a BIAcore 1000 (Amersham Biosciences). γA/γA-Fibrinogen or ovalbumin were covalently linked to separate flow cells of a carboxymethylated dextran (CM-5) biosensor chip using an amine coupling kit. All SPR procedures were done in HBS containing 0.005% Tween 20, and flow cells were regenerated with 1 m NaCl in HBS between runs. γA/γA-Fibrinogen or ovalbumin was injected at a flow rate of 20 μl/min in acetate buffer at pH 5.5 and pH 4.0, respectively, until 10,000 response units (RU) of γA/γA-fibrinogen or 5,500 RU of ovalbumin was adsorbed. To convert immobilized γA/γA-fibrinogen to γA/γA-fibrin, 100 nm α-thrombin was injected in three successive runs of 60 min. To measure binding, 2 μm FPR-α-thrombin was injected at a flow rate of 20 μl/min for 1 min in the presence of 0–20 μm HD22, 0–50 μm γ′-peptide, or 0–100 μm F2. A 1-min wash with HBS was used to monitor dissociation and regeneration of the flow cell. Peak RU values were determined using Scrubber2 (BioLogic Software Co., Campbell, Australia) for each ligand concentration and then corrected by subtracting the RU values from the ovalbumin control.
Streptavidin was immobilized onto a CM-5 chip by injection of 40 μl of a 0.4 mg/ml streptavidin solution in 10 mm sodium acetate, pH 4.5, over all four flow cells at a flow rate of 5 μl/min. After neutralizing any remaining reactive groups by injection of 40 μl of 1 m ethanolamine, flow cells were washed with 100 μl of solution containing 10 mm NaOH and 1 m NaCl at a flow rate of 50 μl/min to minimize nonspecific binding. Biotinylated γ′-peptide was adsorbed to the streptavidin-modified CM-5 chip at a flow rate of 20 μl/min to 500 RU. As a control, a separate flow cell containing only streptavidin was used. For binding studies, aliquots containing 0–20 μm HD22, 0–50 μm γ′-peptide, 0–20 μm HD1 or 0–20 μm hirudin-(54–65), and 1 μm FPR-α-thrombin or 2 μm γ-thrombin were injected over the flow cells. Peak RU values were determined for each concentration of exosite-directed ligand and corrected by subtracting the peak RU values determined from the blank flow cell. The fraction of FPR-α-thrombin or γ-thrombin that bound in the presence of exosite ligand was calculated by dividing by the RU values determined in the absence of competitor. Data were analyzed by nonlinear regression analysis.
Thrombin Diffusion from Intact γA/γA-Fibrin Clots
The effect of exosite ligands on the rate of diffusion of 125I-YPR-α-thrombin from intact fibrin clots was measured as previously described (34). Briefly, 10 μm γA/γA-fibrinogen, 17 nm 125I-YPR-α-thrombin, and 15 nm factor XIII in HBS with 0.005% Tween 20 were added to microcentrifuge tubes containing a plastic inoculation loop (Bac-Loop, Thermo-Fisher Scientific, Waltham, MA). Clotting was initiated by addition of 100 nm α-thrombin. After incubation for 45 min at 23 °C, fibrin clots attached to the plastic loops were removed and counted for radioactivity. Clots were then placed in 50-ml conical tubes containing 5 ml of HBS with 0.005% Tween 20 or buffer containing either 2 m NaCl, 5 μm HD1, or 20 μm HD22. At intervals, clots were removed from bathing buffer, counted for radioactivity, and returned to the respective tubes. The fraction of 125I-YPR-α-thrombin remaining in the clot at each time point was calculated relative to the initial amount bound, and time courses were fit to a two-phase exponential decay curve with a zero end point (34).
Effects of Exosite-directed Ligands on Clot Times
The effects of the exosite-directed ligands on thrombin activity were examined using a fibrinogen clotting assay. To wells of a 96-well plate were added 2 nm α-thrombin and 0–1 μm HD1, 0–1 μm hirudin-(54–65), 0–20 μm HD22, 0–50 μm γ′-peptide, or 0–80 μm F2 in a total volume of 20 μl. After adding an 80-μl aliquot of γA/γA-fibrinogen and CaCl2 to yield final concentrations of 2 μm and 2 mm, respectively, plates were incubated at 23 °C, and turbidity was monitored at 405 nm in a plate reader. The time to half-maximal increase in turbidity was determined both in the absence or presence of exosite-directed ligands, and values were plotted as a function of time. Experiments were performed in duplicate and repeated three times.
Influence of Exosite-directed Ligands on Thrombin Chromogenic Activity
The influence of exosite-directed ligands on the chromogenic activity of α-thrombin was determined by monitoring the hydrolysis of 0–500 μm tosyl-Gly-Pro-Arg-p-nitroaniline acetate, S2238, or S2366 by 2 nm thrombin in the absence or presence of 10 μm HD1, 10 μm HD22, or 10 μm hirudin-(54–65). Km and kcat were determined by nonlinear regression analysis of the Michaelis-Menten equation. All experiments were performed in duplicate three times.
RESULTS
Binding of Exosite-directed Ligands to 5-IAF-Quick I-thrombin
To re-examine the possibility that exosite 2-directed ligands modulate exosite 1, we utilized a thrombin variant fluorescently labeled at exosite 1. Quick I-thrombin is a naturally occurring thrombin variant with Arg-67 in exosite 1 replaced with Cys (37). The fluorescence of 5-IAF-Quick I-thrombin was monitored as the sample was titrated with exosite 2 ligands. Addition of γ′-peptide or unfractionated heparin decreased the fluorescence intensity of 5-IAF-Quick I-thrombin in a dose-dependent and saturable manner (Fig. 1). The Kd for γ′-peptide binding 5-IAF-Quick I-thrombin was 2.5 ± 0.6 μm, a value similar to the Kd of 2.2 μm determined previously when thrombin fluorescently labeled at the active site was titrated with γ′-peptide (32), whereas unfractionated heparin bound 5-IAF-Quick I-thrombin with a Kd of 530 ± 58 nm. In contrast to γ′-peptide or unfractionated heparin, HD22 increased the fluorescence of 5-IAF-Quick I-thrombin and bound with a Kd value of 38 nm (Fig. 1). These findings support the concept that binding of these exosite 2-directed ligands to thrombin elicits a conformational change in exosite 1. As controls, binding of HD1 and hirudin-(54–65) to 5-IAF-Quick I-thrombin also were examined. Both exosite 1 ligands bound 5-IAF-Quick I-thrombin with lower affinity than that observed with α-thrombin (not shown). Thus, HD1 and hirudin-(54–65) bound 5-IAF-Quick I-thrombin with Kd values of 5 μm and 10.8 μm, respectively. To extend the investigation beyond that achieved by fluorescence measurements, we used additional methods to examine the functional consequences of the long range communication between thrombin exosites.
FIGURE 1.

Binding of exosite-directed ligands to fluorescently labeled Quick I-thrombin. After determining the initial fluorescence intensity (Io) of 50 nm 5-IAF-Quick I-thrombin (λex 492, λem 532 nm), fluorescence (I) was monitored as the sample was titrated with aliquots of either 200 μm γ′-peptide (▴), 66.7 μm heparin (♦), or 1 mm HD22 (inset). Values of I/Io were then calculated and plotted as a function of the titrant concentration, and the Kd values were determined by nonlinear regression analysis (lines). Data represent the means ± S.E. of three experiments.
Effect of Exosite Ligands on Clotting Times
To verify the results of previous studies, we first examined the effect of additional exosite 2-directed ligands on thrombin-mediated clotting of γA/γA-fibrinogen, a form of fibrinogen that interacts with thrombin exclusively via exosite 1 (32). Fibrin clot formation was monitored by turbidity and yielded a baseline clotting time of 100.6 ± 8.1 s under control conditions. All three exosite 2-directed ligands prolonged the thrombin clotting times in a concentration-dependent fashion (Fig. 2A). At 37.5 μm, F2 prolonged the clotting time 1.8-fold; results in agreement with previous work (24). HD22 was the most potent effector, producing a 1.8-fold prolongation of the thrombin clot time at a concentration of 20 μm, whereas γ′-peptide had only minimal effects at concentrations up to 50 μm. Although it is more potent, the effect of HD22 on the clotting time appears to saturate more rapidly than that of F2. Whereas potency likely reflects the affinities of the various ligands for exosite 2 on thrombin, with HD22 binding with the highest affinity, the ligands may have divergent effects on the clotting time, because they contact different residues on exosite 2. As positive controls, the effect of exosite 1-directed ligands also was examined. As expected, HD1 and hirudin-(54–65) had a greater effect than the exosite 2-directed ligands, prolonging the clot time >8-fold at concentrations below 1 μm (Fig. 2B). Because exosite 2-directed ligands could modulate the thrombin clotting times by allosterically influencing the active site and/or exosite 1, additional experiments were performed to identify the responsible mechanism.
FIGURE 2.

Effect of exosite-directed ligands on thrombin clotting times. γA/γA-Fibrinogen (2 μm) containing 2 mm CaCl2 was clotted with 2 nm α-thrombin in the absence or presence of exosite 2-directed ligands, HD22 (●), γ'-peptide (▴), or F2 (■) (A), or exosite 1-directed ligands, HD1 (○) or hirudin-(54–65) (▵), exosite 1-directed ligands (B). Absorbance was determined spectrophotometrically at 405 nm, and the time to reach half-maximal turbidity (clot time) was normalized with respect to that measured in the absence of ligands. Symbols represent the means ± S.E. of three experiments, each done in duplicate, while the lines represent non-linear regression analysis of the data.
Effect of Exosite Ligands on Thrombin Chromogenic Activity
The possibility that exosite-directed ligands can allosterically alter the active site of thrombin was tested using chromogenic assays. The rates of hydrolysis of varying concentrations of chromogenic substrates by 2 nm thrombin were determined in the absence or presence of saturating concentrations of exosite ligands. As reported previously for hirudin-(54–65) (7, 20), HD1 and HD22 affected the kinetic parameters for cleavage of chromogenic substrates (Table 1). There was no consistent response to the presence of HD22, with the catalytic efficiency increasing with tosyl-Gly-Pro-Arg-p-nitroaniline acetate and S2366 and decreasing with S2238. The two aptamers also did not elicit the same response. For example, HD22 reduced both the Km and kcat values with tosyl-Gly-Pro-Arg-p-nitroaniline acetate, whereas HD1 had no significant effects on the rate of hydrolysis of this substrate. It is notable that, for exosite 1, HD1 and hirudin-(54–65) had opposite effects on the catalytic efficiency with S2238. This confirms the observation from Fig. 2 that different ligands for the same exosite can elicit different responses. These experiments also reveal that, like protein or polypeptide ligands, aptamers can modulate the active site by binding to either exosite. However, these results do not exclude the possibility that the exosite 2-directed ligands also affect the function of exosite 1 and that this contributes to their inhibitory activity in the clotting assays. We used a fibrin binding assay to explore functional connections between the exosites, because the interaction of thrombin with fibrin is independent of the active site.
TABLE 1.
Km, kcat, and kcat/Km values for thrombin hydrolysis of chromogenic substrates in the presence of exosite ligands
The rates of thrombin cleavage of various chromogenic substrates were used to determine the kinetic parameters by Michaelis-Menten analysis. Experiments were performed in the absence or presence of 10 μm HD1, HD22, or hirudin-(54–65). The experiments were repeated three times in duplicate, and the values represent means ± S.E.
| Substrate | Addition | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | μm−1s−1 | ||
| tGPR-pNA | ||||
| None | 16.4 ± 1.2 | 234.5 ± 4.1 | 14.3 ± 1.0 | |
| HD1 | 14.2 ± 1.4 | 240.3 ± 9.6 | 16.9 ± 1.8 | |
| HD22 | 8.5 ± 0.7 | 192.8 ± 16.3 | 22.7 ± 2.8 | |
| Hirudin-(54–65) | 11.7 ± 2.0 | 251.6 ± 20.3 | 21.5 ± 4.1 | |
| S2238 | ||||
| None | 2.6 ± 0.4 | 101.9 ± 6.8 | 39.8 ± 6.5 | |
| HD1 | 14.3 ± 0.6 | 209.4 ± 6.7 | 14.7 ± 0.8 | |
| HD22 | 12.1 ± 1.7 | 106.8 ± 4.1 | 8.8 ± 1.3 | |
| Hirudin-(54–65) | 3.1 ± 0.1 | 156.5 ± 1.8 | 51.0 ± 2.1 | |
| S2366 | ||||
| None | 282.9 ± 30.6 | 249.5 ± 11.3 | 0.9 ± 0.1 | |
| HD1 | 195.0 ± 18.3 | 217.1 ± 9.4 | 1.1 ± 0.1 | |
| HD22 | 155.4 ± 14.1 | 231.1 ± 13.4 | 1.5 ± 0.2 | |
| Hirudin-(54–65) | 147.1 ± 6.0 | 278.6 ± 3.5 | 1.9 ± 0.1 |
Effect of Exosite 2-Directed Ligands on Thrombin Binding to Fibrin(ogen)
Because thrombin binds to γA/γA-fibrin(ogen) solely via exosite 1, this interaction can be exploited to determine whether exosite 2-directed ligands induce an allosteric change at exosite 1. To assess thrombin binding to γA/γA-fibrin, we measured the amount of 125I-YPR-α-thrombin in supernatants of compacted fibrin clots prepared with a catalytic amount of α-thrombin in the absence or presence of exosite-directed ligands. Although the fibrin was compacted after 45 min of incubation, control experiments demonstrated no change in the concentration of bound 125I-YPR-α-thrombin after the first 10 min of incubation. In the absence of effectors, 125I-YPR-α-thrombin bound γA/γA-fibrin with a Kd of 2.1 ± 0. 7 μm (Fig. 4), a value consistent with the previously reported Kd of 2.3 μm (32). In the presence of HD22, 125I-YPR-α-thrombin binding to γA/γA-fibrin was reduced by 50% with a half-maximal effect (Kiobs) at 673 nm (Fig. 3). Because oligonucleotides have a negatively charged backbone, the potential nonspecific effects were examined using HD23, a scrambled version of HD22. HD23 decreased thrombin binding to γA/γA-fibrin by 10%. This background binding was subtracted from the results obtained with HD22. F2 and γ′-peptide also were tested to determine whether the response was exclusive to HD22. Both γ′-peptide and F2 reduced thrombin binding to γA/γA-fibrin clots, with Kiobs values of 15.6 ± 3.8 μm and 18.5 ± 3.3 μm, respectively. The weaker potencies of γ′-peptide and F2 relative to HD22 are consistent with their lower affinities for thrombin with reported Kd values of 2.2 μm (32) and 4 μm (29), respectively. HD1, the exosite 1-directed oligonucleotide, also was tested as a positive control. As a direct competitor of thrombin binding to γA/γA-fibrin, HD1 had a potent effect and fully inhibited 125I-YPR-α-thrombin binding to the clot with a Kiobs of 157 nm (data not shown). This equates to a Ki of 78.5 nm, which is comparable to the Kd of HD1 for 125I-YPR-α-thrombin of 34 nm (38). Unlike the exosite 1-directed ligand, the exosite 2-directed ligands only partially displace thrombin from fibrin clots, suggesting that they are not acting in a competitive fashion.
FIGURE 4.

Effect of HD22 on the affinity of thrombin for γA/γA-fibrin clots. 20 nm 125I-YPR-α-thrombin and 5 nm α-thrombin were added to a series of microcentrifuge tubes containing 0–12.5 μm γA/γA-fibrinogen and 2 mm CaCl2 in the absence (♦) or presence of 20 μm HD22 (●). After incubation for 45 min, fibrin was pelleted by centrifugation, and free 125I-YPR-α-thrombin in the supernatant was used to calculate the bound fraction. Data are plotted as the relative amount of thrombin bound to the fibrin clot versus the γA/γA-fibrin concentration and represent the means ± S.E. of three experiments, each done in duplicate, while the lines represent non-linear regression analysis of the data. The affinity of 125I-YPR-α-thrombin for γA/γA-fibrin is 6-fold lower in the presence of HD22 than it is in its absence (Kd values of 12.6 ± 1.4 and 2.1 ± 0.7 μm, respectively).
FIGURE 3.

The effect of exosite 2-directed ligands on 125I-YPR-α-thrombin binding to γA/γA-fibrin clots. 20 nm 125I-YPR-α-thrombin and 5 nm α-thrombin were added to microcentrifuge tubes containing 2 μm γA/γA-fibrinogen, 2 mm CaCl2, and HD22 (●), γ′-peptide (▴), or F2 (■) at the indicated concentrations. After incubation for 45 min, clots were pelleted by centrifugation, and free 125I-YPR-α-thrombin in the supernatant was used to calculate the bound fraction. Data are plotted as the relative amount of thrombin bound to the clot versus the concentration of exosite-directed ligand and represent the means ± S.E. of three experiments, each done in duplicate, while the lines represent non-linear regression analysis of the data. HD22 was the most potent of the three ligands, having a Kiobs of 673 ± 83 nm. γ′-Peptide and F2 are less potent than HD22, and reduce thrombin binding with Kiobs values of 15.6 ± 3.8 and 18.5 ± 3.3 μm, respectively.
Because HD22 attenuates thrombin binding to fibrin, it was of interest to determine its effect on the affinity of the interaction. In the presence of 20 μm HD22, the Kd of 125I-YPR-α-thrombin for γA/γA-fibrin was 12.6 ± 1.4 μm (Fig. 4), an affinity 6-fold lower than that measured in the absence of HD22. This result provides further support for the concept that exosite 2-directed ligands can influence interactions mediated by exosite 1.
In a separate experiment, active thrombin was used in place of active site-blocked thrombin to address the possibility that the presence of an inhibitor in the active site could alter the binding characteristics of the exosites. The snake venom Batroxobin also was present to ensure consistent and rapid clot formation, even in the presence of exosite ligands. The amount of thrombin in clot supernatants determined by chromogenic assay was used to calculate thrombin binding. Like its effect on the binding of 125I-YPR-α-thrombin to fibrin, HD22 also reduced the binding of α-thrombin to fibrin in a dose-dependent and saturable manner (data not shown). These results suggest that the presence of the chloromethyl ketone moiety in the active site has no influence on thrombin binding to fibrin or HD22 binding to thrombin.
SPR was used to verify the results obtained in the clot binding assays. Previously, we demonstrated that the binding constants for the thrombin-fibrin interaction measured using SPR are comparable to those determined with fibrin clots (34), thereby validating SPR as a method for monitoring the thrombin-fibrin interaction. For the SPR studies, γA/γA-fibrinogen was immobilized onto a CM-5 biosensor chip, and ovalbumin was immobilized as a control for nonspecific binding. FPR-α-thrombin bound γA/γA-fibrinogen with a Kd of 3.9 ± 1.8 μm, a value comparable to the affinity of 125I-YPR-α-thrombin for γA/γA-fibrin clots (Fig. 4). The exosite 2-directed ligands were subsequently co-injected with 2 μm FPR-α-thrombin. Maximal RU values were used to quantify thrombin binding, and the data were plotted as the relative amount of FPR-α-thrombin bound versus the concentration of exosite 2-directed ligand (Fig. 5A). In the presence of HD22, the amount of thrombin bound to the immobilized γA/γA-fibrinogen decreased in a dose-dependent and saturable manner. HD22 was found to have a Kiobs of 3.9 ± 1.8 μm and, unlike the response with intact clots, was able to fully inhibit thrombin binding. Similar results were obtained with γ′-peptide and F2, yielding Kiobs values of 7.1 ± 1.1 μm and 19.7 ± 5.2 μm, respectively.
FIGURE 5.

SPR analysis of the effect of exosite 2-directed ligands on the binding of thrombin to immobilized γA/γA-fibrinogen or fibrin. A, γA/γA-fibrinogen was immobilized on a CM-5 flow cell to ∼10000 RU. To control for nonspecific binding of thrombin, ovalbumin was immobilized in a separate flow cell. Increasing concentrations of HD22 (●), γ′-peptide (▴), or F2 (■), exosite 2-directed ligands, were injected at flow rates of 20 μl/min in the presence of 2 μm FPR-α-thrombin. Binding of FPR-α-thrombin was determined by the maximum RU values, and data were plotted as the relative amount of thrombin bound versus the concentration of exosite-directed ligand. Symbols represent the means ± S.E. of three experiments, while the lines represent non-linear regression analysis of the data. HD22, γ′-peptide, and F2 disrupt FPR-α-thrombin binding to fibrinogen with Kiobs values of 3.9 ± 1.8, 7.1 ± 1.1, and 19.7 ± 5.2 μm, respectively. B, after γA/γA-fibrinogen was converted to fibrin by treatment with thrombin, 2 μm FPR-α-thrombin was injected in the absence or presence of exosite 2-directed ligands. HD22, γ′-peptide, and F2 disrupt the binding of FPR-α-thrombin to immobilized γA/γA-fibrin with Kiobs values of 2.5 ± 0.2, 10.1 ± 0.2, and 11.7 ± 3.8 μm, respectively.
To examine the effect of the exosite-directed ligands on thrombin binding to γA/γA-fibrin, the flow cell containing immobilized γA/γA-fibrinogen was treated with 100 nm thrombin to convert the fibrinogen to fibrin. FPR-α-thrombin bound to the immobilized γA/γA-fibrin with a Kd of 2.1 μm, comparable to the Kd value of 3.4 μm obtained previously (34). Like the SPR results obtained with γA/γA-fibrinogen, all three exosite 2-directed ligands abrogated FPR-α-thrombin binding to γA/γA-fibrin at the highest concentrations tested (Fig. 5B). The Kiobs for HD22 was 2.5 ± 0.2 μm, which is similar to that obtained with immobilized γA/γA-fibrinogen. F2 and γ′-peptide, having Kiobs values of 10.1 ± 0.2 μm and 11.7 ± 3.8 μm, respectively, also had similar potencies with γA/γA-fibrinogen as they did with intact fibrin clots. Although both techniques demonstrated that exosite 2-directed ligands reduced the exosite 1-mediated interaction of thrombin with fibrin(ogen), quantitative differences were noted. With SPR analysis, exosite 2-directed ligands completely eliminated thrombin binding to immobilized fibrinogen or fibrin, whereas with fibrin clots, 50% of the thrombin remained bound, even at the highest concentrations of exosite 2-directed ligands. The most likely explanation for this difference is the fact that thrombin binding to fibrin clots is assessed under static conditions, whereas SPR analysis is done under flow conditions. It is less likely that the differences reflect conformational changes induced by immobilization of fibrin on the biosensor chip because thrombin binds to immobilized fibrin and fibrin clots with comparable affinities (34).
Thrombin Diffusion from Preformed Fibrin Clots
In the previous experiments, thrombin binding occurred in the presence of the exosite ligands, potentially altering the equilibrium of fibrin binding. To address this potential limitation, we monitored the dissociation of 125I-YPR-α-thrombin from preformed fibrin clots in the absence or presence of exosite ligands to determine whether exosite ligands influence thrombin already bound to fibrin (34). Fibrin clots prepared in the presence of 125I-YPR-α-thrombin were counted for radioactivity and then suspended in buffer that did or did not contain the competitors. Clots suspended in buffer alone exhibited the slowest diffusion, with only 45% of the thrombin dissociating after 5 h. In the presence of 2 m NaCl, 80% of 125I-YPR-α-thrombin dissociated after 5 h. HD1 promoted diffusion of 125I-YPR-α-thrombin to a value comparable to that of high salt, whereas HD22 promoted the dissociation of 70% of 125I-YPR-α-thrombin from the clots (Fig. 6). Thus, the effect of HD22 was less than that of HD1 but more than that of buffer alone. These results lend further support for the concept that an exosite 2-directed ligand can affect binding mediated by exosite 1.
FIGURE 6.

Dissociation of 125I-YPR-α-thrombin from preformed γA/γA-fibrin clots in the absence or presence of exosite-directed ligands. γA/γA-Fibrinogen (9 μm) containing 2 mm CaCl2 was clotted around plastic inoculation loops by addition of 100 nm α-thrombin in the presence of 17 nm 125I-YPR-α-thrombin and 15 nm factor XIII. After incubation for 45 min, clots were counted for radioactivity and placed in 50-ml conical tubes containing either 5 ml of HBS (♦), 2 m NaCl (▴), 5 μm HD1 (○), or 20 μm HD22 (●). At intervals, the clots were removed and counted for residual radioactivity to determine the amount of 125I-YPR-α-thrombin that remained bound. Symbols represent the means ± S.E. of three experiments, while the lines represent non-linear regression analysis of the data by two-component exponential decay.
Effect of Exosite Ligands on Thrombin Binding to γ′-Peptide
Having demonstrated that exosite 2-directed ligands affect the function of exosite 1, it was of interest to determine whether the reverse also is true. To explore this possibility, HD1 and hirudin-(54–65) were examined for their effect on exosite 1-mediated binding of thrombin to biotinylated γ′-peptide. Using SPR, the Kd of thrombin binding to the immobilized γ′-peptide was 0.95 μm (not shown), an affinity comparable to the Kd of 1.0 μm previously obtained in direct binding experiments (32). As a control, FPR-α-thrombin was injected over the flow cells in the presence of exosite 2-directed ligands. HD22 and γ′-peptide inhibited FPR-α-thrombin binding to the immobilized γ′-peptide with Kiobs values of 2.6 ± 0.4 μm (Fig. 7A) and 14.2 ± 0.4 μm (data not shown), respectively. The greater potency of HD22 relative to γ′-peptide is consistent with its higher affinity for thrombin with Kd values of 29 nm and 1 μm, respectively (32, 38). When the experiment was repeated in the presence of exosite 1 ligands, a reduction of thrombin binding also was observed (Fig. 7A). HD1 completely inhibited FPR-α-thrombin binding to immobilized γ′-peptide, whereas hirudin-(54–65) inhibited binding by 80%. The Kiobs values for HD1 and hirudin-(54–65) were 0.7 ± 0.02 μm and 1.3 ± 0.3 μm, respectively. It was evident that HD1 was more potent than HD22, even though they bind thrombin with similar affinities (38). These results are consistent with those obtained for γA/γA-fibrinogen binding, where an exosite ligand can influence the capacity of the reciprocal exosite to bind its ligands.
FIGURE 7.

SPR analysis of the effect of exosite ligands on thrombin binding to immobilized γ′-peptide. Biotinylated γ′-peptide was adsorbed to the flow cell of a streptavidin-CM5 chip to ∼700 RU. To control for nonspecific binding, a flow cell containing only streptavidin was used. A, 1 μm FPR-α-thrombin was injected in the presence of increasing concentrations of HD1 (○), HD22 (●), or hirudin-(54–65) (▵) at a rate of 20 μl/min, and maximal RU values determined at each concentration of exosite ligand were corrected by subtracting the background RU from the control flow cell. Symbols represent the means ± S.E. of three experiments, while the lines represent non-linear regression analysis of the data. HD22, HD1, and hirudin-(54–65) inhibit FPR-α-thrombin binding with Kiobs values of 2.6 ± 0.4, 0.7 ± 0.1, and 1.3 ± 0.3 μm, respectively. B, the experiment was repeated using 2 μm γ-thrombin in place of FPR-α-thrombin. Hirudin-(54–65) has no effect on γ-thrombin binding, whereas the Kiobs values for HD1 and HD22 are 20.8 ± 2.1 and 3.2 ± 0.6 μm, respectively.
To verify that communication occurs between the exosites, the binding experiment was repeated using γ-thrombin. Because it lacks an intact exosite 1, we hypothesized that γ-thrombin would bind to γ′-peptide in an interaction that would not be modulated by exosite 1 ligands. γ-Thrombin bound to γ′-peptide with a Kd of 2.1 μm (data not shown), a value comparable to the affinity of α-thrombin for γ′-peptide. Hirudin-(54–65) had no effect on the binding of γ-thrombin to immobilized γ′-peptide (Fig. 7B), consistent with the observation that it does not bind γ-thrombin (32). In contrast, HD1 weakly reduced γ-thrombin binding to γ′-peptide, displaying a Kiobs 20-fold higher than that observed with FPR-α-thrombin. This may reflect the presence of a residual HD1-binding sub-domain of exosite 1 on γ-thrombin. HD23, which was used as a nonspecific oligonucleotide control, reduced α- or γ-thrombin binding to immobilized γ′-peptide by ∼15% (data not shown). As expected, HD22 reduced γ-thrombin and FPR-α-thrombin binding to γ′-peptide to a similar extent, in keeping with the exosite 2-dependent interaction of γ-thrombin with γ′-peptide. These results provide further confirmation of the allosteric connection between the exosites.
DISCUSSION
Because of thrombin's central role in hemostasis, regulation of its activity is critical. The exosites flanking the active site of thrombin contribute to the regulation of thrombin by directing substrates to the active site or by bringing thrombin into proximity of substrates or inhibitors. Beyond these steering and tethering roles, the exosites also serve to allosterically modulate thrombin's catalytic activity. When PAR1 binds exosite 1, an obstructing surface loop is shifted away from the active site (18). Numerous ligands that bind to either exosite have been shown to modulate thrombin activity with both synthetic and natural substrates (21, 25, 32, 39). Using fluorescently labeled probes, we previously demonstrated that synthetic ligands bound the exosites in a reciprocal manner, suggesting inter-exosite communication (29). Thus, we showed that binding of F2 to exosite 2 on thrombin caused displacement of a hirudin-derived peptide from exosite 1. A subsequent report using similar techniques challenged this observation (25). In the current study, a fluorescent thrombin variant labeled exclusively in exosite 1 was used to examine the effects of ligand binding to exosite 2. HD22, γ′-peptide, and heparin elicited fluorescent changes at exosite 1, and their binding affinities were similar to those obtained by other means. These findings provide additional evidence of an allosteric linkage between the exosites.
To explore the functional consequences of this inter-exosite linkage, additional techniques were used to supplement the fluorescence studies. Unmodified ligands and targets were used in these experiments so that the interaction of native proteins could be examined. By exploiting the observation that thrombin binds the γA/γA form of fibrin(ogen) exclusively via exosite 1 (32), the effect of exosite 2-directed ligands on thrombin binding to fibrin was examined. Three exosite 2-directed ligands of varying affinity and composition were able to saturably reduce the amount of thrombin bound to fibrin by 50%. In particular, HD22 reduced the affinity of thrombin for fibrin clots by 6-fold. These results confirm that there is a structural connection between the exosites. Furthermore, our data show that the reverse also is true. Thus, exosite 1-directed ligands reduce thrombin binding to γ′-peptide, which is an exosite 2-mediated interaction (32). Finally, we demonstrate that HD22 displaces thrombin already bound to preformed fibrin clots, a finding that addresses the concern that the presence of the ligands during the clotting process might influence thrombin binding to fibrin. Thus, these studies provide further evidence that the exosites of thrombin are functionally linked.
The mechanism by which the exosites communicate remains unclear. Ligand interactions are thought to be exclusive to the respective exosites. Although prior to cleavage, fibrinogen is proposed to contact the periphery of exosite 2 via the NH2-terminal fibrinopeptide A, the interaction with fibrin only involves exosite 1 (34, 35). This is also supported by crystallographic evidence from the interaction of thrombin with fibrin fragment E, which shows binding mediated exclusively via exosite 1 (36). The crystal structure of thrombin in complex with HD22 has yet to be reported. Nonetheless, structures of thrombin in complex with an exosite 2-binding RNA aptamer, F2, or heparin reveal sites of contact limited to exosite 2 (40–42). Functional studies also support exclusive occupation of exosite 2 by the ligands used in our study (32, 33, 42). The findings of these crystallographic and functional studies make it unlikely that the reciprocal effects that we observed are the result of ligands bound to one exosite sterically hindering the opposite exosite. Instead, the reciprocal nature of the interactions described here must result from long range structural effects. There is ample evidence, both structural and functional, that occupation of exosite 1 induces changes in the active site. It also is clear that there is an allosteric connection between exosite 1 and the Na+-binding site (43, 44). Na+ is an allosteric effector that modulates the procoagulant and anticoagulant functions of thrombin. An allosteric network can be traced through a series of Trp residues in the core of the thrombin molecule that extend from exosite 1 to the Na+-binding site. For the current study, it is notable that the Na+-binding site resides between the active site and exosite 2. In the crystal structure of thrombin in complex with the γ′-peptide, the ligand binds to exosite 2 and contacts the hydrophobic pocket of the Na+-binding site (45). Therefore, because Na+ has a global effect on thrombin structure, it is possible that the allosteric core extends past the Na+ site to exosite 2 (44). Thus, there appears to be no basis for the competition observed in this study other than indirect effects via thrombin itself. Previously, the absence of observable differences of the structures of thrombin-ligand complexes by crystallography led to the conclusion that the allosteric changes are subtle. However, this may be a consequence of occupation of the active site by tripeptide chloromethyl ketone moieties (44). In support of an allosteric connection between the exosites, NMR studies provide structural evidence that binding of γ′-peptide to exosite 2 alters exosite 1 (30). It follows that if ligands for either exosite can affect the active site, then their effects might extend beyond the active site. This is consistent with the conclusion that thrombin is a globally allosteric enzyme.
Although this study provides evidence for inter-exosite linkage, there are numerous examples of ligands that bind both exosites simultaneously. These include critical regulatory interactions, such as those with thrombomodulin, γA/γ′-fibrin, or GpIbα/PAR1. Although these observations may initially appear at odds with the current results, it is important to note that these targets are bivalent or closely associated such that the ligands for both exosites are tethered to each other. The same is true of the binding of a bivalent aptamer that binds both exosites (46, 47). Bivalent binding places a potentially important limitation on the ability of thrombin to dissociate from either individual ligand.
It is unclear whether the allosteric connection between the exosites has physiological relevance. However, these results reveal that the allosteric connections that exist in thrombin likely extend to all regulatory domains (44). Because occupation of individual exosites can have unique effects on the active site (24, 32), it is possible that simultaneous occupation of both exosites evokes a response distinct from that induced by occupation of individual exosites. For example, thrombin-mediated activation of factors V or VIII (4), which requires both exosites on thrombin for optimal activity, may reflect unique modulation of the enzyme.
In summary, our findings extend the array of mechanisms by which thrombin may be regulated. The observation that an exosite 2 ligand can affect thrombin's interaction with fibrin also provides a novel avenue through which thrombin's activity can be modulated. Thus, it might be possible to utilize exosite 2-directed ligands to displace fibrin-bound thrombin, thereby rendering it more accessible to inhibition by circulating antithrombin. Exosite 2-directed ligands also have the potential to attenuate platelet activation by reducing thrombin binding to glycoprotein 1bα and PAR1. At the same time, hemostatic reactions of thrombin, such as clotting of fibrinogen, which is mediated by exosite 1, would only be modestly impaired. These properties may endow exosite 2-directed ligands with a benefit-to-risk profile superior to that of anticoagulants such as argatroban, which targets the active site of thrombin, or hirudin and bivalirudin, which target exosite 1 and the active site (48).
This work was supported, in part, by the Canadian Institutes of Health Research (Grants MOP 3992 and CTP 79846), the Heart and Stroke Foundation of Ontario (Grants T4729 and T4730), and the Ontario Research and Development Challenge Fund.
- PAR
- protease-activated receptor
- 5-IAF
- 5-iodoacetamidofluorescein
- F1
- prothrombin fragment 1
- F2
- prothrombin fragment 2
- SPR
- surface plasmon resonance
- HBS
- Hepes-buffered saline
- FPRck
- d-Phe-Pro-Arg chloromethyl ketone
- YPRck
- d-Tyr-Pro-Arg chloromethyl ketone
- RU
- response unit(s).
REFERENCES
- 1.Lane D. A., Philippou H., Huntington J. A. (2005) Blood 106, 2605–2612 [DOI] [PubMed] [Google Scholar]
- 2.Meh D. A., Siebenlist K. R., Mosesson M. W. (1996) J. Biol. Chem. 271, 23121–23125 [DOI] [PubMed] [Google Scholar]
- 3.Myles T., Yun T. H., Hall S. W., Leung L. L. (2001) J. Biol. Chem. 276, 25143–25149 [DOI] [PubMed] [Google Scholar]
- 4.Esmon C. T., Lollar P. (1996) J. Biol. Chem. 271, 13882–13887 [DOI] [PubMed] [Google Scholar]
- 5.Ayala Y. M., Cantwell A. M., Rose T., Bush L. A., Arosio D., Di Cera E. (2001) Proteins 45, 107–116 [DOI] [PubMed] [Google Scholar]
- 6.Ye J., Liu L. W., Esmon C. T., Johnson A. E. (1992) J. Biol. Chem. 267, 11023–11028 [PubMed] [Google Scholar]
- 7.Naski M. C., Fenton J. W., 2nd, Maraganore J. M., Olson S. T., Shafer J. A. (1990) J. Biol. Chem. 265, 13484–13489 [PubMed] [Google Scholar]
- 8.Baglin T. P., Carrell R. W., Church F. C., Esmon C. T., Huntington J. A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esmon C. T., Esmon N. L., Harris K. W. (1982) J. Biol. Chem. 257, 7944–7947 [PubMed] [Google Scholar]
- 10.Hayashi T., Zushi M., Yamamoto S., Suzuki K. (1990) J. Biol. Chem. 265, 20156–20159 [PubMed] [Google Scholar]
- 11.Wang W., Nagashima M., Schneider M., Morser J., Nesheim M. (2000) J. Biol. Chem. 275, 22942–22947 [DOI] [PubMed] [Google Scholar]
- 12.Sheehan J. P., Sadler J. E. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 5518–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson S. T., Shore J. D. (1982) J. Biol. Chem. 257, 14891–14895 [PubMed] [Google Scholar]
- 14.Yamagishi R., Koide T., Sakuragawa N. (1987) FEBS Lett. 225, 109–112 [DOI] [PubMed] [Google Scholar]
- 15.Baglia F. A., Badellino K. O., Li C. Q., Lopez J. A., Walsh P. N. (2002) J. Biol. Chem. 277, 1662–1668 [DOI] [PubMed] [Google Scholar]
- 16.De Candia E., Hall S. W., Rutella S., Landolfi R., Andrews R. K., De Cristofaro R. (2001) J. Biol. Chem. 276, 4692–4698 [DOI] [PubMed] [Google Scholar]
- 17.Berny M. A., White T. C., Tucker E. I., Bush-Pelc L. A., Di Cera E., Gruber A., McCarty O. J. (2008) Arterioscler. Thromb. Vasc. Biol. 28, 329–334 [DOI] [PubMed] [Google Scholar]
- 18.Gandhi P. S., Chen Z., Mathews F. S., Di Cera E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1832–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye J., Esmon N. L., Esmon C. T., Johnson A. E. (1991) J. Biol. Chem. 266, 23016–23021 [PubMed] [Google Scholar]
- 20.Hortin G. L., Trimpe B. L. (1991) J. Biol. Chem. 266, 6866–6871 [PubMed] [Google Scholar]
- 21.Liu L. W., Vu T. K., Esmon C. T., Coughlin S. R. (1991) J. Biol. Chem. 266, 16977–16980 [PubMed] [Google Scholar]
- 22.Sakharov D. V., Plow E. F., Rijken D. C. (1997) J. Biol. Chem. 272, 14477–14482 [DOI] [PubMed] [Google Scholar]
- 23.Jakubowski H. V., Kline M. D., Owen W. G. (1986) J. Biol. Chem. 261, 3876–3882 [PubMed] [Google Scholar]
- 24.Liaw P. C., Fredenburgh J. C., Stafford A. R., Tulinsky A., Austin R. C., Weitz J. I. (1998) J. Biol. Chem. 273, 8932–8939 [DOI] [PubMed] [Google Scholar]
- 25.Verhamme I. M., Olson S. T., Tollefsen D. M., Bock P. E. (2002) J. Biol. Chem. 277, 6788–6798 [DOI] [PubMed] [Google Scholar]
- 26.Walker F. J., Esmon C. T. (1979) J. Biol. Chem. 254, 5618–5622 [PubMed] [Google Scholar]
- 27.Bock P. E. (1992) J. Biol. Chem. 267, 14974–14981 [PubMed] [Google Scholar]
- 28.Liu L. W., Ye J., Johnson A. E., Esmon C. T. (1991) J. Biol. Chem. 266, 23633–23636 [PubMed] [Google Scholar]
- 29.Fredenburgh J. C., Stafford A. R., Weitz J. I. (1997) J. Biol. Chem. 272, 25493–25499 [DOI] [PubMed] [Google Scholar]
- 30.Sabo T. M., Farrell D. H., Maurer M. C. (2006) Biochemistry 45, 7434–7445 [DOI] [PubMed] [Google Scholar]
- 31.Lancellotti S., Rutella S., De Filippis V., Pozzi N., Rocca B., De Cristofaro R. (2008) J. Biol. Chem. 283, 30193–30204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pospisil C. H., Stafford A. R., Fredenburgh J. C., Weitz J. I. (2003) J. Biol. Chem. 278, 21584–21591 [DOI] [PubMed] [Google Scholar]
- 33.Lovely R. S., Moaddel M., Farrell D. H. (2003) J. Thromb. Haemost. 1, 124–131 [DOI] [PubMed] [Google Scholar]
- 34.Fredenburgh J. C., Stafford A. R., Leslie B. A., Weitz J. I. (2008) J. Biol. Chem. 283, 2470–2477 [DOI] [PubMed] [Google Scholar]
- 35.Owen W. G., Esmon C. T., Jackson C. M. (1974) J. Biol. Chem. 249, 594–605 [PubMed] [Google Scholar]
- 36.Church W. R., Ouellette L. A., Messier T. L. (1991) J. Biol. Chem. 266, 8384–8391 [PubMed] [Google Scholar]
- 37.Henriksen R. A., Mann K. G. (1988) Biochemistry 27, 9160–9165 [DOI] [PubMed] [Google Scholar]
- 38.Kretz C. A., Stafford A. R., Fredenburgh J. C., Weitz J. I. (2006) J. Biol. Chem. 281, 37477–37485 [DOI] [PubMed] [Google Scholar]
- 39.Duffy E. J., Angliker H., Le Bonniec B. F., Stone S. R. (1997) Biochem. J. 321, 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long S. B., Long M. B., White R. R., Sullenger B. A. (2008) RNA 14, 2504–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter W. J., Cama E., Huntington J. A. (2005) J. Biol. Chem. 280, 2745–2749 [DOI] [PubMed] [Google Scholar]
- 42.Arni R. K., Padmanabhan K., Padmanabhan K. P., Wu T. P., Tulinsky A. (1993) Biochemistry 32, 4727–4737 [DOI] [PubMed] [Google Scholar]
- 43.Pineda A. O., Carrell C. J., Bush L. A., Prasad S., Caccia S., Chen Z. W., Mathews F. S., Di Cera E. (2004) J. Biol. Chem. 279, 31842–31853 [DOI] [PubMed] [Google Scholar]
- 44.Di Cera E. (2008) Mol. Aspects Med. 29, 203–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pineda A. O., Chen Z. W., Marino F., Mathews F. S., Mosesson M. W., Di Cera E. (2007) Biophys. Chem. 125, 556–559 [DOI] [PubMed] [Google Scholar]
- 46.Kim Y., Cao Z., Tan W. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 5664–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Müller J., Freitag D., Mayer G., Pötzsch B. (2008) J. Thromb. Haemost. 6, 2105–2112 [DOI] [PubMed] [Google Scholar]
- 48.Weitz J. I., Buller H. R. (2002) Circulation 105, 1004–1011 [DOI] [PubMed] [Google Scholar]