

Abstract
d-Serine is an endogenous coagonist for the N-methyl-d-aspartate receptor and is involved in excitatory neurotransmission in the brain. Mammalian pyridoxal 5′-phosphate-dependent serine racemase, which is localized in the mammalian brain, catalyzes the racemization of l-serine to yield d-serine and vice versa. The enzyme also catalyzes the dehydration of d- and l-serine. Both reactions are enhanced by Mg·ATP in vivo. We have determined the structures of the following three forms of the mammalian enzyme homolog from Schizosaccharomyces pombe: the wild-type enzyme, the wild-type enzyme in the complex with an ATP analog, and the modified enzyme in the complex with serine at 1.7, 1.9, and 2.2 Å resolution, respectively. On binding of the substrate, the small domain rotates toward the large domain to close the active site. The ATP binding site was identified at the domain and the subunit interface. Computer graphics models of the wild-type enzyme complexed with l-serine and d-serine provided an insight into the catalytic mechanisms of both reactions. Lys-57 and Ser-82 located on the protein and solvent sides, respectively, with respect to the cofactor plane, are acid-base catalysts that shuttle protons to the substrate. The modified enzyme, which has a unique “lysino-d-alanyl” residue at the active site, also exhibits catalytic activities. The crystal-soaking experiment showed that the substrate serine was actually trapped in the active site of the modified enzyme, suggesting that the lysino-d-alanyl residue acts as a catalytic base in the same manner as inherent Lys-57 of the wild-type enzyme.
d-Serine, which is present at a high level in the mammalian brain, serves as an endogenous coagonist for the N-methyl-d-aspartate (NMDA)5 receptor selectively localized on the postsynaptic membrane of the excitatory synapse (1–5) and is involved in excitatory neurotransmission and higher brain functions such as learning and memory (3, 6, 7). Stimulation of the NMDA receptor requires the binding of d-serine as well as the agonist l-glutamate. The major enzyme for d-serine synthesis from l-serine in the brain is considered to be pyridoxal 5′-phosphate (PLP)-dependent serine racemase (SR) (8–10). d-Serine and SR are localized on protoplasmic astrocytes that have the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor. Glutamate released from presynaptic neurons approaches and activates the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor, which in turn induces SR to produce d-serine and is followed by d-serine release from astrocytes that act on the NMDA receptor. Recently, it was shown that not only glia but also neurons synthesize and release d-serine involved in signaling (11). SR also catalyzes α,β-elimination of water from d- or l-serine to form pyruvate and ammonia as well as the conversion of l-serine into d-serine and vice versa and is presumed to link d-serine synthesis and energy metabolism of astrocytes (12) and to control the d-serine level (13). Mg·ATP, which is fully bound to SR under physiological conditions, stimulates racemization and the α,β-elimination reaction catalyzed by SR (12, 14).
SR was first discovered in pupae of the silkworm Bombyx mori (15), which was followed by purification of the enzyme from a rat brain and cloning of the mouse and human genes (8, 9). The primary structure of mammalian SR is distinct from those of racemases from prokaryotes but is similar to those of fold-type II PLP-dependent enzymes (16–18). We have cloned and expressed the Schizosaccharomyces pombe gene homologous to human and mouse SRs, the sequence identities being 35.1 and 37.4%, respectively, in Escherichia coli. The protein product is a bifunctional enzyme that catalyzes racemization and the α,β-elimination reaction of D, l-serine as mammalian SR does. SR from S. pombe (spSR) comprises 322 residues (the N-terminal Met is removed in the purified enzyme) and one PLP per subunit, the subunit molecular weight being 34,917. The mammalian SR homolog, spSR, is an interesting target enzyme for the development of a novel therapeutic compound controlling the d-serine level because d-serine is the product of an SR-catalyzed reaction. In our recent report, the active site of spSR was shown to be modified with its natural substrate serine by mass spectroscopic and x-ray studies (19). Interestingly, the catalytic lysine, which originally forms a Schiff base with PLP, is converted to a lysino-d-alanyl residue through the reaction with the substrate, serine (Fig. 1). The modified enzyme exhibits racemase (54% of the wild-type enzyme) and α,β-elimination (68% of the wild-type enzyme) activities with the amino group of the d-alanyl moiety of the lysinoalanyl residue forming a Schiff base with PLP in place of the lysine (19). In addition, the mammalian SR seems to be possibly modified to have a lysinoalanyl residue at the active site, as observed in spSR (20).
FIGURE 1.

Covalent modification of the active site. The catalytic Lys-57 in spSRw is converted to lysino-d-alanyl residue. The α-amino group (indicated with “α”) of the d-alanyl moiety in the residue acts as a catalytic base in spSRm. The circled P is a phosphate group.
Although the structure of modified spSR (spSRm) has been determined (19), the structure-function relationship of essential wild-type spSR (spSRw), the binding mode of activator Mg·ATP, the catalytic base to shuttle protons to the substrate d-serine, and the substrate recognition of the modified enzyme have not yet been uncovered. We now report the three-dimensional structures of unliganded spSRw in the open form, spSRw·AMP-PCP in the open form, and spSRm·serine in the closed form.
EXPERIMENTAL PROCEDURES
Construction of a Mutant spSR and Enzyme Preparation
The S82A mutation was introduced into the plasmid pETSRF1, which was used to express spSRw (19), using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's recommendations. The primers used for the mutation were: 5′-GGTGTTTTGACGTTTTCTGCTGGTAATCATGCTCAGGCC-3′ and 5′-GGCCTGAGCATGATTACCAGCAGAAAACGTCAAAACACC-3′. The entire coding region was sequenced to confirm the mutation. Expression of spSRw and the mutant spSR in E. coli and purification of the enzymes were performed as described elsewhere (19).
Enzyme Assays
Both the racemization and the α,β-elimination (dehydration) of l-serine and d-serine were performed in a reaction mixture comprising 100 mm Tris-HCl (pH 8.0), 10 mm l- or d-serine, 20 μm PLP, and 1 mg/ml enzyme at 30 °C for 1 h. For the racemization assay, the amounts of l- and d-serine were determined as described previously (21). The limit of detection of our assay was 0.1 μmol/ml d- or l-serine in the reaction mixture. For the α,β-elimination assay, the amount of pyruvate was measured with 2,4-dinitrophenylhydrazine as described previously (22).
Crystallization and Data Collection
Unliganded spSRw was crystallized at 293 K using a vapor diffusion method. Drops were prepared by mixing 3 μl of protein solution (2.2 mg ml−1 protein and 10 mm Tris-HCl buffer, pH 8.0) with an equal volume of reservoir solution (28% (w/v) polyethylene glycol 4000, 200 mm sodium acetate, and 100 mm Tris-HCl, pH 8.5) and equilibrated against 450 ml of the reservoir solution. The crystals are orthorhombic with the C2221 space group and one subunit per asymmetric unit. To prepare the complex with the ATP analog, AMP-PCP, wild-type crystals were soaked in a solution supplemented with 10 mm AMP-PCP and 0.2 m MgCl2 at 293 K. Crystals of spSRm complexed with serine were obtained by soaking spSRm crystals (19) in a solution containing 10 mm l-serine.
The x-ray diffraction data sets for spSRw and spSRm·serine crystals were collected to 1.7 and 2.2 Å resolution, respectively, at 100 K on the BL44B2 station at SPring-8 (Hyogo, Japan), using an x-ray beam with a wavelength 1.0 Å and a MAR CCD165 detector system. The x-ray diffraction data set for spSRw·AMP-PCP crystal was collected to 1.9 Å resolution at 100 K on the NW12 station at the Photon Factory, KEK (Tsukuba, Japan) using an x-ray beam with a wavelength of 1.0 Å and an ADSC quantum 4R CCD camera. All data were processed using HKL2000 (23).
Structure Solution and Refinement
The initial structures for spSRw, spSRw·AMP-PCP, and spSRm·serine were determined with AMoRe (24), using the structure of spSRm as the search model (19). Modeling of the polypeptide chains was performed with the program O (25). The structures were subjected to simulated annealing and positional and thermal parameter refinement with the program CNS (26). The AMP-PCP and serine were introduced into peaks on a simulated annealing 2Fo − Fc map. The R-factor values for spSRw, spSRw·AMP-PCP, and spSRm·serine are 22.0, 23.0, and 16.9% to 1.7, 1.9, and 2.2 Å resolution with Rfree values of 24.9, 26.6, and 21.6%, respectively. The resulting subunit model of spSRw comprises 318 amino acids (Val-6–Gln-323), one PLP, one metal ion, and 256 water molecules. The spSRw·AMP-PCP subunit comprises 318 amino acids (Val-6–Glu-323), one PLP, one metal ion, one Mg·AMP-PCP, and 328 water molecules. The spSRm·serine subunit comprises 319 amino acids (Leu-5–Gln-323), one PLP-d-alanyl Schiff base, one metal ion, one serine, and 103 water molecules. The models exhibit good stereochemistries (Table 1), as verified with PROCHECK (27). Structure diagrams were drawn using the programs Molscript (28), PyMOL (29), Povscript+ (30), and Raster3D (31).
TABLE 1.
Data collection and refinement statistics
| spSRw | spSRw·AMP-PCP | spSRm·serine | |
|---|---|---|---|
| Data collection | |||
| PDB ID | 1V71 | 1WTC | 2ZR8 |
| Space group | C2221 | C2221 | C2 |
| Unit cell | |||
| a | 69.47 | 69.43 | 59.54 |
| b | 148.17 | 148.09 | 72.88 |
| c | 64.20 | 64.66 | 64.81 |
| β | 90 | 90 | 101.73 |
| Diffraction data | |||
| Resolution (Å) | 1.70 | 1.90 | 2.20 |
| No. of reflections | |||
| Unique | 35,931 | 26,679 | 13,894 |
| Completeness (%) | 97.4 (85.4)a | 100 (100)a | 99.2 (94.7)a |
| Rmerge (%)b | 5.5 (30.3)a | 7.2 (25.8)a | 8.6 (28.6)a |
| I/σ(I) | 28.1 (2.4)a | 7.1 (2.2)a | 15.2 (3.3)a |
| Refinement | |||
| Resolution limits (Å) | 10.0–1.70 | 10.0–1.90 | 10.0–2.20 |
| R-factor (%) | 21.96 | 22.84 | 17.05 |
| Rfree (%) | 24.85 | 26.63 | 21.68 |
| Deviations | |||
| Bond lengths (Å) | 0.006 | 0.007 | 0.006 |
| Bond angles (deg) | 1.20 | 1.24 | 1.28 |
| Mean B-factors | |||
| Main chain atoms (Å2) | 21.54 | 21.62 | 20.89 |
| Side-chain atoms (Å2) | 25.13 | 23.79 | 23.99 |
| Hetero atoms (Å2) | 15.70 | 17.36 | 21.70 |
| Water atoms (Å2) | 32.91 | 29.88 | 29.06 |
| Procheck | |||
| Favored | 91.3 | 90.6 | 93.5 |
| Additionally allowed | 8.3 | 9.4 | 7.6 |
| Generously allowed | 0.4 | 0.0 | 0.4 |
| Disallowed | 0.0 | 0.0 | 0.0 |
a The values in the parentheses are for highest resolution shells (1.76-1.70, 1.97-1.90, and 2.28-2.20 Å) in spSRw, spSRw·AMP-PCP, and spSRm·serine, respectively.
bRmerge = 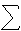 hkl i |Ihkl,i − 〈Ihkl〉|/hkl i Ihkl,i, where I = observed intensity and 〈I〉 = average intensity for multiple measurements.
hkl i |Ihkl,i − 〈Ihkl〉|/hkl i Ihkl,i, where I = observed intensity and 〈I〉 = average intensity for multiple measurements.
RESULTS AND DISCUSSION
Overall and Subunit Structure
The asymmetric unit has one subunit, which comes from a dimer lying along the crystallographic two-fold axis (Fig. 2, A and B). The subunit comprises two domains, a large domain (1–55, 153–323) and a small domain (56–152). The surface area of the subunit interface was calculated to be 873 Å2, of which 408 Å2 is hydrophobic, 279 Å2 is hydrophilic, and 187 Å2 is ionic. We note that the enzyme forms a biological dimer, which is consistent with the molecular weight of 72,000 estimated on gel filtration through a Superose 12 column. The subunit interface consists of the large domain residues only, although the small domain is adjacent to the large domain of the other subunit at the ATP-binding site. Eight of the 12 residues involved in the subunit interface are conserved or strongly conserved in human and mouse SRs. The molecule has a rectangular shape with a long groove formed at the domain and subunit interfaces connecting the top and bottom of the molecule (Fig. 2). The groove starts from the top of the molecule, passes through the PLP binding site (active site) and the ATP binding site located on the right side of the molecule, turns to the back, runs to the left side of the molecule through the two-fold axis, and then reaches the bottom of the molecule.
FIGURE 2.

Dimeric and monomeric structures of spSR. A, stereo view of the spSRw complex with AMP-PCP in the open form along the two-fold axis. The small and large domains of one subunit are shown in light and deep blue, respectively, and the other subunit is shown in green. A large deep groove is formed at the domain interface. The cofactor PLP shown by a space-filling model (yellow) is bound to the bottom of the groove. The groove extends to the subunit interface. AMP-PCP (red) is bound to the groove located at the boundary between the domain interface and the subunit interface on the right or left side of the molecule. N-ter, N terminus; C-ter, C terminus. B, side view of the molecule perpendicular to the two-fold axis. The image structure is turned 90° counterclockwise around the vertical axis relative to the image in Fig. 1A. The groove embracing AMP-PCP is further extended to the subunit interface formed at the back of the molecule. Binding of AMP-PCP induces the rotation of both subunits to widen the back groove. C, stereo view of the subunit in the open form with the secondary structure assignments. The small and the large domains are shown in light and deep blue, respectively. The cofactor PLP-Lys-57 Schiff base located at the bottom of the groove is drawn as the stick model. Besides the cofactor, a metal ion (gray circle) is bound to the large domain to stabilize the active site folding. The metal ion is coordinated by the side-chain carboxylates of Glu-208 and Asp-214. D, large (lower half) domain fitting between spSRw in the open form (red) and spSRm in the closed form (green). The large domain superimposition reveals the rotation of the small domain toward the large domain to close the active site.
The subunit structure is presented with secondary structure assignments made with the program DSSP (Fig. 2C) (32). The large domain is an α/β one with an open twisted five-stranded β-sheet (S1, S5–S8) surrounded by nine α-helices (H1, H2, H8–H14) (Fig. 2C). Many of the active site residues that interact with the cofactor are situated at or near one end of the five-stranded β-sheet forming the base of the active site cavity. The small domain also assumes an α/β-structure with a three-stranded parallel β-sheet (S2–S4) surrounded by four α-helices (H3–H7). The program DALI was used to search the Protein Data Bank (PDB) data base for enzymes possessing subunit structures similar to that of spSR (33). The highest Z scores (strength of structural similarity) were calculated to be 46.8 with 38% sequence identity for threonine dehydratase from Salmonella typhimurium (34), 41.9 with 30% for threonine dehydratase from E. coli (35), 36.2 with 24% for serine dehydratase like-1 from human cancer cells (36), 34.4 with 24% for serine dehydratase from rat (37), 34.2 with 19% for threonine synthase from Thermus thermophilus HB8 (38), 32.6 with 17% for O-acetylserine sulfhydrylase from S. typhimuriumin (39), and 30.2 with 18% for tryptophan synthase from E. coli (40), indicating that the overall fold of spSR is similar to those of the PLP-dependent fold-type II enzymes and is almost the same as those of threonine dehydratases from S. typhimurium and E. coli.
Open-Closed Conformational Change
PLP-dependent aspartate aminotransferases belonging to the fold-type I are known to show movement of the small domain to close the active site upon binding of the substrate (41–43). Ligand-induced movement of the small domain has also been observed in the fold-type II enzymes threonine synthase and O-acetylserine sulfhydrylase, with the 25 and 15° rotation of the small domains, respectively (38, 39). Large domain Cα atom superimposition of spSRw onto spSRm·serine clearly indicates that the small domain undergoes a 20° rotation toward the large domain as compared with that of spSRw without a change in the conformation of the large domain dimer to enclose the ligand in the active site formed at the domain interface (Fig. 2D). The molecular surface of the monomer unit in the closed form is reduced from 13,166 Å2 in the open form to 12,455 Å2. The domain movement plays an important role in the formation of the substrate recognition site and the catalysis of the enzyme.
Active Site of spSRw in the Open Form
The large deep groove of spSR formed at the domain interface is a part of the long groove connecting the top and bottom of the molecule (Fig. 2A). The groove embraces the cofactor PLP in the central region and is filled with water molecules. The active site structure of spSRw in the open form is shown in Fig. 3A. Many of the interactions between PLP and active-site residues reflect those observed in PLP-dependent fold-type II enzymes (37, 38, 44). The cofactor PLP forms a typical Schiff base bond (internal aldimine) with the catalytic Lys-57 and interacts with the large domain residues with its si-face directed toward the protein side. The pyridine ring of PLP is sandwiched by Gly-236 and Phe-56 from the re- and si-face sides, respectively. Gly-236 comes from the loop between α-helices H10 and H11, and Phe-56 comes from the loop between β-strands S1 and H3. The tetra-glycine loop (Gly-183–Gly-184–Gly-185–Gly-186–Leu-187) located at the N-terminal end of α-helix H9 forms a binding site for the phosphate group. The N-terminal loop (Ser-81–Ser-82–Gly-83–Asn-84–His-85) of α-helix H5, which is called the asparagine loop (44), resides on the O3′ side of PLP and acts as the recognition site for the substrate carboxylate in the closed form. The polar hydroxy group of Ser-308 forms a neutral hydrogen bond with the N-1 atom of the pyridine ring of PLP (37, 38, 45). The side chain of Asn-84 forms a hydrogen bond with the O3′ of PLP to play a role in fine-tuning of the electronic state of the PLP-Schiff base conjugate together with the hydrogen bond to the N-1 atom (46–48). These active-site residues are completely conserved in human and rat SRs except for Leu-187 (Met in human and rat), suggesting that the side chain arrangements as well as the active site folds are quite similar in these SRs in which the substrate serine is catalyzed through the same mechanism.
FIGURE 3.

Stereo view of the active site in spSR. The front and back of each figure show the solvent side (entrance of the active site) and the protein side (bottom of the active site), respectively. The side chains of the active site residues are depicted as stick models. The cofactor is shown in yellow with phosphate in red. The secondary structures of the small and large domains are drawn in light and deep blue, respectively, and the loops are drawn in gray. Water molecules are represented by red circles. A, close-up view of the active site of spSRw in the open form. A simulated annealing omit map contoured at the 1.0 σ level shows the PLP-Lys-57 internal Schiff base structure. The active site is exposed to the solvent and is filled with many water molecules. The water molecules numbered W1, W2, and W3 are conserved in spSRm in the closed form. B, close-up view of the spSRm complex with the substrate serine. PLP and the lysino-d-alanyl residue form a Schiff base. l-Serine (pink) is tentatively modeled into the active site. Arg-133, which is far from the active site in the open form, approaches the re-face side of PLP to form a salt bridge with the carboxylate of the substrate. The asparagine loop (Ser-81–Ser-82–Gly-83–Asn-84–His-85) moves to the active site center to recognize the carboxylate of the d-alanyl portion of the lysino-d-alanyl residue.
A metal ion, which was assumed to be Mg2+ based on the height of the residual electron density, the atomic displacement parameter, and the coordination geometry, is located beside the tetra-glycine loop and coordinated by the carboxylates of Glu-208 and Asp-214 (Fig. 2C), the carbonyl group of Gly-212, and three water molecules (Fig. 4). spSR loses its activity on treatment with EDTA and recovers its activity on the addition of Mg2+ or Ca2+. However, the divalent cation is considered not to be directly involved in the catalytic action but in stabilization of the folding of the protein because the ion is located outside the active-site center. Mammalian brain SR was activated in the presence of divalent cations (12, 14). Glu-208 and Asp-214 are conserved in human and rat SRs, suggesting that a divalent cation is similarly bound to human and rat SRs. The Schiff base bond (C4′=N) is roughly coplanar with the pyridine ring of PLP (torsional angle of C3-C4-C4′-n = 32°) to form the conjugated π-system of the PLP-protonated Schiff base.
FIGURE 4.

Schematic diagram showing hydrogen bond and salt bridge interactions of the active site residues in spSRm·serine. Putative interactions are shown by dotted lines if the acceptor and donor are less than 3.3 Å apart.
Active Site of spSRm·Serine in the Closed Form
In our previous report, it was shown that spSRm catalyzes the racemase and α,β-elimination reactions, although the essential Lys-57 inherently forming a Schiff base with PLP in spSRw is converted to lysino-d-alanyl residue (19). This indicates that the α-amino group of lysino-d-alanyl residue acts as a catalytic base in the same manner as the ϵ-amino group of Lys-57 in spSRw (Fig. 1). To elucidate the interactions of spSRm with the substrate, a spSRm crystal soaked in a solution containing 10 mm l-serine was subjected to x-ray analysis. The omit electron density map exhibited the residual electron density corresponding to d- or l-serine or mixture of the two on the re-face side of PLP (Fig. 3B). The fact that spSRm forms a complex with serine is consistent with the residual activity of the modified enzyme.
The small domain rotates by 20° to the large domain as compared with that of spSRw to close the active site. The α-helix H5 and its N-terminal loop (the residues from Ser-81 to His-85) approach the PLP-lysinoalanine Schiff base with a large conformational change in the Ser-81–Ser-82–Gly-83 region forming the binding site for the carboxylate of the lysino-d-alanyl residue. The carboxylate interacts with Ser-81, Ser-82, Asn-84, and His-85 (Fig. 4). The negative charge of the carboxylate is partially compensated for by the dipole of α-helix H5.
The substrate enters the water tunnel connecting the re-face side of the PLP-lysinoalanine Schiff base and the molecular surface and resides on the cofactor. The carboxylate of the substrate serine forms an end-on type salt bridge with the guanidino group of the N-terminal Arg-133 of α-helix H7 (49) and a hydrogen bond with the main-chain NH group of Ser-82 of the asparagine loop. Intriguingly, Arg-133, which is a key residue for recognition of the substrate in spSRm, is conserved in human and mouse SRs but not in other fold-type II enzymes. The amino group of the substrate forms a hydrogen bond with the main-chain C O group of Gly-236 situated on the pyridine ring of PLP. The distance between the amino nitrogen of the substrate and the C4′ of PLP is 3.6 Å, suggesting that the cofactor PLP may form a new Schiff base (external aldimine) with the substrate serine to release the lysinoalanine residue (Fig. 1). The α-amino group of the lysinoalanine residue might serve as a base that shuttles protons to the substrate similarly to that of Lys-57 in spSRw. Thereby, the PLP-lysinoalanine Schiff base might play a critical role in the reactions catalyzed by spSRm.
O group of Gly-236 situated on the pyridine ring of PLP. The distance between the amino nitrogen of the substrate and the C4′ of PLP is 3.6 Å, suggesting that the cofactor PLP may form a new Schiff base (external aldimine) with the substrate serine to release the lysinoalanine residue (Fig. 1). The α-amino group of the lysinoalanine residue might serve as a base that shuttles protons to the substrate similarly to that of Lys-57 in spSRw. Thereby, the PLP-lysinoalanine Schiff base might play a critical role in the reactions catalyzed by spSRm.
Binding Mode of AMP-PCP
Mg·ATP is a physiological activator and is considered to be fully coordinated to the ATP binding site of mouse SR in vivo (5, 12). The racemization and α,β-elimination catalyzed by mouse SR are stimulated upon the addition of Mg·ATP. Similarly, in the presence of 1 mm Mg·ATP, spSRw is activated by 1.2 and 1.8 times for racemization and α,β-elimination of l-serine, respectively. The lower stimulation observed for the racemization reaction might be due to the degradation of the produced d-serine through the increased α,β-elimination activity. To clarify the binding site of ATP and its interaction with spSRw, complex crystals of spSRw with AMP-PCP were prepared by soaking wild-type crystals in a solution containing 10 mm AMP-PCP.
Mg·AMP-PCP is bound to the groove formed at the intersection between the domain interface and the subunit interface (Figs. 2, A and B, and 5). The binding of Mg·AMP-PCP to spSRw in the open form does not induce a subunit conformational change, but, interestingly, changes the relative orientation between the two subunits. Each subunit rotates around the axis that goes through the adenyl rings of two AMP-PCPs to enlarge the width of the back groove (Fig. 2B). The ATP binding site is surrounded by Ala-115 (helix H6) and Tyr-119 (C-terminal loop of H6) of the small domain, Asn-25 (loop between H1 and H2), Phe-50, Asn-51, Lys-52, and Met-53 (loop between β-strand S1 and H3), and Asn-311 (loop between S8 and H14) of the large domain, and Ser-32*,6 Ser-33*, and Thr-34* (N-terminal part of H2) and Arg-275*, Met-276*, and Lys-277* (C-terminal part of H12) of the large domain of the other subunit (Fig. 5). The adenine ring of AMP-PCP is sandwiched by Ala-115 and the guanidino plane of Arg-275*. The O3′ of the ribose moiety interacts with the side chains of Asn-311 and Lys-277*. The triphosphate group interacts with Asn-25, Phe-50–Met-53, Tyr-119, Thr-31*–Thr-34*, and many water molecules. Mg2+ is coordinated by two oxygen atoms of the terminal pyrophosphate and four water molecules with octahedral geometry. Out of the 14 residues interacting with ATP, 11 are conserved in human and rat SRs; the exceptions are Asn-25 (His in human and rat SRs), Thr-34 (Ile), and Met-53 (Thr). On the other hand, only 2 residues are conserved in threonine dehydratase from E. coli, and no residues are conserved in O-acetylserine sulfhydrylase from S. typhimurium or tryptophan synthase from E. coli. Possibly, ATP is coordinated to mammalian SR in a mode similar to that in spSR.
FIGURE 5.

Interactions of AMP-PCP with spSRw in the open form. The simulated annealing omit map is contoured at the 1.0 σ level. AMP-PCP interacts with the small and large domain residues (gray sticks with red oxygen and blue nitrogen) of one subunit and the large domain residues (orange sticks) of the other subunit. Hydrogen bonds are shown by dotted lines (green). Met-53, Asn-84, Gln-87, Glu-281, and Asn-311 (blue sticks) form a hydrogen bond network (red) to link PLP and AMP-PCP. The magnesium ion is surrounded by β- and γ-phosphate and four water molecules. The water molecules are involved in direct hydrogen bonds with the neighboring amino acid residues.
It seems that no significant change in the cofactor or side-chain orientation occurs on binding of AMP-PCP in the active site of spSRw. However, the O3′ of PLP is linked to the ribose hydroxy groups and γ-phosphate of AMP-PCP through a hydrogen bond network formed by Met-53, Asn-84, Gln-87, Glu-281, Asn-311, and water molecules (Fig. 5). It should be noted that Asn-84 is a constituent of the mobile asparagine loop that recognizes the substrate carboxylate, and the side chain of Gln-87 interacts with the loop through water molecules. Upon binding of the substrate to the spSRw complexed with ATP, the enzyme changes its conformation from the open form to the closed form similar to that observed in spSRm·serine. In the closed form, the hydrogen bond network between PLP and ATP might be reorganized to fine-tune the active site structure, resulting in the stimulation of the enzyme activity. In effect, the side chain of Gln-87 is within direct hydrogen bond distances from the ribose hydroxy group of AMP-PCP and the side chain of Asn-84 if the large domain Cα atoms of the enzyme in the closed form are superimposed on those of spSRw·AMP-PCP. In addition, the adenine moiety of AMP-PCP interacts with the small domain of one subunit as well as with the large domain of the other subunit. The interactions of ATP with both domains would have effects on the mode of the open-closed conformational change (small domain movement) leading to the enhancement of the enzyme activity.
Structural Model of External Aldimine of spSRw
spSRw crystals soaked in a solution supplemented with 10 mm l-serine were subjected to x-ray structure analysis. However, the omit electron density map of the active site showed that no substrate or product was bound to the active site. The co-crystallization method was employed to produce the enzyme-substrate complex. X-ray analysis of the crystal thus obtained indicated that during the co-crystallization process in the presence of l-serine, which took much longer than the soaking method, spSRw was converted to spSRm.
To clarify the substrate recognition mechanism and the reaction mechanism, a computer graphics model of the external aldimine form of spSRw and l-serine was designed by utilizing the active site structure of the rat liver serine dehydratase complexed with O-methyl-l-serine in the closed form in which PLP forms a Schiff base with O-methyl-l-serine (external aldimine form) (37). The active site of spSRm including the cofactor PLP is essentially superimposable on that of serine dehydratase except for the d-alanyl moiety of the PLP-lysinoalanine Schiff base. The cofactor PLP in spSRm assumes the same orientation as that in serine dehydratase, whereas the d-alanyl portion moves slightly toward the protein side as compared with O-methyl-l-serine as d-alanine forms a covalent bond with Lys-57. The PLP-lysinoalanine Schiff base in spSRm was replaced by the PLP-O-methyl-l-serine Schiff base and the catalytic Lys in serine dehydratase, respectively. The O-methyl group was then removed, and the hydroxy group of l-serine was positioned so as to form a hydrogen bond with PLP phosphate producing the external aldimine model with l-serine (Fig. 6). The external aldimine model with d-serine was constructed by replacing l-serine of the PLP-l-serine Schiff base model by d-serine assuming that the Cα atom, the amino nitrogen, and the α-carboxylate of d-serine have positions similar to those of l-serine. The β-oxygen atom of d-serine was placed so as to form a hydrogen bond with the PLP phosphate oxygen atom. In either the d-serine or the l-serine complex model, the pyridine ring of PLP, Schiff base bond, Cα, and α-carboxylate group are approximately coplanar, ensuring that the p-orbital at the Cα atom formed after α-proton elimination is perpendicular to the PLP π-plane making substrate Cα-proton elimination easy.
FIGURE 6.

Superposition of external aldimine models between spSRw·d-serine and spSRw·l-serine. The models were designed by taking advantage of the active site superimposition of the rat serine dehydratase complex with O-methyl-l-serine onto spSRm. The substrates, l-serine and d-serine, are shown in green and pink, respectively. The catalytic bases, Lys-57 and Ser-82 (blue), reside on the si- and re-faces of PLP, respectively, and are directed toward the Cα hydrogen atoms. The putative hydrogen bonds between PLP-phosphate and substrate are shown by dotted lines.
The substrate l-serine, which forms a Schiff base with PLP, directs its α-hydrogen to the protein side (si-face side) of PLP with its β-hydroxy group hydrogen-bonded to the PLP phosphate oxygen as shown in Fig. 6 (37). The side-chain amino group of Lys-57 is within a distance of 3.3 Å from the substrate Cα atom. The substrate d-serine directs its α-hydrogen to the solvent side (re-face side) of PLP with its β-hydroxy group interacting with the PLP phosphate group. The hydroxy group of Ser-82 is at a distance of 3.0 Å from the Cα atom of the substrate d-serine. Lys-57 is considered to act as an acid-base catalyst to add a proton to, or abstract a proton from, the Cα atom of the substrate on the si-face side of PLP because the orientation of Lys-57 with respect to PLP is quite similar to those of fold type II enzymes such as serine dehydratase, threonine synthase, and tryptophan synthase (37, 38, 40). The external aldimine model with d-serine shows that Ser-82 is a candidate that shuttles protons to the substrate on the re-face side PLP (Fig. 6). To examine the involvement of Ser-82 in the catalysis, the S82A mutant was subjected to the enzyme assay. The assay showed that the racemase activity was not detected and that d-serine α,β-elimination activity of the S82A mutant was almost completely lost (<0.01% of the activity of spSRw), whereas the l-serine α,β-elimination activity of the mutant was retained (Km = 45 mm, Vmax = 450 nmol·min−1·mg−1) as compared with that of wild-type enzyme (Km = 36 mm, Vmax = 870 nmol·min−1·mg−1). A similar result was reported for the mutant as to the corresponding residue of serine racemase from Dictyostelium discoideum (50). It is thus reasonable to assume that Ser-82 is the proton carrier to the substrate Cα atom on the re-face side of the cofactor.
Mechanistic Implication of Wild-type Enzyme
The pyridine N-1 atom of the cofactor forms a hydrogen bond with the hydroxy group of Ser-308, acting as a hydrogen bond acceptor, as shown in the x-ray structures of threonine dehydratase (35), O-acetylserine sulfhydrylase (44, 51–53), threonine synthase (38, 54, 55), tryptophan synthase (40, 56), and serine dehydratase (N-1–H–S hydrogen bond). The involvement of the N-1 atom in the neutral hydrogen bond with the hydroxy group of Ser or Thr or the sulfhydryl group of l-Cys as an acceptor is one of the characteristics of the fold-type II enzymes. In the fold-type I or IV enzymes, the positively charged protonated N-1 atom of PLP forms a salt bridge with the negatively charged carboxylate of aspartate or glutamate. The neutral hydrogen bond reduces the electron-withdrawing effect of the pyridine ring of PLP as compared with the salt bridge, destabilizing the quinonoid intermediate produced on Cα proton elimination from the substrate amino acid (37, 38, 45, 46). The reaction may proceed through the carbanion intermediate after substrate α-proton elimination rather than the quinonoid intermediate, which requires the protonated N-1 atom (Fig. 7).
FIGURE 7.

The proposed reaction mechanism of spSRw and spSRm with the substrate l- or d-serine.
The substrate l- or d-serine approaches the re-face side of PLP with its α-amino group directed toward the Schiff base of PLP to induce the small domain movement forming a Michaelis complex. The amino group of the substrate must be in an unprotonated state to make a nucleophilic attack on C4′ of the PLP Schiff base to form an external aldimine (Fig. 7). This form is generated either through binding of the substrate with a free amino group or through deprotonation of the ammonium group after binding of the substrate with a protonated amino group. The nucleophilic attack produces an external aldimine to release the neutral side chain of Lys-57. The next step is the α-proton elimination of the substrate. Judging from the PLP-l-serine external aldimine model, Lys-57 directs its side-chain amino group toward the α-proton of the substrate l-serine, whereas Ser-82 directs its hydroxy group toward the α-proton of the substrate d-serine. Ser-82 is one of the key residues for the enzyme to act as a SR because Ser-82 is conserved in mammalian SRs but not in other enzymes of fold-type II.
The reaction catalyzed by spSR is explained by the classic two-base mechanism (46, 57) (Fig. 7). The arrangement of two bases (Lys-57 and Ser-82) and the PLP-substrate Schiff base in spSRw is reminiscent of that observed in alanine racemase, although the catalytic lysine is on the re-face side of the cofactor (46). The neutral amino group of Lys-57 or the deprotonated hydroxy group of Ser-82 abstracts the substrate α-proton of the PLP-l-serine or PLP-d-serine Schiff base, respectively, to yield the carbanion intermediate. The addition of the proton to the Cα atom of the carbanion intermediate from the reverse side of PLP results in the racemization reaction. The hydroxy group of Ser-82 or the protonated amino group of Lys-57 adds its proton to the Cα atom of the carbanion from the re- or si-face side to produce the PLP-d-serine or PLP-l-serine Schiff base, respectively.
After the carbanion intermediate is formed through Cα proton elimination, the protonation on the substrate β-hydroxy group results in the β-elimination reaction, producing the cofactor-aminoacrylate Schiff base and a water molecule. The PLP phosphate is a possible candidate for the proton carriers to the substrate β-hydroxy group if the substrate hydroxy group is in a favorable position to form a hydrogen bond with the PLP phosphate, as shown in external aldimine models (Fig. 6) (37). Finally, the cofactor recovers the Schiff base with Lys-57 to release the aminoacrylate, which is nonenzymatically converted to pyruvate and ammonia. Alternatively, a portion of the α-aminoacrylate intermediate is converted into spSRm, i.e. PLP-lysinoalanine Schiff base, by the Michael addition reaction of Lys-57 with the α,β-unsaturated product (Fig. 7). spSRm thus formed also participates in the overall catalytic cycle in the same manner as spSRw.
In summary, we have determined the three-dimensional structures of spSRw, spSRw·AMP-PCP, and spSRm·serine by means of x-ray crystallographic methods. On binding of the substrate, the small domain rotates toward the large domain with a local conformational change to close and complete the active site. The activator (ATP) is bound to the intersection between the domain and the subunit interface. Lys-57 and Ser-82 are considered to be acid-base catalysts that shuttle protons to the substrate Cα atoms. The substrate serine trapped in the active site of spSRm reasonably interacts with active site residues.
This work was supported by research grants from the Japan Society for the Promotion of Science and National Project on Protein Structural and Functional Analyses (to N. E. and to K. H.).
The atomic coordinates and structure factors (codes 1V71, 1WTC, and 2ZR8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
In residues, the asterisk indicates a residue from another subunit of the dimer unit.
- NMDA
- N-methyl-d-aspartate
- PLP
- pyridoxal 5′-phosphate
- SR
- serine racemase
- spSR
- SR from S. pombe
- spSRw
- wild-type spSR
- spSRm
- modified spSR
- AMP-PCP
- 5′-adenylyl methylenediphosphate.
REFERENCES
- 1.Hashimoto A., Nishikawa T., Oka T., Takahashi K. (1993) J. Neurochem. 60, 783–786 [DOI] [PubMed] [Google Scholar]
- 2.Schell M. J., Molliver M. E., Snyder S. H. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 3948–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mothet J. P., Parent A. T., Wolosker H., Brady R. O., Jr., Linden D. J., Ferris C. D., Rogawski M. A., Snyder S. H. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolosker H., Panizzutti R., De Miranda J. (2002) Neurochem. Int. 41, 327–332 [DOI] [PubMed] [Google Scholar]
- 5.Boehning D., Snyder S. H. (2003) Annu. Rev. Neurosci. 26, 105–131 [DOI] [PubMed] [Google Scholar]
- 6.Danysz W., Parsons C. G. (1998) Pharmacol. Rev. 50, 597–664 [PubMed] [Google Scholar]
- 7.Yang Y., Ge W., Chen Y., Zhang Z., Shen W., Wu C., Poo M., Duan S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15194–15199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolosker H., Sheth K. N., Takahashi M., Mothet J. P., Brady R. O., Jr., Ferris C. D., Snyder S. H. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 721–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolosker H., Blackshaw S., Snyder S. H. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13409–13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue R., Hashimoto K., Harai T., Mori H. (2008) J. Neurosci. 28, 14486–14491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolosker H. (2006) Sci. STKE 2006, pe41. [DOI] [PubMed] [Google Scholar]
- 12.De Miranda J., Panizzutti R., Foltyn V. N., Wolosker H. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 14542–14547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foltyn V. N., Bendikov I., De Miranda J., Panizzutti R., Dumin E., Shleper M., Li P., Toney M. D., Kartvelishvily E., Wolosker H. (2005) J. Biol. Chem. 280, 1754–1763 [DOI] [PubMed] [Google Scholar]
- 14.Baumgart F., Rodríguez-Crespo I. (2008) FEBS J. 275, 3538–3545 [DOI] [PubMed] [Google Scholar]
- 15.Uo T., Yoshimura T., Shimizu S., Esaki N. (1998) Biochem. Biophys. Res. Commun. 246, 31–34 [DOI] [PubMed] [Google Scholar]
- 16.Grishin N. V., Phillips M. A., Goldsmith E. J. (1995) Protein Sci. 4, 1291–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta P. K., Christen P. (2000) Adv. Enzymol. Relat. Areas Mol. Biol. 74, 129–184 [DOI] [PubMed] [Google Scholar]
- 18.Schneider G., Käck H., Lindqvist Y. (2000) Structure 8, R1–6 [DOI] [PubMed] [Google Scholar]
- 19.Yamauchi T., Goto M., Wu H. Y., Uo T., Yoshimura T., Mihara H., Kurihara T., Miyahara I., Hirotsu K., Esaki N. (2009) J. Biochem. 145, 421–424 [DOI] [PubMed] [Google Scholar]
- 20.Panizzutti R., De Miranda J., Ribeiro C. S., Engelender S., Wolosker H. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5294–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto A., Nishikawa T., Oka T., Takahashi K., Hayashi T. (1992) J. Chromatogr. 582, 41–48 [DOI] [PubMed] [Google Scholar]
- 22.Uo T., Yoshimura T., Nishiyama T., Esaki N. (2002) Biosci. Biotechnol. Biochem. 66, 2639–2644 [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 24.Navaza J. (1994) Acta Crystallogr. Sect. A 50, 157–163 [Google Scholar]
- 25.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 26.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 27.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 28.Kraulis P. J. (1991) J. Appl. Crystallogr. 24, 946–950 [Google Scholar]
- 29.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 30.Fenn T. D., Ringe D., Petsko G. A. (2003) J. Appl. Crystallogr. 36, 944–947 [Google Scholar]
- 31.Merritt E. A., Murphy M. E. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 869–873 [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W., Sander C. (1983) Biopolymers. 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 33.Holm L., Sander C. (1996) Science 273, 595–603 [DOI] [PubMed] [Google Scholar]
- 34.Simanshu D. K., Savithri H. S., Murthy M. R. (2006) J. Biol. Chem. 281, 39630–39641 [DOI] [PubMed] [Google Scholar]
- 35.Gallagher D. T., Gilliland G. L., Xiao G., Zondlo J., Fisher K. E., Chinchilla D., Eisenstein E. (1998) Structure 6, 465–475 [DOI] [PubMed] [Google Scholar]
- 36.Yamada T., Komoto J., Kasuya T., Takata Y., Ogawa H., Mori H., Takusagawa F. (2008) Biochim. Biophys. Acta 1780, 809–818 [DOI] [PubMed] [Google Scholar]
- 37.Yamada T., Komoto J., Takata Y., Ogawa H., Pitot H. C., Takusagawa F. (2003) Biochemistry 42, 12854–12865 [DOI] [PubMed] [Google Scholar]
- 38.Omi R., Goto M., Miyahara I., Mizuguchi H., Hayashi H., Kagamiyama H., Hirotsu K. (2003) J. Biol. Chem. 278, 46035–46045 [DOI] [PubMed] [Google Scholar]
- 39.Burkhard P., Rao G. S., Hohenester E., Schnackerz K. D., Cook P. F., Jansonius J. N. (1998) J. Mol. Biol. 283, 121–133 [DOI] [PubMed] [Google Scholar]
- 40.Rhee S., Parris K. D., Hyde C. C., Ahmed S. A., Miles E. W., Davies D. R. (1997) Biochemistry 36, 7664–7680 [DOI] [PubMed] [Google Scholar]
- 41.Jansonius J. N., Vincent M. G. (1987) in Biological Macromolecules and Assemblies, Vol. 3 ( Jurnak F. A., McPherson A., eds) pp. 187–285, J. Wiley & Sons, Inc., New York [Google Scholar]
- 42.Jäger J., Moser M., Sauder U., Jansonius J. N. (1994) J. Mol. Biol. 239, 285–305 [DOI] [PubMed] [Google Scholar]
- 43.Okamoto A., Higuchi T., Hirotsu K., Kuramitsu S., Kagamiyama H. (1994) J. Biochem. 116, 95–107 [DOI] [PubMed] [Google Scholar]
- 44.Burkhard P., Tai C. H., Ristroph C. M., Cook P. F., Jansonius J. N. (1999) J. Mol. Biol. 291, 941–953 [DOI] [PubMed] [Google Scholar]
- 45.Daum S., Tai C. H., Cook P. F. (2003) Biochemistry 42, 106–113 [DOI] [PubMed] [Google Scholar]
- 46.Shaw J. P., Petsko G. A., Ringe D. (1997) Biochemistry 36, 1329–1342 [DOI] [PubMed] [Google Scholar]
- 47.Inoue K., Kuramitsu S., Okamoto A., Hirotsu K., Higuchi T., Morino Y., Kagamiyama H. (1991) J. Biochem. 109, 570–576 [DOI] [PubMed] [Google Scholar]
- 48.Goldberg J. M., Swanson R. V., Goodman H. S., Kirsch J. F. (1991) Biochemistry 30, 305–312 [DOI] [PubMed] [Google Scholar]
- 49.Mitchell J. B., Thornton J. M., Singh J., Price S. L. (1992) J. Mol. Biol. 226, 251–262 [DOI] [PubMed] [Google Scholar]
- 50.Yoshimura T., Goto M. (2008) FEBS J. 275, 3527–3537 [DOI] [PubMed] [Google Scholar]
- 51.Huang B., Vetting M. W., Roderick S. L. (2005) J. Bacteriol. 187, 3201–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claus M. T., Zocher G. E., Maier T. H., Schulz G. E. (2005) Biochemistry 44, 8620–8626 [DOI] [PubMed] [Google Scholar]
- 53.Heine A., Canaves J. M., von Delft F., Brinen L. S., Dai X., Deacon A. M., Elsliger M. A., Eshaghi S., Floyd R., Godzik A., Grittini C., Grzechnik S. K., Guda C., Jaroszewski L., Karlak C., Klock H. E., Koesema E., Kovarik J. S., Kreusch A., Kuhn P., Lesley S. A., McMullan D., McPhillips T. M., Miller M. A., Miller M. D., Morse A., Moy K., Ouyang J., Page R., Robb A., Rodrigues K., Schwarzenbacher R., Selby T. L., Spraggon G., Stevens R. C., van den Bedem H., Velasquez J., Vincent J., Wang X., West B., Wolf G., Hodgson K. O., Wooley J., Wilson I. A. (2004) Proteins 56, 387–391 [DOI] [PubMed] [Google Scholar]
- 54.Garrido-Franco M., Ehlert S., Messerschmidt A., Marinkoviæ S., Huber R., Laber B., Bourenkov G. P., Clausen T. (2002) J. Biol. Chem. 277, 12396–12405 [DOI] [PubMed] [Google Scholar]
- 55.Mas-Droux C., Biou V., Dumas R. (2006) J. Biol. Chem. 281, 5188–5196 [DOI] [PubMed] [Google Scholar]
- 56.Lee S. J., Ogasahara K., Ma J., Nishio K., Ishida M., Yamagata Y., Tsukihara T., Yutani K. (2005) Biochemistry 44, 11417–11427 [DOI] [PubMed] [Google Scholar]
- 57.Sun S., Toney M. D. (1999) Biochemistry 38, 4058–4065 [DOI] [PubMed] [Google Scholar]