

Abstract
The conformational dynamics of cytochrome P450 2B1 (CYP2B1) were investigated through the introduction of a disulfide bond to link the I- and K-helices by generation of a double Cys variant, Y309C/S360C. The consequences of the disulfide bonding were examined both experimentally and in silico by molecular dynamics simulations. Under high hydrostatic pressures, the partial inactivation volume for the Y309C/S360C variant was determined to be −21 cm3mol−1, which is more than twice as much as those of the wild type (WT) and single Cys variants (Y309C, S360C). This result indicates that the engineered disulfide bond has substantially reduced the protein plasticity of the Y309C/S360C variant. Under steady-state turnover conditions, the S360C variant catalyzed the N-demethylation of benzphetamine and O-deethylation of 7-ethoxy-trifluoromethylcoumarin as the WT did, whereas the Y309C variant retained only 39% of the N-demethylation activity and 66% of the O-deethylation activity compared with the WT. Interestingly, the Y309C/S360C variant restored the N-demethylation activity to the same level as that of the WT but decreased the O-deethylation activity to only 19% of the WT. Furthermore, the Y309C/S360C variant showed increased substrate specificity for testosterone over androstenedione. Molecular dynamics simulations revealed that the engineered disulfide bond altered substrate access channels. Taken together, these results suggest that protein dynamics play an important role in regulating substrate entry and recognition.
Liver microsomal cytochromes P450 (CYP or P450)2 metabolize a large number of clinically used drugs that have diverse steric and functional moieties. Despite low sequence homology among CYPs from different families, all P450s invariably contain a heme cofactor that is coordinated to a thiolate and catalyze the oxidative metabolism, mostly through hydroxylation, of substrates. However, production of reactive intermediates by P450s is often associated with drug toxicity and carcinogenesis, and inhibition or induction of a specific P450 isoform may lead to adverse drug-drug interactions (1). From a clinical and pharmacological perspective, it is important to understand the structure, function, and dynamics of P450s.
Structural studies of P450s by x-ray crystallography in the past decade have provided us with a wealth of information regarding the structural organization, critical active site residues, and proton delivery pathways of P450s (2–4). In particular, these structural analyses have consistently shown that certain regions of the P450 structures such as the F/G and B/B′-C loops are extremely flexible and can undergo large conformational changes to accommodate substrates of various sizes, although the overall folding pattern of all P450s is conserved. For instance, an open conformation was observed in the ligand-free CYP2B4 crystal structure, whereas a closed conformation was reported for the CPI-bound CYP2B4 (3, 5). The open-to-closed conformational change involves large motions of the F- and G-helices and the F/G and B/B′-C loops. Based on comparisons of the crystal structures of CYP2B4 bound with inhibitors of different sizes, Zhao et al. (6) identified five plastic regions in P450s, including the B/B′-C loop (PR2) and F/G loop (PR4). Binding of ketoconazole or erythromycin to CYP3A4 led to a large increase in the active site volume (>80% increase) because of conformational changes primarily in the PR4, but interestingly the F- and G-helices moved in the opposite direction (7). These authors proposed that the extreme flexibility of CYP3A4 accounts for its promiscuity, as CYP3A4 metabolizes nearly ∼50% of all clinically used drugs. The complexity of the conformational flexibility and dynamics are also revealed in an MD simulation study of CYP3A4, 2C9 and 2A6 (8). Importantly, this molecular dynamics (MD) simulation study shows that the three-dimensional structure of P450s is more flexible in solution than was observed in the crystal structure.
Despite intensive studies of the crystal structures of microsomal P450s, insights into the conformational dynamics of P450s in solution, particularly in relation to their functional importance, are lacking. A laser flash photolysis study of CO rebinding to CYP2E1 in solution revealed that the binding of substrates such as ethanol, pyrazole, and acetaminophen restricts the conformational flexibility of CYP2E1, as the kinetics for the rebinding of CO to ligand-bound CYP2E1 are significantly slower than those for the ligand-free CYP2E1 (9). A solution thermodynamics study of CYP2B4 supports the notion that CYP2B4 is remarkably flexible, as the entropy substantially decreases upon inhibitor binding resulting from reduction of the hydrophobic surface (10). In this study, a de novo disulfide bond is engineered into CYP2B1 and the consequences resulting from the disulfide bonding are examined both experimentally and in silico using MD simulations. To discern the effect of the de novo disulfide bond apart from the Cys mutagenesis, both the single and double Cys variants were characterized in detail. To our knowledge, this is the first report that investigates the consequences of limiting conformational dynamics in a P450 by incorporating a disulfide bond. Our results demonstrate that protein dynamics play an important role in regulating substrate entry/product egress channels and substrate recognition and provide insights that will be valuable for rational drug design and protein engineering.
EXPERIMENTAL PROCEDURES
Chemicals
All chemicals used are of ACS grade unless otherwise specified. Benzphetamine, NADPH, sodium dithionite, and dilauroylphosphatidylcholine were purchased from Sigma. Trifluoroacetic acid, N-ethylmaleimide, and Tris(carboxyethyl)phosphine (TCEP) were purchased from Pierce Chemicals. 7-Ethoxy-4-trifluoromethylcoumarin (7-EFC) was purchased from Invitrogen (Molecular Probes® products). Carbon monoxide (purity > 99.5%) was purchased from Cryogenic Gases Inc. (Detroit, MI).
Site-directed Mutagenesis and Overexpression and Purification of CYP2B1 WT and Variants
Single and double Cys mutants were prepared using the QuikChange site-directed mutagenesis kit according to the manufacturer's protocol (Stratagene). The forward mutagenic primers for Y309C and S360C are 5′-GCACCACACTCCGCTGTGGTTTCCTGCTGATG-3′ and 5′-CGAGATTCAGAGGTTTTGCGATCTTGTCCCTATTG-3′, respectively. The double Cys mutant Y309C/S360C was prepared using the plasmid of Y309C as the template and the mutagenic primer of S360C. The site-specific mutation was confirmed by DNA sequencing at the University of Michigan Biomedical Core Facilities.
CYP2B1 WT and variants were overexpressed in Escherichia coli C41(DE3) cells and purified as described previously (11). The expressed proteins contained a His4 tag at the C terminus.
Confirmation of the Disulfide Bond in the Y309C/S360C Variant by ESI-LC/MS
Formation of the disulfide bond in Y309C/S360C was confirmed by ESI-LC/MS after the protein sample had been alkylated with N-ethylmaleimide in the presence of TCEP. Typically, 1 volume of the Y309C/S360C sample (∼1 nmol) was mixed with 2 volumes of 0.1 m Tris/8 m guanidine (pH 7.0) to denature the protein sample. The free thiols in the denatured P450 sample were alkylated by addition of a 250-fold molar excess of N-ethylmaleimide followed by incubation of the reaction mixture for 2 h at room temperature. The sample was then divided into two parts, into one of which were added 50 mm TCEP and 5 mm EDTA. Following the reduction of the disulfide bond by TCEP, both samples were subjected to further alkylation with a 250-fold molar excess of N-ethylmaleimide for 2 h at room temperature. The molecular masses of the alkylated samples were then analyzed using an LCQ ion trap mass spectrometer (Thermo Finnigan, Inc.) as described elsewhere (12).
Effect of Hydrostatic Pressure on Conformational Flexibility of CYP2B1 WT and Variants
The effects of incorporating the disulfide bond on the conformational flexibility of CYP2B1 were studied using a high hydrostatic pressure technique as described previously (13, 14). The optical spectra of the P450-CO complexes were measured in 50 mm Tris-HCl (pH 7.0) at ambient temperature as a function of hydrostatic pressure up to 300 MPa. Tris buffer was chosen because it is rather insensitive to pressure variation (ΔVTris ≈ −1 cm3mol−1) as opposed to potassium phosphate buffer (ΔVKP ≈ −10 cm3mol−1). All samples were corrected for the compressibility of the water solvent, which is initially 4% per 100 MPa and a total of 15% at 600 MPa.
Determination of the pressure-induced changes is based on the pressure dependence of the equilibrium (P450 ↔ P420) as expressed in Equation 1,
 |
where K and ΔVO represent the pressure-induced equilibrium constant and partial volume of the system in cm3mol−1, respectively.
Determination of Steady-state Turnover Rates for the Metabolism of Benzphetamine and 7-EFC by CYP2B1 WT and Variants
The steady-state rate for the N-demethylation of benzphetamine was determined at 30 °C in a reconstituted system as described previously (15), whereas the O-deethylation rate of 7-EFC was measured according to the method of Reed and Hollenberg (16) except that the reaction was carried out on a 96-well microplate at 30 °C. Each well contained 0.15 ml of 50 mm potassium phosphate buffer (pH 7.4), 22.5 pmol of P450 and NADPH-dependent cytochrome P450 reductase (CPR) each, 0.1 mm 7-EFC, and 0.3 mm NADPH. The reaction was terminated with 50 μl of ice-cold acetonitrile. The fluorescent emission from the 7-hydroxy-4-trifluoromethylcoumarin product was recorded at 510 nm with excitation at 410 nm.
Binding of Benzphetamine and 7-EFC to CYP2B1 WT and Variants
The dissociation constants of benzphetamine (KdBP) for the WT and variants were determined spectrophotometrically by measurement of the substrate-induced type I spectral changes. Equal volumes of the P450 solutions containing 1 μm P450, 0.1 m potassium phosphate (pH 7.4), and 0.1 mg/ml dilauroylphosphatidylcholine were aliquoted into the sample and reference cuvettes. A base line was recorded after thermal equilibration at 30 °C for 5 min. Aliquots of 20 mm benzphetamine stock solution in water were added to the sample cuvette, and an equal volume of water was added to the reference cuvette. The difference spectra were recorded from 350 to 500 nm, and the observed ΔA(382–420 nm) at various benzphetamine concentrations (20–560 μm) were fit to Equation 2 to obtain Kd.
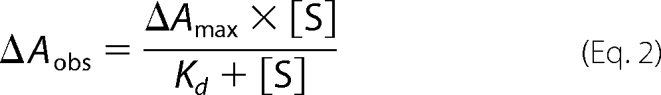 |
The binding of 7-EFC was evaluated by measurement of the Km for 7-EFC (KmEFC) under steady-state turnover conditions, as 7-EFC interferes with the spectrophotometric titration. The steady-state activity measurements were performed in a 96-well microplate as mentioned previously except that the concentrations of 7-EFC were varied from 30 to 300 μm. KmEFC was obtained by fitting the turnover rates at the various concentrations to the Michaelis-Menten equation using SigmaPlot software (Systat Inc.).
HPLC Analysis of Metabolites of Testosterone Produced by CYP2B1 WT and Variants
The metabolism of testosterone by CYP2B1 WT and variants was carried out in a reconstituted system that contained 50 mm potassium phosphate (pH 7.4), 1 μm P450, 1 μm CPR, 50 μm testosterone, and 0.3 mg/ml dilauroylphosphatidylcholine. NADPH was added to 1 mm final concentration to initiate the reaction after the reaction mixture had been reconstituted at 4 °C for 60 min. The reaction mixture was incubated at 30 °C for 30 min and then terminated by the addition of 4 ml of ethyl acetate. Testosterone and its metabolites were extracted into ethyl acetate, and the extracts were dried under a stream of nitrogen gas. The dried extract was redissolved in 70 μl of 65% methanol, and a 50-μl aliquot was injected onto the HPLC for further analysis.
Testosterone and its metabolites were analyzed using a Shimadzu HPLC system and separated on a reverse-phase Zorbax SB C18 column (3.1 × 150 mm, 5 μm, Agilent Technologies) at a flow rate of 0.4 ml/min. The binary mobile phase consisted of solvent A (10% methanol, 90% water) and solvent B (90% methanol, 10% water). Testosterone and its metabolites were eluted with the following gradient: 10% B for 10 min and then increasing linearly to 30% B in 20 min; then to 55% B in 30 min; then held at 55% B for 8 min followed by a linear increase to 100% B in 45 min; and then held at 100% B for 5 min. The molar ratios of the testosterone metabolites were calculated as the ratios of the areas under the curve that were integrated using LCSolution software (Shimadzu).
Determination of the Rate of Electron Transfer from CPR to CYP2B1 WT and Variants
The rates of electron transfer from CPR to the WT and variants were determined in a stopped-flow spectrophotometer as described previously (17). The kinetic traces at 450 nm were fit with two or three exponentials to obtain the apparent rate constants.
Homology Modeling of CYP2B1 and Rational Design of the Disulfide Bond
The rational design of a disulfide bond requires a homology model of CYP2B1. Because the crystal structure of CYP2B1 is not available, a homology model of CYP2B1 was constructed using the Modeler9v5 program (18). The crystal structure of CYP2B4 in a closed conformation (Protein Data Bank code 1SUO) was used as the template (5). The amino acid sequences of CYP2B1 and CYP2B4 were aligned with ClustalW (19) after the first 27 amino acid residues of CYP2B1 were deleted, as they were absent from the template of CYP2B4. The aligned sequences were 78% identical. Because of the relatively high sequence homology, a reliable homology model could be built. The heme was incorporated into the homology model as a rigid body. A total of 50 homology models were built with the md_level and repeat_optimization set to refine.slow and 3, respectively. The top 10 models were selected on the basis of a combination of scores and were further evaluated with the Procheck and ProQ software (20, 21) to choose the best model.
The rational design of the disulfide bond in CYP2B1 was based on the algorithm developed by Dr. Alan Dombkowski (22) and available in Disulfide by Design software, version 1.2. With default parameters, the program generated a list of amino acid pairs that would likely form a disulfide bond once they were mutated to cysteines. Taking into account the energy required to form a disulfide bond and the positions of specific residues, we chose the residues Y309C and S360C to engineer a disulfide bond into CYP2B1.
Conformational Flexibility of CYP2B1 WT and Y309C/S360C Variant as Studied by MD Simulations
MD simulations were performed under periodic boundary conditions in explicit water solvent using NAMD software, version 2.6 (23). As the heme moiety plays an essential role in P450 catalysis, the coordinates of the heme were included in the MD simulations. The heme is linked to Cys436 with a patch included in the CHARMM topology files. For consistency, the identical MD simulation protocol was followed to examine the conformational flexibility of the WT and Y309C/S360C variant. The aforementioned CYP2B1 homology model was solvated in a water box with protein being 10 Å away from the periodic boundary. Counterions were added to neutralize the charge of the whole system. Prior to MD simulations, the energy of the CYP2B1 homology model was minimized for 5000 steps using the conjugate gradient minimization algorithm to remove bad contacts and steric clashes. The energy-minimized CYP2B1 model was then equilibrated at 300 K for 2 ns. After equilibrium, unconstrained MD simulation was performed on the equilibrated CYP2B1 model at 300 K as an NPT ensemble for 2 ns. Protein cavities and tunnels were identified by analyses of a series of MD trajectory files with the Caver program (24) and visualized with PyMOL.
RESULTS
Rational Design of a Disulfide Bond in CYP2B1
The rational design was based on two specific criteria: the first was to examine the energy required to form a disulfide bond and the second to introduce the disulfide bond at a location that is potentially important for P450 catalysis while avoiding inactivation of P450. Based on thermodynamic calculations by the program Disulfide by Design, the energy required to form a disulfide bond between Cys309 and Cys360 is 0.3 kcal/mol. The mean energy for disulfide bond formation is 1.07 kcal/mol (22). Thus, formation of a disulfide bond between Y309C and S360C is favored thermodynamically. This disulfide bond between the Cys309 and Cys360 would cross-link the I-helix to the C-terminal end of the K-helix, both of which are known to be important for P450 catalysis (25–28). Several lines of evidence suggest that the cross-linking site around the Cys309 and Cys360 may constitute part of a substrate access channel. According to Gotoh (29), the substrate recognition site 4 (SRS4) involves the I-helix where Tyr309 is located. Ser360 is located at the SRS5 that begins with the highly conserved EXXR motif (residues 355–358) and spans ∼11 residues to the C terminus (30). This hypothesis is supported by results from MD simulations, mutagenesis studies, x-ray crystallography, and high pressure spectroscopy (31–33). Modification around these sites would allow us to identify the elusive substrate access channels of P450 in solution.
Confirmation of the Disulfide Bond in the Y309C/S360C Variant by ESI-LC/MS
Initial characterization of the three Cys variants showed that their CO-ferrous complexes absorb at 450 nm, like the WT, and that no intermolecular disulfide bonds were formed in any of three variants as revealed by SDS-PAGE (data not shown). The formation of a disulfide bond in the Y309C/S360C variant was confirmed by analyses of the molecular masses of the denatured Y309C/S360C variant that had been reacted with N-ethylmaleimide in the absence and presence of the reducing reagent, TCEP. The control Y309C/S360C sample has a mass of 56,407.0 Da as shown in Fig. 1A, which is in excellent agreement with the theoretical mass of 56,386.0 Da based on its amino acid sequence and is within the experimental error of 0.04%. In the absence of TCEP, the alkylated Y309C/S360C gives a mass of 57,116.0 Da (Fig. 1B). This mass increase of 709.0 Da over that of the control sample indicates that ∼6 of the 8 free thiols in the Y309C/S360C variant have been alkylated. In the presence of TCEP, the alkylated Y309C/S360C shows a mass increase of 1,011.0 Da over the control sample (57,418.0 minus 56,407.0 Da), which corresponds to alkylation of 8.1 free thiols. Thus, reduction of the Y309C/S360C variant by TCEP leads to the alkylation of two additional cysteines. These results demonstrate unequivocally that a disulfide bond is formed in the Y309C/S360C variant.
FIGURE 1.

Molecular masses for the alkylated Y309C/S360C variant of CYP2B1 in the absence and presence of TCEP as determined by ESI-LC/MS. Alkylation was performed by reacting the denatured Y309C/S360C with N-ethylmaleimide, and the mass of the alkylated Y309C/S360C was analyzed by ESI-LC/MS as described under “Experimental Procedures.” A, untreated Y309C/S360C; B, alkylated Y309C/S360C in the absence of TCEP; C, alkylated Y309C/S360C following reduction by TCEP.
Effect of the Disulfide Bond on the Conformational Flexibility of CYP2B1
High pressure is a powerful technique for probing protein dynamics and conformational stability and has been used to study P450s (31, 34–36). Therefore, we applied this technique to investigate whether the mutations and formation of a disulfide bond could have altered the conformational flexibility of CYP2B1. The absorbance of the ferrous P450-CO complex at 450 nm decreases under increasing hydrostatic pressure with a concomitant increase in absorbance at 420 nm. Because of turbidity resulting from protein denaturing and unfolding at > 300 MPa, which is not unusual for microsomal P450s, we analyzed the P450-to-P420 transition at ≤ 300 MPa. Based on a P450 → P420 transition model without the formation of intermediate species, the inactivation volumes were calculated by plotting the lnK as a function of the applied pressure (see supplemental Fig. S1). The partial molar inactivation volume (ΔVO) for the WT was determined to be −51 cm3mol−1, similar to a reported value of −50 cm3mol−1 for CYP2B4 (34). Mutation of Ser360 to a cysteine does not have any impact on the inactivation volume (−50 cm3mol−1), whereas mutation of Tyr309 to a cysteine leads to an increase in the absolute volume change (−62 cm3mol−1), indicating that Y309C is more compressible than the WT. Alternatively, the higher inactivation volume could be consistent with the fact that the Y309C variant may have a heme that is more exposed to the solvent water (31). In striking contrast, the disulfide bonded Y309C/S360C variant exhibits an inactivation volume of −24 cm3mol−1. The absolute partial inactivation volume of Y309C/S360C decreases by more than 2-fold compared with the WT. This is indicative of a more rigid heme pocket and protein structure (36). Thus, it can be concluded that the cross-linking between the I-helix and the K-helix has substantially decreased the plasticity of the Y309C/S360C variant.
Steady-state Turnover Rates for the Metabolism of Benzphetamine and 7-EFC by CYP2B1 WT and Variants
The effects of the disulfide bond on P450 catalysis were studied by measurement of the steady-state turnover rates for benzphetamine and 7-EFC, two commonly used substrates for CYP2B1, and the results are presented in Table 1. As shown, mutation of Ser360 to a Cys has no impact on benzphetamine metabolism, whereas mutation of Y309C to a Cys decreases the activity by 61%. Interestingly benzphetamine activity is restored to the level of the WT in the Y309C/S360C variant, presumably because of formation of the disulfide bond.
TABLE 1.
Steady-state turnover rates for the metabolism of benzphetamine (BP) and 7-EFC by CYP2B1 WT and variants
The specific activities are expressed as nmol of product (formaldehyde (HCHO) for benzphetamine and 7-hydroxy-4-trifluoromethylcoumarin (7-HFC) for 7-EFC)/min/nmol of P450. Refer to the “Experimental Procedures” for incubation times and analyses of product formation.
| CYP2B1 | Specific activity for BP | Specific activity for 7-EFC |
|---|---|---|
| nmol HCHO/min/nmol P450 | nmol 7-HFC/min/nmol P450 | |
| WT | 24.5 ± 0.9 | 2.3 ± 0.2 |
| Y309C | 9.5 ± 0.7 | 1.5 ± 0.1 |
| S360C | 24.7 ± 1.8 | 2.8 ± 0.2 |
| Y309C/S360C | 23.9 ± 1.2 | 0.44 ± 0.01 |
As observed for the metabolism of benzphetamine, the 7-EFC O-deethylation activity of the Y309C variant is decreased to 66% of the WT. The O-deethylation activity of the S360C variant is increased slightly. Unlike the benzphetamine metabolism, however, the O-deethylation activity of the Y309C/S360C variant is decreased dramatically to only ∼19% of the WT. It is intriguing that the disulfide bond has different effects on the catalytic oxidation of benzphetamine and 7-EFC. It seems that conformational dynamics may affect P450 catalysis in a substrate-dependent manner.
Binding of Benzphetamine and 7-EFC to CYP2B1 WT and Variants
To evaluate the effect of mutation and disulfide bond formation on substrate binding, the dissociation constant of benzphetamine, KdBP, was determined by spectral titration, and the Km of 7-EFC was determined under the steady-state turnover conditions. The results are presented in Table 2. The two single variants exhibit a ∼44% increase in KdBP compared with the WT, indicative of a weaker binding of benzphetamine. Interestingly, the binding of benzphetamine to the Y309C/S360C variant is completely restored to the level of WT with a KdBP of 145 μm, consistent with the regain of the catalytic activity. In the case of 7-EFC, CYP2B1 WT catalyzes the O-deethylation of 7-EFC with a Km of 38 μm, similar to the previously reported Km of 25 μm (37). The Y309C and S360C variants show slightly increased KmEFC. In clear contrast to benzphetamine, formation of a disulfide bond in the Y309C/S360C variant does not restore its KmEFC to the level of the WT, but rather it results in a 2.8-fold increase in the KmEFC. The differential effect of the disulfide bond on the substrate binding may provide evidence for possible existence of multiple substrate access channels.
TABLE 2.
Kd of benzphetamine (BP) and Km of 7-EFC (EFC) for CYP2B1 WT and variants at 30 °C
KdBP was determined spectrophotometrically by titration of the type I spectral change as described under “Experimental Procedures.” KmEFC was determined by fitting the steady-state turnover rates of 7-EFC obtained at various 7-EFC concentrations to the Michaelis-Menten equation as described under “Experimental Procedures.”
| CYP2B1 | KdBP | KmEFC |
|---|---|---|
| μm | μm | |
| WT | 184 ± 19 | 38 ± 4.9 |
| S360C | 265 ± 45 | 49 ± 6.5 |
| Y309C | 239 ± 26 | 52 ± 6.2 |
| Y309C/S360C | 145 ± 21 | 110 ± 12 |
Metabolism of Testosterone by CYP2B1 WT and Variants
To study substrate stereo- and regiospecificity, we analyzed the metabolism of testosterone by the WT and Cys variants. As shown in Fig. 2, CYP2B1 WT and the three variants metabolize testosterone to yield five major metabolites: 16β- and 16α-OH androstenedione, 16α- and 16β-OH testosterone, and androstenedione, which elute at 38.2, 39, 41, 44, and 46.2 min, respectively. As shown in Table 3, the molar ratio of 16β/16α-OH testosterone formed by the WT is 1.0, consistent with results reported by Halpert and co-workers (27). S360C shows a slight decrease in the ratio of 16β/16α-OH testosterone, whereas the ratio for Y309C/S360C remains essentially the same as WT. Thus, formation of the disulfide bond does not result in significant shifts in stereo- and regioselectivity for the hydroxylation of testosterone. However, the molar ratio of 16β-OH-T/16β-OH-A increases by ∼2-fold for the Y309C variant and ∼4-fold for the Y309C/S360C variant compared with the WT. It is clear that the disulfide bond linkage in the Y309C/S360C variant has enhanced its specificity for testosterone.
FIGURE 2.

HPLC analysis of the testosterone metabolites by CYP2B1 WT and variants. The five major metabolites are 16β-OH-A (38.2 min), 16α-OH-A (39 min), 16α-OH-T (41 min), 16β-OH-T (44 min), and androstenedione (46.2 min). YS, Y309C/S360C.
TABLE 3.
Stereo- and regioselectivity observed in the metabolism of testosterone by CYP2B1 WT and variants
T and A represent testosterone and androstenedione, respectively. The reconstitution, extractions, and analyses of metabolites are described under “Experimental Procedures.”
| CYP2B1 | 16β/16α-OH-T | 16β-OH-T/16β-OH-A |
|---|---|---|
| molar ratio | molar ratio | |
| WT | 1.0 | 2.3 |
| Y309C | 0.8 | 4.5 |
| S360C | 0.7 | 1.6 |
| Y309C/S360C | 1.1 | 9.0 |
Electron Transfer from CPR to CYP2B1 WT and Variants
To investigate whether the mutation and disulfide bond affect the interaction of CYP2B1 with its redox partner, we measured the rate of electron transfer from CPR to CYP2B1. The results are summarized in Table 4. S360C shows an enhanced rate of electron transfer as its apparent rate constant for the fast phase, k1, increases by ∼60% compared with that of WT. Y309C, however, shows a decrease in the rate of electron transfer as its k1 value declines by 62%. It is of note that this decrease is not due to a slow CO binding to the ferrous Y309C, as CO binds to the ferrous Y309C with a bimolecular rate of 4.6 μm−1s−1.3 Y309C/S360C exhibits a rate of electron transfer comparable to that of the WT. It seems that the disulfide bond compensates for the loss in the rate of electron transfer resulting from the mutation of Y309 to a Cys residue.
TABLE 4.
The rates of electron transfer from NADPH-cytochrome P450 reductase to CYP2B1 WT and Cys variants at 22 °C
The kinetics were determined using stopped-flow spectrophotometry as described under “Experimental Procedures.” The apparent rate constants for the three kinetic phases are k1, k2, and k3, and the relative amplitudes for each respective phase are expressed as %A.
| CYP2B1 | Kinetic parameters |
|||||
|---|---|---|---|---|---|---|
| A1 | k1 | A2 | k2 | A3 | k3 | |
| % | s−1 | % | s−1 | % | s−1 | |
| WT | 20.1 | 0.94 | 34.8 | 0.17 | 45.2 | 0.031 |
| Y309C | 26.2 | 0.36 | 35.1 | 0.16 | 38.7 | 0.033 |
| S360C | 21.1 | 1.5 | 35.5 | 0.33 | 43.4 | 0.10 |
| Y309C/S360C | 45.4 | 1.3 | 54.6 | 0.044 | ||
MD Simulations and Analysis of Access Channels of CYP2B1 WT and Y309C/S360C Variant
The effect of incorporating a disulfide bond on the conformational dynamics of CYP2B1 was further explored in silico by MD simulations. The whole system for the MD simulations consists of ∼51,000 atoms, including the P450, counterions, and water molecules. The system reached equilibrium in approximately ∼700 ps, and the simulation was stable for 2 ns (see supplemental Fig.S2). Calculations of the root mean square deviation of the Cα carbon of each residue reveal that the backbone of CYP2B1 is flexible and can undergo significant conformational change in solution (see supplemental Fig. S3). The flexible regions include the F- and G-helices, B/B′, D/E, F/G, J/J′, K′/K″, and β3-1/β3-2 loops. The flexible regions observed in the MD simulations are consistent with the plastic regions identified by Halpert's group (6) and resemble the flexible regions of CYP2C9 identified previously by MD simulations (8). Our MD simulations also show that the conserved regions in the vicinity of the heme such as the I- and K-helices and β-bulge are less mobile. Such consistencies validate our MD simulations approach to examining whether the disulfide bond incorporation would alter the conformational dynamics of CYP2B1.
The Y309C/S360C variant shares dynamic features very similar to the WT; the five plastic regions observed in the WT are retained (supplemental Fig. S3). The average root mean square deviations for all the residues are 1.24 and 1.35 Å for the Y309C/S360C and WT, respectively, indicating that there is a decrease in the overall conformational flexibility in Y309C/S360C due to the presence of a disulfide bond. The structural differences between the WT and Y309C/S360C variant are highlighted in Fig. 3 by overlapping the average structures of these two proteins. It can be seen that the C-terminal end of the K-helix and the following loop-(360–365) are pulled inward to the active site by the disulfide bond.
FIGURE 3.

Comparison of the average backbone structures of CYP2B1 WT and the Y309C/S360C variant obtained from a 2-ns MD simulation. Both structures were averaged from 1.2 to 2 ns when the system had fully reached equilibrium. The backbone of the WT is a thin green ribbon, and the backbone of the Y309C/S360C is depicted as a thick ribbon and colored by the Cα displacement from the WT from blue (zero) to red (maximum). The heme and side chains of Cys309 and Cys360 are depicted in stick and ball form, sulfur, carbon, and oxygen atoms are colored yellow, cyan, and red, respectively. The FG-loop, β1-1 sheet, and eight helices (A, D, E, F, G, I, K, and J) are labeled.
To examine whether the reduced conformational flexibility resulting from disulfide bonding has any impact on the access channels, we analyzed a series of MD trajectories sampled between 1.2 and 2 ns, using the Caver program to identify potential cavities and tunnels connecting the active site void to the protein exteriors. Fig. 4 shows the three access channels identified for the WT (Fig. 4A) and the Y309C/S360C variant (Fig. 4B). In the WT, channel 1 consists of the void adjacent to the F/G and B′-C loops and β1. This access channel was also identified in the crystal structure of CYP2E1 (38). In CYP2E1, Leu103 and Phe478 form the bottleneck with a distance of 3.7 Å. In CYP2B1, this access channel is wider as the bottleneck is 5.1 Å between two Ile residues (Ile101 and Ile477). Channel 2 connects the active site void to the exterior around the B/B′-C loop. This seems to be one of the more common access channels, as it has been found in several bacterial and mammalian P450s (8, 33, 39). Channel 3a passes between the E-, F-, and I-helices and β3-2. This access channel has been proposed to be the putative access channel for androgen in human aromatase (CYP19A1) based on a crystallographic study of androstenedione-bound CYP19A1 (32). It also resembles the solvent channels found in CYP102A1, CYP2D6, and 2C5 that provide conduits for rapid access to the active site from the protein surface (8, 33, 40).
FIGURE 4.

Access channels identified from the average structures of CYP2B1 WT and Y309C/S360C using the Caver program. A, access channels for WT; channels 1, 2, and 3a are shown as white, magenta, and blue spheres, respectively. B, access channels for the Y309C/S360C variant. Channels 1, 2, and 3b are shown as white, magenta, and blue spheres, respectively. The protein backbones are depicted as green ribbons and the disulfide bond as a yellow stick.
Channel 1 observed in the WT remains essentially unchanged in the Y309C/S360C variant as shown in Fig. 4B. Due to the high flexibility of the B/B′-C loop, the exact pathway of channel 2 varies slightly around the B/B′-C loop. However, the overall direction of channel 2 does not change. The most striking difference in the access channels between the WT and Y309C/S360C variant is the absence of channel 3a in the double variant. Instead, a new channel, termed channel 3b, appears, and it exits from the protein interior toward the proximal side of the heme. It is clear that one of the direct consequences of the disulfide bond formation is the alteration of the ligand access channel.
DISCUSSION
In this study, we successfully engineered a disulfide bond into CYP2B1, which allowed us to investigate the role of conformational dynamics in P450 function. CYP2B1 was chosen for this purpose because it has been extensively studied by site-directed mutagenesis and the consequences of mutations are more predictable than other P450 isoforms. It is well documented that mutations that are not in the vicinity of the active site of P450s may still have substantial impact on P450 catalysis (41–44). This unwanted impact should be minimized in the de novo design of a disulfide bond. To differentiate the catalytic effect of mutations from those of protein dynamics, we characterized both the single and double variants under identical conditions.
The Ser360 to Cys mutation seems to have little effect on the metabolism of the three substrates tested (Tables 1 and 3), nor does it affect the protein flexibility of CYP2B1. However, the S360C variant enhances the first electron transfer from CPR and weakens the benzphetamine binding. Mutation of Tyr309 to a Cys has more profound effects, as it decreases the catalytic activities for benzphetamine and 7-EFC, reduces the rate of electron transfer from CPR, and increases the protein plasticity. To better understand P450 catalysis, it is important to take into account the protein dynamics.
It is interesting that the lost activity for the metabolism of benzphetamine in the Y309C variant is fully restored in the Y309C/S360C variant. This effect can be ascribed solely to the formation of the disulfide bond because S360C has the same benzphetamine activity as the WT does and Y309C exhibits impaired activity for the benzphetamine metabolism. This result also demonstrates that Tyr309 itself is not critical for P450 catalysis, as its contribution can be substituted for by a disulfide bond. However, the bulky side chain of Tyr309 may stabilize the I-helix during catalysis. This hypothesis is consistent with the observation that the absolute inactivation volume of Y309C increases by ∼20% compared with the WT, indicating that Y309C is more flexible than the WT. It has been demonstrated in a number of instances that the I-helices of P450s, particularly the middle section of the I-helix, possess a certain degree of flexibility. For example, the I-helix of P450cam moves outwards to produce a more exposed active site under high hydrostatic pressure, and displacement of the associated residues may alter the ligand access channels (31). In particular, displacement of the critical Thr302 residue of the I-helix, which has been proposed to be involved in proton relay, would have a detrimental effect on P450 catalysis. Therefore, it is important to maintain the optimal conformation of the I-helix for efficient catalysis. Tyr309 is in van der Waals contact with Phe359 located in the K-helix, an interaction that may contribute to the stability of the I-helix. Mutation of Tyr309 to a smaller residue such as Cys would negate this stabilizing interaction. Thus, it seems that a disulfide bond between Cys309 and Cys360 may play a role in maintaining the stability of the I-helix, similar to that between Tyr309 and Phe359 in the WT.
In contrast, formation of the disulfide bond in Y309C/S360C does not restore 7-EFC activity but rather decreases the activity further to only 19% of the WT. This decrease is not due to impaired electron transfer from CPR to the P450s (Table 4). A plausible explanation is that the disulfide bond alters the conformational dynamics and consequently modifies the ligand access channel for 7-EFC.
The direct consequence of the disulfide bonding is a reduction in the conformational flexibility as evidenced by a 2-fold decrease in the absolute inactivation volume observed for the Y309C/S360C variant. The disulfide bond also increases the Km value for 7-EFC, leading to diminished O-deethylation activity (Table 1). Taking into account the results from our MD simulations, we propose that 7-EFC enters the active site through channel 3a (Fig. 4A). Channel 3a is situated on top of the heme on the distal side and most accessible to the active site. This channel is the preferred access channel for CYP2C9 and the only channel seen in CYP2D6 based on two independent MD simulations (8, 33). Based on a recent x-ray crystallographic study of the three-dimensional structure of human aromatase, Ghosh et al. (32) proposed that this channel is the main transport route for steroids and most likely is in close contact with lipid membranes for transporting lipophilic substrates like steroids. The residues that line the channel in human aromatase are a salt bridge pair, Arg192-Glu483, at the protein surface and Asp309 and Ser478 close to the active site. The corresponding residues in CYP2B1 are replaced by a hydrogen-bonded Asp166-Gln485 pair and Glu301 and Gly478. A similar channel to channel 3a has been proposed to be the entry channel for camphor based on an NMR study of the camphor binding in isotopically labeled P450cam (45). Interestingly, our MD simulation shows that the disulfide bond alters the direction of this channel in the Y309C/S360C variant. As a result, channel 3b exits the active site almost in parallel rather than perpendicular to the I-helix. This detour may be sufficient to alter the access of 7-EFC to the active site and hence the catalytic activity.
Alteration of the access channel resulting from the disulfide bond formation may also explain the fact that the O-deethylation activity of 7-EFC is substantially decreased in the Y309C/S360C variant, whereas the N-demethylation activity of benzphetamine is not affected if benzphetamine enters the active site via different access channels than 7-EFC does. Based on sequence alignment and structural analyses of P450s by x-ray crystallography and MD simulations, there exist multiple substrate access channels (8, 29, 38, 39, 46). It is unlikely that a particular substrate would access multiple channels equally. Our results are consistent with the notion that the preferred access channel for 7-EFC is partially or completely blocked by the disulfide bond without affecting the preferred access channel for benzphetamine. The differential selection of specific channels for substrate entry will depend on protein dynamics as well as the size, polarity, and flexibility of the substrates. Benzphetamine and 7-EFC have significantly different conformational flexibility. The structure of benzphetamine is more flexible and can adopt multiple conformations with a “butterfly” conformation as the lowest energy configuration (47). In contrast, the structure of 7-EFC has a rigid planar ring structure with a much lower degree of rotation. As such, it is conceivable that 7-EFC is more selective for substrate entry channels, and hence its metabolism may be more susceptible to the adverse impact resulting from changes in the protein dynamics such as disulfide bonding.
Alternatively protein conformational dynamics may also alter the substrate binding in the active site, which in turn could affect P450 catalysis. A more rigid structure, as found in Y309C/S360C, may be less favorable to adapt to different substrates. This is a less likely scenario, although it cannot be ruled out. Our MD simulations revealed that the regions affected by the disulfide bond are not in the active site per se. A rigid active site may have a more global effect on the substrate binding. The selective inhibition of the 7-EFC activity by the disulfide bond supports the notion that the access channel for 7-EFC is altered.
From a bioengineering view point, a potential beneficial effect of engineering a disulfide bond into P450s would be an increase in the substrate specificity, as the Y309C/S360C variant is less promiscuous with respect to 7-EFC and androstenedione. It is remarkable that at the center of most microsomal P450s lies a well conserved core region that is designed for optimally efficient catalysis and functions in unison with significant conformational flexibility at the periphery, all of which is critical for substrate entry, recognition, and regiospecificity.
In conclusion, we have investigated the conformational dynamics of CYP2B1 by incorporating a de novo disulfide bond. This disulfide bond formed between Cys309 and Cys360 cross-links the I-helix to the K-helix. Detailed characterization of both the single and double Cys variants demonstrates that: 1) the disulfide-bonded CYP2B1 exhibits higher substrate specificity due to reduced conformational flexibility; 2) there is likely more than one substrate access channel in P450s, and benzphetamine and 7-EFC may utilize different channels for entry into the active site; 3) channel 3a, comprising E-, F-, and I-helices and β3-2, is most likely the entry channel for substrates with rigid structures like 7-EFC and steroids. In addition, we conclude that Tyr309, although not essential for P450 catalysis, plays an important structural role in maintaining the stability of the I-helix required for optimal catalysis.
Supplementary Material
Acknowledgments
We thank the Center for Advanced Computing at the University of Michigan for access to the computer clusters for the molecular dynamics simulations and Drs. Mike Tarasev and David Ballou for help with stopped-flow experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant CA16954 (to P. F. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
H. Zhang, C. Kenaan, D. Hamdane, G. Hui Bon Hoa, and P. F. Hollenberg, unpublished data.
- CYP or P450
- cytochrome P450
- MD
- molecular dynamics
- 7-EFC
- 7-ethoxy-4-trifluoromethylcoumarin
- CPR
- NADPH-dependent cytochrome P450 reductase
- ESI-LC/MS
- electrospray ionization liquid chromatography mass spectrometry
- TCEP
- Tris(2-carboxyethyl)phosphine
- MPa
- megapascal
- HPLC
- high pressure liquid chromatography
- WT
- wild type.
REFERENCES
- 1.Hollenberg P. F., Kent U. M., Bumpus N. N. (2008) Chem. Res. Toxicol. 21, 189–205 [DOI] [PubMed] [Google Scholar]
- 2.Williams P. A., Cosme J., Sridhar V., Johnson E. F., McRee D. E. (2000) J. Inorg. Biochem. 81, 183–190 [DOI] [PubMed] [Google Scholar]
- 3.Scott E. E., He Y. A., Wester M. R., White M. A., Chin C. C., Halpert J. R., Johnson E. F., Stout C. D. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13196–13201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. (2000) Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 5.Scott E. E., White M. A., He Y. A., Johnson E. F., Stout C. D., Halpert J. R. (2004) J. Biol. Chem. 279, 27294–27301 [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y., White M. A., Muralidhara B. K., Sun L., Halpert J. R., Stout C. D. (2006) J. Biol. Chem. 281, 5973–5981 [DOI] [PubMed] [Google Scholar]
- 7.Ekroos M., Sjögren T. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skopalík J., Anzenbacher P., Otyepka M. (2008) J. Phys. Chem. B. 112, 8165–8173 [DOI] [PubMed] [Google Scholar]
- 9.Smith S. V., Koley A. P., Dai R., Robinson R. C., Leong H., Markowitz A., Friedman F. K. (2000) Biochemistry 39, 5731–5737 [DOI] [PubMed] [Google Scholar]
- 10.Muralidhara B. K., Negi S., Chin C. C., Braun W., Halpert J. R. (2006) J. Biol. Chem. 281, 8051–8061 [DOI] [PubMed] [Google Scholar]
- 11.Hanna I. H., Roberts E. S., Hollenberg P. F. (1998) Biochemistry 37, 311–318 [DOI] [PubMed] [Google Scholar]
- 12.Kent U. M., Mills D. E., Rajnarayanan R. V., Alworth W. L., Hollenberg P. F. (2002) J. Pharmacol. Exp. Ther. 300, 549–558 [DOI] [PubMed] [Google Scholar]
- 13.Hamdane D., Kiger L., Hoa G. H., Dewilde S., Uzan J., Burmester T., Hankeln T., Moens L., Marden M. C. (2005) J. Biol. Chem. 280, 36809–36814 [DOI] [PubMed] [Google Scholar]
- 14.Hui Bon Hoa G., Douzou P., Dahan N., Balny C. (1982) Anal. Biochem. 120, 125–135 [DOI] [PubMed] [Google Scholar]
- 15.Zhang H., Im S. C., Waskell L. (2007) J. Biol. Chem. 282, 29766–29776 [DOI] [PubMed] [Google Scholar]
- 16.Reed J. R., Hollenberg P. F. (2003) J. Inorg. Biochem. 93, 152–160 [DOI] [PubMed] [Google Scholar]
- 17.Zhang H., Hamdane D., Im S. C., Waskell L. (2008) J. Biol. Chem. 283, 5217–5225 [DOI] [PubMed] [Google Scholar]
- 18.Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 19.Thompson J. D., Higgins D. G., Gibson T. J. (1994) Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laskowski R. A., MacAthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 21.Wallner B., Elofsson A. (2003) Protein Sci. 12, 1073–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dombkowski A. A. (2003) Bioinformatics 19, 1852–1853 [DOI] [PubMed] [Google Scholar]
- 23.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrek M., Otyepka M., Banás P., Kosinová P., Koca J., Damborský J. (2006) BMC Bioinformatics 7, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y. Q., He Y. A., Halpert J. R. (1994) Chem. Res. Toxicol. 8, 574–579 [DOI] [PubMed] [Google Scholar]
- 26.Luo Z., He Y. A., Halpert J. R. (1994) Arch. Biochem. Biophys. 309, 52–57 [DOI] [PubMed] [Google Scholar]
- 27.He Y., Luo Z., Klekotka P. A., Burnett V. L., Halpert J. R. (1994) Biochemistry 33, 4419–4424 [DOI] [PubMed] [Google Scholar]
- 28.Honma W., Li W., Liu H., Scott E. E., Halpert J. R. (2005) Arch. Biochem. Biophys. 435, 157–165 [DOI] [PubMed] [Google Scholar]
- 29.Gotoh O. (1992) J. Biol. Chem. 267, 83–90 [PubMed] [Google Scholar]
- 30.Seifert A., Pleiss J. (2009) Proteins 74, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 31.Tschirret-Guth R. A., Hui Bon Hoa G., Ortiz de Montellano P. (1998) J. Am. Chem. Soc. 120, 3590–3596 [DOI] [PubMed] [Google Scholar]
- 32.Ghosh D., Griswold J., Erman M., Pangborn W. (2009) Nature 457, 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cojocaru V., Winn P. J., Wade R. C. (2007) Biochim. Biophys. Acta 1770, 390–401 [DOI] [PubMed] [Google Scholar]
- 34.Davydov D. R., Deprez E., Hoa G. H., Knyushko T. V., Kuznetsova G. P., Koen Y. M., Archakov A. I. (1995) Arch. Biochem. Biophys. 320, 330–344 [DOI] [PubMed] [Google Scholar]
- 35.Davydov D. R., Halpert J. R., Renaud J. P., Hui Bon Hoa G. (2003) Biochem. Biophys. Res. Commun. 312, 121–130 [DOI] [PubMed] [Google Scholar]
- 36.Hui Bon Hoa G., McLean M. A., Sligar S. G. (2002) Biochim. Biophys. Acta 1595, 297–308 [DOI] [PubMed] [Google Scholar]
- 37.Von Weymarn L. B., Sridar C., Hollenberg P. F. (2004) J. Pharmacol. Exp. Ther. 311, 71–79 [DOI] [PubMed] [Google Scholar]
- 38.Porubsky P. R., Meneely K. M., Scott E. E. (2008) J. Biol. Chem. 283, 33698–33707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W., Liu H., Scott E. E., Gräter F., Halpert J. R., Luo X., Shen J., Jiang H. (2005) Drug Metab. Dispos. 33, 910–919 [DOI] [PubMed] [Google Scholar]
- 40.Haines D. C., Tomchick D. R., Machius M., Peterson J. A. (2001) Biochemistry 40, 13456–13465 [DOI] [PubMed] [Google Scholar]
- 41.Lin H. L., Zhang H., Waskell L., Hollenberg P. F. (2003) J. Pharmacol. Exp. Ther. 306, 744–751 [DOI] [PubMed] [Google Scholar]
- 42.Lin H. L., Kent U. M., Zhang H., Waskell L., Hollenberg P. F. (2003) Chem. Res. Toxicol. 16, 129–136 [DOI] [PubMed] [Google Scholar]
- 43.He Y. A., Balfour C. A., Kedzie K. M., Halpert J. R. (1992) Biochemistry 31, 9220–9226 [DOI] [PubMed] [Google Scholar]
- 44.Domanski T. L., He Y. Q., Scott E. E., Wang Q., Halpert J. R. (2001) Arch. Biochem. Biophys. 394, 21–28 [DOI] [PubMed] [Google Scholar]
- 45.Yao H., McCullough C. R., Costache A. D., Pullela P. K., Sem D. S. (2007) Proteins 69, 125–138 [DOI] [PubMed] [Google Scholar]
- 46.Nguyen T. A., Tychopoulos M., Bichat F., Zimmermann C., Flinois J. P., Diry M., Ahlberg E., Delaforge M., Corcos L., Beaune P., Dansette P., André F., de Waziers I. (2008) Mol. Pharmacol. 73, 1122–1133 [DOI] [PubMed] [Google Scholar]
- 47.Harris D. L., Park J. Y., Gruenke L., Waskell L. (2004) Proteins 55, 895–914 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.