

Abstract
Human transthyretin (TTR) is an amyloidogenic protein whose mild amyloidogenicity is enhanced by many point mutations affecting considerably the amyloid disease phenotype. To ascertain whether the high amyloidogenic potential of TTR variants may be explained on the basis of the conformational change hypothesis, an aim of this work was to determine structural alterations for five amyloidogenic TTR variants crystallized under native and/or destabilizing (moderately acidic pH) conditions. While at acidic pH structural changes may be more significant because of a higher local protein flexibility, only limited alterations, possibly representing early events associated with protein destabilization, are generally induced by mutations. This study was also aimed at establishing to what extent wild-type TTR and its amyloidogenic variants are intrinsically prone to β-aggregation. We report the results of a computational analysis predicting that wild-type TTR possesses a very high intrinsic β-aggregation propensity which is on average not enhanced by amyloidogenic mutations. However, when located in β-strands, most of these mutations are predicted to destabilize the native β-structure. The analysis also shows that rat and murine TTR have a lower intrinsic β-aggregation propensity and a similar native β-structure stability compared with human TTR. This result is consistent with the lack of in vitro amyloidogenicity found for both murine and rat TTR. Collectively, the results of this study support the notion that the high amyloidogenic potential of human pathogenic TTR variants is determined by the destabilization of their native structures, rather than by a higher intrinsic β-aggregation propensity.
Protein misfolding and aggregation are involved in the pathogenesis of particularly relevant human deposition diseases, known as amyloidoses. In such diseases, normally soluble proteins undergo misfolding and become insoluble, causing the extracellular deposition of fibrillar aggregates (for reviews, see Ref. 1, 2). To date, more than 40 distinct human proteins have been associated with amyloidoses. For some of such proteins, including transthyretin (TTR),4 lysozyme, gelsolin, ApoAI, and ApoAII, fibrinogen A α-chain and cystatin C, the amyloidogenic potential is induced, or is enhanced as in the case of TTR (see below), by specific mutations. The most frequent hereditary amyloidoses are caused by the genetic variants of human TTR (2).
TTR is a homotetramer of about 55 kDa involved in the transport of thyroxine in the extracellular fluids and in the co-transport of vitamin A, by forming a macromolecular complex with retinol-binding protein, the specific plasma carrier of retinol (3–5). Its three-dimensional structure is known at high resolution (6, 7). The structure is characterized by a large predominance of β-strands, and its four monomers are arranged according to a 222 symmetry, where one of the 2-fold symmetry axes of the molecule coincides with a long channel that transverses the entire tetramer and harbors two symmetrical binding sites for the thyroid hormone thyroxine. Each monomer contains eight β-strands (A-H), arranged in a β-sandwich of two four-stranded β-sheets, with a short α-helix connecting two of the eight β-strands. In the tetramer, the four monomers are organized as a dimer of dimers. Two monomers are held together, forming a stable dimer through a net of H-bond interactions involving the two external β-strands H and F. The two dimers associate back to back and form the tetramer, by interacting mostly through hydrophobic contacts between residues of the AB and GH loops.
Normal TTR possesses an inherent potential, albeit low, to generate amyloid fibrils, giving rise to Senile Systemic Amyloidosis (SSA) in ∼25% of the population aged over 80 years (8). More than 100 point mutations are described for human TTR. Most of them are involved in the hereditary amyloidoses known as familial amyloidotic polyneuropathy (FAP) or cardiomyopathy (FAC) (9). Single point mutations enhance the amyloidogenicity of TTR, so that patients show an earlier age of onset and a faster disease progression compared with SSA patients. The observation that single point mutations can drastically influence the disease phenotype is particularly relevant. In fact, the study of pathogenic TTR variants may provide clues to the mechanism of their abnormal behavior leading to amyloid formation. Although amyloidogenic proteins in general may be structurally unrelated to each other, and lead to various pathological phenotypes in humans, the amyloid fibrils originating from different proteins share the common cross-β structure, consisting in continuous β-sheets lying parallel to the longitudinal axis of the fibril, with the constituent β-strands running perpendicular to this axis. Therefore, the amyloidogenic proteins have to undergo structural alterations to be able to generate the cross-β structure, i.e. new β-pairing interactions have to be established on the way to fibril formation. However, the molecular mechanisms underlying protein misfolding and aggregation into highly ordered fibrillar structures are not clarified definitely, although significant progress is recently been made toward their elucidation (1, 10, 11).
Based on the seminal observation that the rates of aggregation into amyloid fibrils in vitro correlate with simple physico-chemical amino acid features (12), several algorithms were introduced in recent years to predict, with good success, the intrinsic β-aggregation propensities of protein and peptide sequences (for a review, see Ref. 13). The intrinsic β-aggregation propensity is a measure of the tendency polypeptide chains may have to aggregate into the amyloid structure, provided that aggregation proceeds from unstructured monomers. The prediction of intrinsic propensities to β-aggregation for amyloidogenic or non-amyloidogenic variants of the same sequence was used to explain in several instances their relative ability to speed up/slow down in vitro fibrillogenesis or the enhancement/reduction of their amyloidogenic potential in vivo (14). However, a high intrinsic aggregation propensity may not result in an actual aggregation, due to the protecting role of the ordered native structure (15, 16). Therefore, the amyloidogenic potential in the TTR variants may depend further on the change of stability in the native TTR tetramer induced by mutations. In particular, it remains to be clarified to what extent human TTR possesses an intrinsic propensity to β-aggregation, and whether amyloidogenic mutations enhance such a propensity, or only destabilize the TTR tetramer, thereby facilitating the misfolding and misassembly of a protein which is in itself prone to β-aggregation.
With regard to the pathway from native to misfolded TTR and to amyloid aggregation, the results of a number of in vitro studies are consistent with the rate-limiting dissociation of the TTR tetramer, followed by misfolding of TTR monomers and their downhill polymerization to generate pathological aggregates (17–25). The crystal structures of amyloidogenic TTR variants are generally well conserved (26–30). Accordingly, the functional properties of the variants, such as the ability to interact with retinol-binding protein (5), are maintained, being consistent with the fact that large conformational changes are not induced by amyloidogenic mutations, at least under native-like conditions (11). In vitro studies have shown that a moderately acidic medium (pH 4–5) facilitates TTR fibrillogenesis (17) and that the extent of fibril formation is remarkably enhanced for amyloidogenic TTR variants in comparison to wild-type TTR (31). Recently, it has been shown by x-ray analysis that an acidic pH (4.6) causes a large local conformational change in an amyloidogenic TTR variant (I84S) affecting two subunits within the tetramer, which probably destabilizes the TTR tetramer (32). In contrast, no significant structural changes for wild-type TTR at pH 4.6 and for I84S TTR at neutral pH were found, suggesting that conformational changes associated with a destabilization of the TTR native state may be induced or enhanced in amyloidogenic TTR variants by partially denaturing conditions (32). Pursuing these observations, we extend here our investigation to include other amyloidogenic TTR variants in comparison to the wild-type protein, with the aim to unravel structural alterations that are possibly associated with an enhanced amyloidogenic potential, according to the conformational change hypothesis (11). In addition, we report the results of a computational analysis of the mutational effects on both the intrinsic propensity to β-aggregation and the stability of the native β-structure. The same analysis is performed on murine and rat TTRs, whose structural organizations are very similar to that of the human protein (33, 34).
EXPERIMENTAL PROCEDURES
Structure Determination and Refinement for Human TTR Variants
Crystals of the recombinant human TTR variants V30M, L55P, L58H, T60A, and Y114H, prepared as described (5, 32), were grown either at pH 7.5 or 4.6 as reported elsewhere (32). Only for L55P TTR, crystals obtained at neutral pH were soaked overnight in the pH 4.6 precipitant solution, containing 50 mm sodium acetate. The datasets concerning the different variants were collected at the synchrotron radiation facility ESRF in Grenoble, using several beamlines (ID23-1, ID29, ID14-1, ID14-4). All crystals were briefly soaked in a cryoprotectant (essentially 10 to 25% ethylene glycol) added to the precipitant solution, before freezing and data collection. Datasets were collected for all variants at slightly different resolutions, between 1.8 Å to 2.4 Å. The crystals of four of the TTR variants belong to the orthorhombic space group P21212 as the wild-type TTR, with two monomers in the asymmetric unit. Only the T60A variant produced crystals belonging to the monoclinic space group P21, with 2 tetramers (8 monomers) in the asymmetric unit. Datasets were processed with the software Mosflm (35) and scaled with Scala (36) contained in the CCP4 suite (37). The structures of the mutants were determined by molecular replacement, applying the automatic full procedure in Phaser (38) and using a dimer of the native TTR as a template (PDB code 1F41, Ref. 7). The models were initially refined by restrained molecular dynamics, minimization, and refinement steps with CNS (39), and in the final cycles by Refmac (40) or Shelxl software (41). Map visualization and manual adjustment of the models were performed using the Coot graphic interface (42). Statistics concerning the data collections and the quality of the refined models are summarized in Table 1.
TABLE 1.
Statistics on data collection and refinement
| L58H pH 4.6 | L58H pH 7.5 | V30M pH 4.6 | L55P pH 7.5 | L55P pH 4.6 | Y114H pH 4.6 | T60A pH 4.6 | |
|---|---|---|---|---|---|---|---|
| Space group, cell parameters (a, b, c, β, Å, °) | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 | P21a |
| 43.85, 85.69, 64.48 | 42.88, 84.14, 64.42 | 43.78, 86.06, 64.23 | 42.73, 83.63, 66.60 | 42.63, 84.78, 65.42 | 42.34, 85.78, 62.52 | 60.59, 75.91, 107.27, 93.1 | |
| Resolutionb (Å) | 64.42-1.80 | 64.42-2.02 | 86.07-2.20 | 66.67-1.82 | 51.3-1.85 | 85.75-2.35 | 107.21-2.40 |
| (1.90-1.80) | (2.11-2.02) | (2.32-2.20) | (1.92-1.82) | (1.95-1.85) | (2.48-2.35) | (2.53-2.40) | |
| Independent reflections | 23088 (3343) | 15678 (1609) | 12491 (1716) | 21935 (3157) | 19735 (2554) | 9985 (1409) | 38055 (5558) |
| Multiplicity | 6.7 (7.0) | 6.7 (3.5) | 3.3 (3.0) | 6.9 (7.1) | 6.3 (4.2) | 6.6 (6.9) | 4.0 (4.1) |
| Completeness (%) | 99.4 (100) | 95.4 (69.5) | 97.6 (94.9) | 99.4 (99.3) | 98.1 (89.7) | 99.8 (100) | 99.6 (99.8) |
| <I/σ(I)> | 16.4 (5.3) | 22.6 (4.1) | 12.0 (5.5) | 21.3 (4.4) | 19.8 (3.5) | 17.1 (3.8) | 14.3 (2.9) |
| Rmergec | 0.103 (0.342) | 0.057 (0.349) | 0.094 (0.326) | 0.054 (0.414) | 0.062 (0.434) | 0.069 (0.394) | 0.069 (0.486) |
| Refinement statistics | |||||||
| Protein atoms | 1796 | 1782 | 1834 | 1894d | 1854 | 1862d | 7161 |
| Water molecules/ligands | 184 | 62 | 138 | 126 | 133 | 105 | 131/20 |
| Rcryste/Rfree | 0.204/0.217 | 0.202/0.228 | 0.215/0.253 | 0.197/0.219 | 0.208/0.223 | 0.217/0.247 | 0.205/0.267 |
| R.m.s.d. from ideality | |||||||
| Bond lengtds (Å) | 0.013 | 0.017 | 0.025 | 0.019 | 0.010 | 0.005 | 0.027 |
| Bond angles (°) | 1.5 | 1.6 | 1.8 | 1.6 | 1.0 | 1.6 | 2.2 |
| Ramachandran plot: | |||||||
| Favored/allowed/disallowed (%) | 90.6/9.4/0 | 89/11/0 | 87.6/12.4/0 | 89.9/10.1/0 | 89.0/11.0/0 | 85.6/14.4/0 | 83.7/16.2/0.1 |
| Overall G factor | 0 | 0 | −0.3 | 0 | 0.1 | 0 | −0.4 |
a Two tetramers in the asymmetric unit.
b Values in parentheses are for the outer resolution shell.
c Rmerge = Σhkl|Ihkl − <Ihkl>|/ΣIhkl.
d Some residues were refined in double conformation.
e Rcryst = Σhkl||F0| − k|Fc||/Σhkl|F0| where |Fo| and |Fc| are the observed and calculated structure factor amplitudes for reflection hkl, applied to the work (Rcryst) and test (Rfree) (7% omitted from refinement) sets, respectively.
Computational Analysis of the Propensity to β-Structure Formation for Wild-type TTR and TTR Variants
To evaluate the intrinsic β-aggregation propensities of the TTR sequences analyzed in this study we employed the algorithm PASTA. It was recently proposed (43) to predict (i) which portions of a given protein or peptide sequence forming amyloid fibrils are stabilizing the corresponding cross-β structure and (ii) the specific intermolecular pattern of hydrogen bonded amino acids. The algorithm is based on an energy function for specific β-aggregation, which was derived from a large group of non-redundant high resolution x-ray structures of globular proteins (44). The assumption that information regarding the structure of amyloid fibrils can be deduced by studying the structures of globular proteins rests on the well established observation (45, 46) that the aggregation mediated by nonnative β-pairings is a possible mode that polypeptide chains may adopt against insufficient folding stability. By assuming that the globular protein data base represents a system in thermodynamic equilibrium at a single temperature, which is supposed to be roughly constant for all proteins in the data bank, it then follows that the propensities pabP(A) of finding a given residue pair ab in the ensemble, with a and b facing each other in neighboring parallel (P) or antiparallel (A) β-strands, are given by pabP(A) = exp(−EabP(A)), where −EabP(A) are effective adimensional energies, and both a and b run over the 20 different amino acids (47). Propensities are defined as the ratio of the observed over the expected probabilities. They tell whether there is a higher (EabP(A) < 0) or a lower (EabP(A) > 0) probability of finding the residue pair ab in parallel (or antiparallel) β-pairing in comparison to what is found in the reference state, built by considering all possible residues. Based on these effective energies we can associate an energy score to the β-pairing of any two sequence stretches, which can be chosen from distinct protein chains sharing an identical sequence. The pairing is specific since only pairs of residues facing each other in the corresponding register contribute to the energy score. All possible aggregation patterns are then defined in terms of the positions along the sequence i,j, the length L, and the relative orientation (either parallel or antiparallel) of the two sequence stretches participating in the pairing. An entropic term is included in the energy score to take into account the total entropy loss due to the β-ordering of the L residue pairs upon aggregation. PASTA assumes that the pattern which minimizes this energy score becomes the aggregating unit at the basis of the assembly of polypeptide chains into amyloid fibrils.
A novel feature of the method is the ability to predict the registry of the intermolecular hydrogen bonds formed between amyloidogenic sequence stretches (43). We employ this unique ability by computing the stability Enat of the native β-structure based on the hydrogen bond registry found within β-sheets in the native TTR structure, as follows in Equation 1.
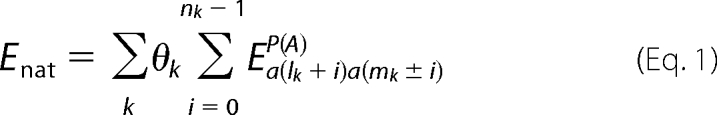 |
The first sum runs over all pairings between β-strands, whereas the second sum runs over all nk residue pairs involved in pairing k. Each pairing is further identified by its first residue pair (lk, mk). Energy contributions depend on the residue types a(lk) and a(mk) which are paired and on the parallel/antiparallel nature of the pairing. The energy contribution from pairing k also depends on its intra(inter)-subunit character, according to θk = l(0.5), so that Enat is an energy score per monomer. Native pairings for the human TTR native structure are taken from the PDB structure 1F41 (7) and are summarized in Table 2.
TABLE 2.
Summary of structural information related to native β-pairings within the human TTR dimer structure (PDB: 1F41)
For each pairing between two β-strands the orientation parallel (p) or antiparallel (a), the weight θk of its contribution to the total stability score, the number of involved residue pairs nk and the first and last residue pair are listed. Pairings 1–5 and 6–9 compose two separate β-sheets. Pairings 5 and 9 are established between different monomers.
| Pairing | Orientation | θk | nk | First pair | Last pair |
|---|---|---|---|---|---|
| 1 | a | 1 | 2 | 14–55 | 15–54 |
| 2 | a | 1 | 2 | 17–24 | 18–23 |
| 3 | p | 1 | 7 | 12–105 | 18–111 |
| 4 | a | 1 | 9 | 104–123 | 112–115 |
| 5 | a | 0.5 | 5 | 115–119 | 119–115 |
| 6 | a | 1 | 7 | 29–48 | 35–42 |
| 7 | a | 1 | 6 | 30–73 | 35–68 |
| 8 | a | 1 | 7 | 67–97 | 73–91 |
| 9 | a | 0.5 | 7 | 89–95 | 95–89 |
By using PASTA it is possible to rationalise the observed tendency of proteins to assemble into parallel β-sheets in which the individual strands are in-register contributing to form stackings of the same residue type along the fibril axis. The algorithm is also able to correctly discriminate the orientation between intermolecular β-strands, either parallel or antiparallel. Moreover, PASTA is able to recognize the sequence forming the cross β-core of the fibrils, in good agreement with the experimentally determined amyloid structures (43, 48). The energy functions used by PASTA are published in Ref. 43, and the algorithm can be utilized in its standard form (49); the analysis presented here requires different options in providing input sequences and native β-pairings which are presently not available on the web-server. Notice that the native structure of human TTR is present in the dataset used to obtain PASTA energy functions (43). We obtained a new set of potentials by removing the TTR structure from the dataset, but the results did not significantly change (data not shown). To favor validation by other groups, we use in this work the already published energy functions.
RESULTS AND DISCUSSION
Crystal Structures of Amyloidogenic TTR Variants at Neutral and Acidic pH
The crystal structure of five TTR amyloidogenic variants, at neutral and/or acidic pH, were refined (Table 1), and the results of this analysis are presented below.
V30M TTR Variant
The crystal structure of the V30M TTR variant at acidic pH (pH 4.6) is for both monomers A and B, and for the equivalent monomers C and D (for the designation of TTR subunits, see Fig. 1A), virtually identical to those of the same variant at pH 5.3 (PDB: 1TTC, (26)) and of the wild-type protein at neutral pH (pH 7.0) (PDB: 1F41, (7)). The region of the mutation is also unaltered, since the hydrophobic wild-type Val residue, which points toward the interior of the protein and is there surrounded by hydrophobic residues (Val-14, Val-16, Val-28, Val-49, Leu-55, Val-71, Ile-73), is substituted by a similarly hydrophobic Met residue. The only difference between the pH 4.6 V30M TTR structure and the pH 7.0 wild-type structure is that in the first the distances between the CE atom of Met-30 and Val-14 atoms are around 3.5 Å, which roughly corresponds to the van der Waals distance, while the Val-30 side chain atoms are more distant from those of nearby residues (around 4.3 Å from the Val-14 atoms and 3.7 Å from the Val-71 atoms). This difference is at least in part due to a steric hindrance, being the side chain of the Met residue slightly larger than the one of Val-30, and as such is in closer contact with nearby residues. Although the structure of the V30M TTR variant is apparently not significantly affected by the mutation relative to the wild-type TTR, one can assume that too close contacts between Met-30 and nearby residues may have a destabilizing effect on the TTR structure.
FIGURE 1.


A, schematic view of the TTR structure, seen looking through the central channel that transverses the entire tetramer. The four monomers, shown in different colors, are labeled from A to D. Monomers A and B (or C and D) form the asymmetric unit in the crystal cell for the P21212 space group. The eight β-strands forming the two β-sheets are labeled with small capitals from A to H. B, stereoview of the regions 12–16 and 52–59 for the L55P TTR variant after crystal soaking at acidic pH (pH 4.6) superimposed on the same regions of wild-type TTR at neutral pH (PDB: 1F41, magenta). Because of the presence of Pro-55 and possibly of the protonation of His-56, the stretch 52–55 does not interact any more with β-strand A. C, stereoview of the region 52–62 of the L58H TTR variant at acidic pH (pH 4.6) (green), neutral pH (light blue) superimposed on the same region of wild-type TTR (magenta). D, stereoview of a portion of the chain trace of monomer A of the T60A TTR variant at acidic pH (pH 4.6) (green) superimposed on the same region of the variant crystallized at pH 5.5 (PDB: 1TSH, cyan) and of wild-type TTR (red). The side chains of residues 56–64 for our structure are shown explicitly. The N-terminal region 1–9, which is usually disordered, can be seen in the PDB 1THS structure and appears to be partially superposed on residues 59–61 of our structure. E, r.m.s.d. of α-carbon atoms of variant structures relative to wild-type TTR (PDB: 1F41). For clarity, only data for subunit A are reported. Data set labels are: PDB: 3DJR, L58H at pH 7.5; PDB: 3DJS, L58H at pH 4.6; PDB: 3DJT, V30M at pH 4.6; PDB: 3DJZ, L55P at pH 7.5; PDB: 3DK0, L55P at pH 4.6; PDB: 3DK2, Y114H at pH 4.6; PDB: 3DO4, and T60A at pH 4.6.
L55P TTR Variant
The crystal structure of the L55P TTR variant (space group P21212) at neutral pH has been compared with that of the wild-type protein crystallized isomorphously at neutral pH (PDB: 1F41) and to the same variant in a different crystal form also at neutral pH (space group C2; PDB: 5TTR, (28)). The same small, but significant, differences relative to the wild-type protein that were already observed for the C2 crystal form are visible in the mutated region of the structure we have determined. The L55P mutation introduces an irregularity in the β-strand in which the mutation is present, causing a loss of H-bond interactions and, consequently, a likely destabilization of the monomer conformation. Specifically, in both subunits of the dimer, though in a slightly different way, residues from 54 to 56 are slightly shifted away (the maximum shift of Cα atoms involves residue 56 and is of about 2.6–2.8 Å), so that two H bond interactions, between Leu-55 (β-strand D) and Val-14 (β-strand A) (O55-N14 and O14-N55, Fig. 1B), are lost. Because of the fact that L55P TTR did not crystallize at acidic pH, the structure at low pH could be obtained by soaking crystals, grown at neutral pH, in a solution at pH 4.6 (see “Experimental Procedures”). After soaking, the main chain atoms kept their original positions relative to the variant at neutral pH. Differences could be observed in the orientation of surface exposed ionizable side chains, in particular Glu and Arg residues, similar to those found for wild-type TTR at moderately acidic pH (32), before more substantial conformational changes occurred at lower pH values (50). Interestingly, the side chain of His-56 of monomer B was disordered, whereas in monomer A, it showed a good definition in the electron density, in accordance with a certain degree of structural flexibility in the mutated region.
L58H TTR Variant
By comparing the crystal structures of L58H TTR and wild-type TTR at neutral pH, only a small difference is visible in the region of the mutation in monomer A. In particular, the His-58 side chain rotates slightly compared with the Leu-58 side chain position, pointing toward the solvent. This movement is more drastic in the variant at pH 4.6, and because of the protonation of His-58, in both monomers A and B its side chain points toward the solvent; the entire main chain from residues 56 to 59 rearranges as well, so that the side chain of His-56 also changes its orientation (Fig. 1C). Because of these movements at acidic pH, one H-bond interaction per subunit is lost (O55-N14, 3.1 Å and 3.2 Å in monomers A and B, respectively).
T60A TTR Variant
This is the only variant, among those investigated here, where the low pH (pH 4.6) induces the crystallization in a different crystal form (space group P21, with two tetramers in the asymmetric unit). Monomers were refined imposing restraints on non-crystallographic symmetries, with the exception of regions 56–64 and 100–103, which assume a different conformation in some of the subunits. In particular, for the first tetramer (subunits labeled A, B, C, E), the electron density for the region from residues 56 to 64, comprising the mutated residue, is well visible in subunit A, B, C, while it presents a small break at residues 58–59 in subunit E. The electron density is more disordered in the subunits of the second tetramer (subunits labeled D, F, G, H), with the exception of subunit G, in which there is a short break at residues 60–62. In the pH 4.6 structure subunit A assumes a completely different conformation, subunit E a quite different, and subunits B and C a similar one compared with the wild-type protein. A superposition of this area is illustrated in Fig. 1D. It should be noted that the structure of human TTR in complex with thyroxine crystallized in a monoclinic crystal form with two tetramers in the asymmetric unit (PDB: 1ICT (51)) shows more similarities to the wild-type protein (PDB: 1F41) in comparison to T60A variant. Moreover, the latter variant crystallized at pH 5.5 (PDB: 1TSH (29)) is also similar to the wild-type protein; the only significant difference is the presence of the N-terminal residues (residues 1–9), which are not visible in most of the TTR structures present in the PDB.
Y114H TTR Variant
In all subunits, His-114 in one subunit is positioned in the loop connecting strands G and H and is close to strand H of the other subunit of the dimer. It is exposed to the solvent and a change in its protonation state does not influence the protein conformation in both subunits. In fact, in both subunits of the dimer the structure of the Y114H variant crystallized at pH 7 is practically superimposable on that of the same variant at pH 4.6 (present report) or to the wild-type protein (PDB: 1F41). However, because the side chain of His-114 is close to hydrophobic residues (to Phe-87 of the same subunit and to Val-122 of another subunit, the shortest distance being 3.6 and 4.2 Å, respectively), it is reasonable to assume that its protonation at acidic pH destabilizes the tetrameric assembly, favoring its dissociation.
A graph summarizing the r.m.s.d. of the Cα atoms for the various TTR variants relative to the wild-type protein is illustrated in Fig. 1E. Major differences in the main chain fold are grouped in the area comprising residues from 51 to 61, since there are most of the examined mutations. Among the TTR mutations examined here, two are located in β-strands (V30M, L55P) and the others in loop regions (L58H, T60A, Y114H). Three mutations (L55P, L58H, T60A) induce small, but significant, conformational changes within single monomers. It is rather difficult to summarize the influence on the protein structure in the case of the mutation T60A, partly, because it induces a change in the crystal form, with subsequent different packing contacts, and partly for the presence of different conformations of this region in the subunits of the two tetramers present in the asymmetric unit. Overall, it can be stated that the T60A mutation induces a flexibility in the loop 57–62, which is accentuated by the decrease of the pH. Such an effect is reminiscent of that observed for I84S and I84A TTR variants, for which a large conformational change in the region hosting the mutation was induced by the lowering of the pH (32). Although limited, at least at neutral pH, the above described structural perturbations induced in TTR by amyloidogenic mutations possibly represent early events correlated with a destabilization of the TTR structure, in accordance with several lines of evidence indicating that amyloidogenic TTR variants are more prone to denaturation and amyloid aggregation relative to wild-type TTR (22, 52–62).
Computational Analysis of the Propensity to Native and Non-native β-Aggregation of Wild-type TTR and Amyloidogenic TTR Variants
As mentioned above, the intrinsic propensity to β-aggregation of an amino acid chain and its dependence on specific point mutations can be analyzed by computational techniques (13). To predict the intrinsic β-aggregation propensities for both wild-type and several amyloidogenic variants of TTR, we employed the algorithm PASTA (43), which associates an energy score to the β-pairing, either parallel or antiparallel, of any two sequence segments of length L (see “Experimental Procedures”). The energy function used by PASTA was determined by looking at preferential pairings between different residue types within β-strands in a data set of globular proteins. Hence, the algorithm is suitable to evaluate both the stability of the native β-structure and the intrinsic propensity to form a generic nonnative β-structure, typically as a result of the aggregation of different chains.
The intrinsic β-aggregation propensity of a given sequence is simply estimated as the lowest energy score E0 which is found among all possible β-pairings, with L ≥ 4, obtained by sliding the sequence along itself. Energies lower than the threshold Et = −4.0 are good predictors of fibril forming sequences (49). For wild-type human TTR, E0 is estimated to be −9.41, a value much lower than Et and lower than those of other known amyloidogenic proteins and peptides analyzed previously (43), thereby indicating an extremely high intrinsic β-aggregation propensity. Remarkably, for both wild-type murine and rat TTR, which are not amyloidogenic (33, 63), E0 is estimated to be −5.83. The predicted intrinsic aggregation propensities of murine and rat TTR are thus strongly reduced relative to human TTR, yet remain rather high in absolute terms.
Based on the set of specific β-pairings which are present in the native TTR structure (see “Experimental Procedures” and Table 2), the stability of the native β-structure can be evaluated by neglecting the presence of different entropy loss contributions due to the formation of intrachain pairings. For a given TTR variant, the change in native β-stability relative to the wild-type protein ΔEnat can obviously be computed only when the mutated residue is located in a β-strand. In addition, the variation in the intrinsic aggregation propensity, relative to the wild-type protein, is obtained as the change ΔE0 in the lowest energy score found among all β-pairings obtained when a 7 residue segment, with the mutated residue at its center, is slid along the whole 127 residue sequence. In this way it is possible to assess the mutational effect on all possible β-pairing interactions involving small sequence portions that contain the mutated residue. Only β-pairings with 4 ≤ L ≥ 7 were considered, according to the fact that the smallest peptide known to undergo fibrillogenesis is a tetra-peptide (64). The above approach is made possible by the unique ability of PASTA to take into account explicitly β-pairings between different sequence segments, at variance with other algorithms predicting aggregation propensities. The changes ΔEnat in native β-stabilities and ΔE0 in the intrinsic β-aggregation propensities relative to wild-type TTR are shown in Table 3, for the amyloidogenic TTR variants investigated here and for the I84S TTR variant already investigated structurally (32). A further test was carried out by considering all the 97 amyloidogenic TTR variants reported in (9), and computing the average intrinsic propensity changes <ΔE0> separately, for mutations located in β-strands and in loop regions. One obtains <ΔE0> = 0.3 ± 1.2 for β-strands and <ΔE0> = 0.1 ± 0.9 for loop regions, whereas <ΔEnat> = 0.54 ± 0.86 is determined for mutated residues in β-strands. By considering single TTR variants, we find that ΔEnat and ΔE0 are positively correlated (r = 0.60), as shown in Fig. 2A. The individual values are shown in Fig. 2B on a residue by residue basis.
TABLE 3.
Change in the stabilities of the native β-structure (ΔEnat) and in the intrinsic propensities to β-aggregation (ΔE0) of TTR variants
The fourth column denotes the location of the amino acid replacement in the native structure for TTR variants. ΔE0 is an adimensional energy, since the Boltzmann factor is implicit in its definition (“Experimental Procedures”). The last rows refer to murine and rat TTRs compared to human TTR with which they share an amino acid sequence identity of about 80% (Fig. 2C).
| TTR variant | ΔEnat | ΔE0 | Native structure |
|---|---|---|---|
| V30M | +1.54 | +1.95 | β strand B |
| L55P | +1.80 | +2.94 | β strand D |
| L58H | +0.62 | Loop d-E | |
| T60A | −0.12 | Loop d-E | |
| I84S | +0.59 | Loop α-F | |
| Y114H | +0.64 | Turn G-H | |
| Murine TTR | +0.04 | +3.57 | |
| Rat TTR | −0.57 | +3.57 |
FIGURE 2.

A, correlation of changes ΔEnat in native β-stability (y axis) and ΔE0 in the β-aggregation propensity (x axis) relative to wild-type TTR for the 65 amyloidogenic TTR variants whose mutated residue is part of a β-strand in the native structure (blue circles) (9). The (x,y) axes are shown as dashed lines to emphasize that virtually no mutation is present in the lower right quadrant where both effects (ΔEnat < 0 and ΔE0 > 0) would cause a decrease in the amyloidogenicity of the TTR variant. Because the considered mutations are indeed amyloidogenic, this further validates our theoretical approach. The red (gold) circle shows the values of ΔEnat and ΔE0 found by comparing murine (rat) and human TTR. The green and magenta circles refer to the two protective mutations T119M and R104H, respectively. B, changes ΔEnat in the native β-stabilities (light blue) and ΔE0 in the β-aggregation propensities (red), relative to wild-type TTR, are shown for all the 97 amyloidogenic TTR variants considered in Ref. 9 as a function of the index of the residue involved in the mutation. The variant corresponding to the deletion of Val-122 was not considered in the whole analysis. C, multiple amino acid sequence alignment of human, murine (GenBankTM NP_038725) and rat (NP_036813) TTR. Identical and chemically similar residues in the three sequences are shaded in red and denoted by red characters, respectively (similarity groups are: HKR, DE, STNQ, AVLIM, FYW, PG, C). Numbering and secondary structure elements are based on the structure of human TTR (PDB: 1F41).
A positive ΔE0 implies that the mutated TTR variant is predicted to be intrinsically less prone to non-native β-aggregation than the wild-type protein. A positive ΔEnat implies that the native β-structure is predicted to be destabilized relative to the wild-type protein, which would favor instead the process of β-aggregation by reducing the protective role played by the native structure. If both quantities ΔE0 and ΔEnat are positive, the two effects may compete with each other. However, if the intrinsic propensity to β-aggregation is very high for the wild-type protein, as in the case of human TTR, its reduction due to a mutation is not abolishing its intrinsic ability to aggregate. This fact, coupled to the destabilization of the native structure produced by the mutation, will induce, as a net effect, an increment of the resulting amylodogenic potential. It should be pointed out, however, that a mutated variant could be more or less amyloidogenic because other determinants of native structure stability, not correlated with PASTA predictions, may also be affected by the mutation. It can be noted that among the TTR variants inducing the biggest effect on ΔE0 (and hence on ΔEnat), one finds V30M and L55P, whose crystallographic structure has been determined in this work (Table 3). In such cases, the positive signs of ΔE0 and ΔEnat are consistent with the notion that the mutation is amyloidogenic because it destabilizes the native β-structure rather than because it enhances the intrinsic β-aggregation propensity. The other mutations listed in Table 3 involve loop regions and could promote amyloidogenicity either by destabilizing the native structure, a case which is not expected to be correlated with PASTA predictions, or alternatively by stabilizing nonnative β-aggregation, a case which would result in negative values of ΔE0. Our data are consistent with the former hypothesis, and with other theoretical predictions previously reported for a few TTR variants (15).
The overall data on all amyloidogenic TTR variants known from the literature show that the average intrinsic propensity change <ΔE0> for mutations in loop regions predicted by PASTA is zero. The same thing basically holds for mutations in β-strands, because both <ΔE0> and <ΔEnat> are found to be smaller than the large standard deviations found. Yet, the fact that <ΔEnat> > <ΔE0> > 0 points to a destabilization of the native β-structure as the preferred mechanism, promoting amyloidogenicity for mutated residues in native β-strands. The comparison of the standard deviations, obtained in computing <ΔE0> and <ΔEnat> with the β-aggregation propensity changes (predicted in Table 3) reinforces the observation that the predicted values are significant only for the mutations V30M and L55P, which affect residues located in β-strands.
We have computed PASTA predictions also for two variants, R104H and T119M, which have been shown to play a protective role against fibril aggregation, when coupled to amyloidogenic mutations. Our results (ΔE0 = 0 and ΔEnat = −0.03 for R104H; ΔE0 = +0.27 and ΔEnat = +0.52 for T119M, see also Fig. 2A) correctly predicts that these variants do not increase the intrinsic aggregation propensity but also that they maintain essentially the same native β-stability as wild-type TTR. One would expect that protective mutations should rather increase the native stability of TTR. Indeed, it was experimentally shown that several new stabilizing interactions are established within the T119M variant, involving various other residues besides the mutated one (57). PASTA, however, considers only the contributions from residue pairs that involve the mutated residue.
A good test case for PASTA predictions is given by the non-amyloidogenic murine and rat TTRs. It was recently shown that murine TTR is not amyloidogenic because its tetrameric native structure is kinetically stabilized relative to human TTR (33). Additionally, a recombinant murine TTR variant engineered to prevent native tetramer formation was shown to form amyloid fibrils in vitro, albeit to a lesser extent relative to human TTR in the same conditions (33). The changes ΔEnat in native β-stabilities and ΔE0 in the intrinsic propensities to form non-native β-structure relative to wild-type human TTR were computed for both murine and rat TTRs, as shown in Table 3. Contributions from different native β-pairings are summed to get ΔEnat (see “Experimental Procedures”), as they are affected by 9 out of the total amino acid replacements present in the sequences of murine (25 replacements) and rat (24 replacements) TTR (Fig. 2C). This is made possible by the conservation of native β-pairings in the native structures of different TTR proteins (33). ΔE0 was defined as the change in the lowest energy score found among all β-pairings which are obtained when the whole 127 residue sequence is slid along itself. PASTA predicts that both murine and rat TTRs sequences exhibit some intrinsic propensity to aggregate into β-structure, but quite lower than for human TTR (ΔE0 = +3.57 for both murine and rat TTRs, Table 3), and remarkably predicts also that the stability of the native β-structure is maintained (ΔEnat = +0.04, Table 3) for murine TTR, and even slightly increased (ΔEnat = −0.57, Table 3) for rat TTR. Notably, PASTA predictions are consistent with the low amount of fibril formation displayed by the murine TTR variant engineered to remain monomeric (the two mutations engineered for that purpose do not affect ΔE0). The prediction of tetramer stabilization is out of the reach of our analysis, because it is not due to β-structure formation. Our results instead reveal that sequence changes are concerted to maintain the same stability of the native β-structure for the three sequences. The inclusion of inter-subunit β-pairings within the dimer in computing ΔEnat is essential for this result. It should be noted, however, that 8 out of 9 residues replaced in native β-strands are chemically similar in human and either murine or rat TTR. Tetramer stabilization protecting murine TTR against amyloid formation would then be caused by other sequence changes not affecting the native β-structure. The significance of the values of ΔE0 and ΔEnat found for murine and rat TTRs can be appreciated when plotted together with the corresponding values found for single variants of human TTR (Fig. 2A).
Atomic coordinates and structure factors have been deposited at the Protein Data Bank (http://www.rcsb.org) for immediate release: PDB ID codes 3DJR (L58H, pH 7.5), 3DJS (L58H, pH 4.6), 3DJT (V30M, pH 4.6), 3DJZ (L55P, pH 7.5), 3DK0 (L55P, pH 4.6), 3DK2 (Y114H, pH 4.6), and 3DO4 (T60A, pH 4.6).
Concluding Remarks
Based on the evidence presented here, it can be inferred that human TTR represents a case in which the main effect of the amyloidogenic replacement of an amino acid residue is not that of further promoting the intrinsic propensity to β-structure formation, which is already quite high for the wild-type sequence. Rather, the main effect of amyloidogenic mutations is consistent with the known destabilization of the tertiary and/or quaternary structure of TTR. In this respect, quite significant is the case of mutations that involve residues placed in β-strands within the native structure, such as V30M and L55P, for which the intrinsic β-aggregation propensity is even reduced as compared with wild-type TTR. Also, the results of the x-ray analysis of amyloidogenic TTR variants have revealed limited but significant conformational changes, which may be accentuated by partially denaturing conditions. The present study provides further support to the therapeutic strategy based on the use of drugs consisting of TTR ligands able to bind to the TTR tetramer and to stabilize it to prevent or reduce amyloid aggregation (65).
Acknowledgments
We thank Anke Seydel for carefully reading the manuscript and the technical assistance of the staff of beamlines ID23-1, ID29, ID14-1, ID14-4 of the ESRF synchrotron (Grenoble).
This work was supported by the University of Padua and Progetto di Ateneo n. CPDA083702, by the University of Parma and by the PRIN Project 2007 No. 2007B57EAB.
- TTR
- transthyretin
- r.m.s.d.
- root mean-square deviation
- PDB
- Protein Data Bank.
REFERENCES
- 1.Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2.Pepys M. B. (2006) Annu. Rev. Med. 57, 223–241 [DOI] [PubMed] [Google Scholar]
- 3.Monaco H. L., Rizzi M., Coda A. (1995) Science 268, 1039–1041 [DOI] [PubMed] [Google Scholar]
- 4.Naylor H. M., Newcomer M. E. (1999) Biochemistry 38, 2647–2653 [DOI] [PubMed] [Google Scholar]
- 5.Zanotti G., Folli C., Cendron L., Alfieri B., Nishida S. K., Gliubich F., Pasquato N., Negro A., Berni R. (2008) FEBS J. 275, 5841–5854 [DOI] [PubMed] [Google Scholar]
- 6.Blake C. C., Geisow M. J., Oatley S. J., Rérat B., Rérat C. (1978) J. Mol. Biol. 121, 339–356 [DOI] [PubMed] [Google Scholar]
- 7.Hörnberg A., Eneqvist T., Olofsson A., Lundgren E., Sauer-Eriksson A. E. (2000) J. Mol. Biol. 302, 649–669 [DOI] [PubMed] [Google Scholar]
- 8.Westermark P., Sletten K., Johansson B., Cornwell G. G., 3rd (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 2843–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benson M. D., Kincaid J. C. (2007) Muscle Nerve 36, 411–423 [DOI] [PubMed] [Google Scholar]
- 10.Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 11.Chiti F., Dobson C. M. (2009) Nat. Chem. Biol. 5, 15–22 [DOI] [PubMed] [Google Scholar]
- 12.Chiti F., Stefani M., Taddei N., Ramponi G., Dobson C. M. (2003) Nature 424, 805–808 [DOI] [PubMed] [Google Scholar]
- 13.Fernàndez-Busquets X., de Groot N. S., Fernandez D., Ventura S. (2008) Curr. Med. Chem. 15, 1336–1349 [DOI] [PubMed] [Google Scholar]
- 14.Luheshi L. M., Tartaglia G. G., Brorsson A. C., Pawar A. P., Watson I. E., Chiti F., Vendruscolo M., Lomas D. A., Dobson C. M., Crowther D. C. (2007) PLoS Biol. 5, e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Escamilla A. M., Rousseau F., Schymkowitz J., Serrano L. (2004) Nat. Biotechnol. 22, 1302–1306 [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F., Vendruscolo M. (2008) J. Mol. Biol. 380, 425–436 [DOI] [PubMed] [Google Scholar]
- 17.Colon W., Kelly J. W. (1992) Biochemistry 31, 8654–8660 [DOI] [PubMed] [Google Scholar]
- 18.Lai Z., Colón W., Kelly J. W. (1996) Biochemistry 35, 6470–6482 [DOI] [PubMed] [Google Scholar]
- 19.Quintas A., Saraiva M. J., Brito R. M. (1999) J. Biol. Chem. 274, 32943–32949 [DOI] [PubMed] [Google Scholar]
- 20.Liu K., Cho H. S., Lashuel H. A., Kelly J. W., Wemmer D. E. (2000) Nat. Struct. Biol. 7, 754–757 [DOI] [PubMed] [Google Scholar]
- 21.Quintas A., Vaz D. C., Cardoso I., Saraiva M. J., Brito R. M. (2001) J. Biol. Chem. 276, 27207–27213 [DOI] [PubMed] [Google Scholar]
- 22.Jiang X., Smith C. S., Petrassi H. M., Hammarström P., White J. T., Sacchettini J. C., Kelly J. W. (2001) Biochemistry 40, 11442–11452 [DOI] [PubMed] [Google Scholar]
- 23.Hammarström P., Wiseman R. L., Powers E. T., Kelly J. W. (2003) Science 299, 713–716 [DOI] [PubMed] [Google Scholar]
- 24.Hurshman A. R., White J. T., Powers E. T., Kelly J. W. (2004) Biochemistry 43, 7365–7381 [DOI] [PubMed] [Google Scholar]
- 25.Foss T. R., Kelker M. S., Wiseman R. L., Wilson I. A., Kelly J. W. (2005) J. Mol. Biol. 347, 841–854 [DOI] [PubMed] [Google Scholar]
- 26.Hamilton J. A., Steinrauf L. K., Braden B. C., Liepnieks J., Benson M. D., Holmgren G., Sandgren O., Steen L. (1993) J. Biol. Chem. 268, 2416–2424 [PubMed] [Google Scholar]
- 27.Damas A. M., Ribeiro S., Lamzin V. S., Palha J. A., Saraiva M. J. (1996) Acta Crystallogr. D Biol. Crystallogr. 52, 966–972 [DOI] [PubMed] [Google Scholar]
- 28.Sebastião M. P., Saraiva M. J., Damas A. M. (1998) J. Biol. Chem. 273, 24715–24722 [DOI] [PubMed] [Google Scholar]
- 29.Schormann N., Murrell J. R., Benson M. D. (1998) Amyloid 5, 175–187 [DOI] [PubMed] [Google Scholar]
- 30.Neto-Silva R. M., Macedo-Ribeiro S., Pereira P. J., Coll M., Saraiva M. J., Damas A. M. (2005) Acta Crystallogr. D Biol. Crystallogr. 61, 333–339 [DOI] [PubMed] [Google Scholar]
- 31.Bonifácio M. J., Sakaki Y., Saraiva M. J. (1996) Biochim. Biophys. Acta 1316, 35–42 [DOI] [PubMed] [Google Scholar]
- 32.Pasquato N., Berni R., Folli C., Alfieri B., Cendron L., Zanotti G. (2007) J. Mol. Biol. 366, 711–719 [DOI] [PubMed] [Google Scholar]
- 33.Reixach N., Foss T. R., Santelli E., Pascual J., Kelly J. W., Buxbaum J. N. (2008) J. Biol. Chem. 283, 2098–2107 [DOI] [PubMed] [Google Scholar]
- 34.Wojtczak A. (1997) Acta Biochim. Pol. 44, 505–517 [PubMed] [Google Scholar]
- 35.Leslie A. G. W. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 36.Evans P. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, Number 4. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 38.McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 39.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 40.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 41.Sheldrick G. M., Schneider T. R. (1997) Methods Enzymol. 277, 319–343 [PubMed] [Google Scholar]
- 42.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 43.Trovato A., Chiti F., Maritan A., Seno F. (2006) PLoS Comput. Biol. 2, e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 45.Dobson C. M. (2004) Science 304, 1259–1262 [DOI] [PubMed] [Google Scholar]
- 46.Hoang T. X., Marsella L., Trovato A., Seno F., Banavar J. R., Maritan A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 6883–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samudrala R., Moult J. (1998) J. Mol. Biol. 275, 895–916 [DOI] [PubMed] [Google Scholar]
- 48.Trovato A., Maritan A., Seno F. (2007) J. Phys. Condens. Matter 19, 285221 [Google Scholar]
- 49.Trovato A., Seno F., Tosatto S. C. (2007) Protein Eng. Des. Sel. 20, 521–523 [DOI] [PubMed] [Google Scholar]
- 50.Palaninathan S. K., Mohamedmohaideen N. N., Snee W. C., Kelly J. W., Sacchettini J. C. (2008) J. Mol. Biol. 382, 1157–1167 [DOI] [PubMed] [Google Scholar]
- 51.Wojtczak A., Cody V., Luft J. R., Pangborn W. (2001) Acta Crystallogr. D Biol. Crystallogr. 57, 1061–1070 [DOI] [PubMed] [Google Scholar]
- 52.McCutchen S. L., Colon W., Kelly J. W. (1993) Biochemistry 32, 12119–12127 [DOI] [PubMed] [Google Scholar]
- 53.McCutchen S. L., Lai Z., Miroy G. J., Kelly J. W., Colón W. (1995) Biochemistry 34, 13527–13536 [DOI] [PubMed] [Google Scholar]
- 54.Jenne D. E., Denzel K., Blätzinger P., Winter P., Obermaier B., Linke R. P., Altland K. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 6302–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quintas A., Saraiva M. J., Brito R. M. (1997) FEBS Lett. 418, 297–300 [DOI] [PubMed] [Google Scholar]
- 56.Shnyrov V. L., Villar E., Zhadan G. G., Sanchez-Ruiz J. M., Quintas A., Saraiva M. J., Brito R. M. (2000) Biophys. Chem. 88, 61–67 [DOI] [PubMed] [Google Scholar]
- 57.Sebastião M. P., Lamzin V., Saraiva M. J., Damas A. M. (2001) J. Mol. Biol. 306, 733–744 [DOI] [PubMed] [Google Scholar]
- 58.Hammarstrom P., Jiang X., Hurshman A. R., Powers E. T., Kelly J. W. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, Suppl. 4, 16427–16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hammarström P., Sekijima Y., White J. T., Wiseman R. L., Lim A., Costello C. E., Altland K., Garzuly F., Budka H., Kelly J. W. (2003) Biochemistry 42, 6656–6663 [DOI] [PubMed] [Google Scholar]
- 60.Ferrão-Gonzales A. D., Palmieri L., Valory M., Silva J. L., Lashuel H., Kelly J. W., Foguel D. (2003) J. Mol. Biol. 328, 963–974 [DOI] [PubMed] [Google Scholar]
- 61.Altland K., Benson M. D., Costello C. E., Ferlini A., Hazenberg B. P., Hund E., Kristen A. V., Linke R. P., Merlini G., Salvi F., Saraiva M. J., Singer R., Skinner M., Winter P. (2007) Electrophoresis 28, 2053–2064 [DOI] [PubMed] [Google Scholar]
- 62.Hurshman, Babbes A. R., Powers E. T., Kelly J. W. (2008) Biochemistry 47, 6969–6984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tajiri T., Ando Y., Hata K., Kamide K., Hashimoto M., Nakamura M., Terazaki H., Yamashita T., Kai H., Haraoka K., Imasato A., Takechi K., Nakagawa K., Okabe H., Ishizaki T. (2002) Clin. Chim. Acta 323, 129–137 [DOI] [PubMed] [Google Scholar]
- 64.Reches M., Porat Y., Gazit E. (2002) J. Biol. Chem. 277, 35475–35480 [DOI] [PubMed] [Google Scholar]
- 65.Johnson S. M., Wiseman R. L., Sekijima Y., Green N. S., Adamski-Werner S. L., Kelly J. W. (2005) Acc. Chem. Res. 38, 911–921 [DOI] [PubMed] [Google Scholar]