

Abstract
Sensing DNA damage and initiation of genetic responses to repair DNA damage are critical to cell survival. In E. coli, RecA polymerizes on ssDNA produced by DNA damage creating a RecA-DNA filament that interacts with the LexA repressor inducing the SOS Response. RecA filament stability is negatively modulated by RecX and UvrD. recA730 (E38K) and recA4142 (F217Y) constitutively express the SOS Response. recA4162 (I298V) and recA4164 (L126V) are intragenic suppressors of the constitutive SOS phenotype of recA730. Herein, it is shown that these suppressors are not allele specific and can suppress SOSC expression of recA730 and recA4142 in cis and in trans. recA4162 and recA4164 single mutants (and the recA730 and recA4142 derivatives) are Rec+, UVR and are able to induce the SOS response after UV treatment like wild type. UvrD and RecX are required for the suppression in two (recA730,4164 and recA4142,4162) of the four double mutants tested. To explain the data, one model suggests that recAC alleles promote SOSC expression by mimicking RecA filament structures that induce SOS and the suppressor alleles mimic RecA filament at end of SOS. UvrD and RecX are attracted to these latter structures to help dismantle or destabilize the RecA filament.
Keywords: RecA, SOS Response, Recombination, DNA Repair
INTRODUCTION
Organisms have the ability to detect and respond to DNA damage. In Escherichia coli, RecA and LexA regulate, at the level of transcription, the cellular reaction to DNA damage called the SOS Response. RecA is the sensor for the response and LexA is the repressor (Walker, 1996). RecA also repairs DNA damage through recombination (Lusetti and Cox, 2002). The SOS Response is activated when DNA damage generates ssDNA to which RecA binds and forms a protein-DNA filament. The RecA-DNA filament then increases the rate of LexA auto-proteolysis (Little, 1991) and this, in turn, increases the rate of transcription of SOS genes (Courcelle et al., 2001; Fernandez De Henestrosa et al., 2000). The loading of RecA onto DNA requires either the RecBCD enzyme or the RecFOR complex depending on the substrate (Clark and Sandler, 1994). RecA filaments are dynamic structures and grow by adding monomers at the 3′ end and losing them from the 5′ end (Bork et al., 2001; Shan et al., 1997).
The stability of the RecA-DNA filaments is of critical importance for its ability to function in recombination, DNA repair and induction of the SOS response. Of several proteins known to influence RecA filament stability (reviewed in (Cox, 2007)); RecX and UvrD both have negative impacts on the stability of RecA-DNA filaments and both are part of the SOS regulon. Biochemical (Drees et al., 2004a; Drees et al., 2004b; Lusetti et al., 2006; Stohl et al., 2003) and structural analysis (Mishra et al., 2003; Ragone et al., 2008; VanLoock et al., 2003) suggest that RecX binds either at the 3′ end of the RecA filament and or in the major groove where it promotes local dissociation, creating new 5′ and 3′ ends, while inhibiting the addition of monomers at the 3′ end. RecX mediated destabilization is further complicated by the ability of RecFOR to antagonize this activity (Long et al., 2008; Lusetti et al., 2006). Recently, it has been suggested that ssDNA binding may also be critical for RecX function (Baitin et al., 2008). The recX gene is co-transcribed with the recA gene (Pages et al., 2003).
The UvrD helicase can remove RecA from ssDNA in vitro (Veaute et al., 2005). RecA and UvrD’s eukaryotic homologs, RAD51 and SRS2, share a similar relationship (reviewed in (Macris and Sung, 2005) and (Veaute et al., 2003)). The absence of these helicases often leads to hyper-recombinogenic phenotypes under conditions of normal growth and the inability to suppress stressful situations caused by recombination (Kerrest et al., 2009; Zieg et al., 1978). In E. coli, uvrD mutants have two-fold more RecA-GFP foci and these foci are on average two-fold brighter than wild type strains (Centore and Sandler, 2007). The cell uses UvrD to remove RecA from the DNA. For instance, UvrD removes RecA from stopped replication forks in dnaNts and dnaEts strains where RecA’s activity is detrimental ((Flores et al., 2005) and reviewed in (Michel et al., 2007)).
recA constitutive (recAC) mutants have lost the ability to properly regulate the SOS Response and show high levels of SOS expression in log phase cells in the absence of external DNA damage {(Kirby et al., 1967; Nastri et al., 1997; Skiba and Knight, 1994; Tessman and Peterson, 1985; Witkin et al., 1982) and reviewed in (McGrew and Knight, 2003)}. Two recAC mutants discussed here are recA730 (E38K) and recA4142 (F217Y) (McGrew and Knight, 2003; Skiba and Knight, 1994; Witkin et al., 1982). Structurally these two mutations change amino acids in different parts of the RecA protein (Figure 1). recA730 changes an amino acid located on the outside of the RecA-DNA filament and recA4142 changes an amino acid at the monomer-monomer interface. These two mutants have been recently characterized for the mechanism by which they cause constitutive SOS (SOSC) expression in log phase cells (Long et al., 2008). Several differences were seen. The first is that SOSC expression in recA4142 mutants is sensitive to mutations in RecA loading and stability factors: recBCD, recX and dinI; whereas the SOSC expression in a recA730 mutant is not. Another difference is that SOSC expression in a recA730 strain is sensitive to the hyper-helicase mutant uvrD303, whereas the SOSC expression in a recA4142 mutant is not (Centore et al., 2009). The initial level of transcription in recA4142 cells (and not recA730 cells) is critical for SOSC expression. In recAo+ recA4142 mutants, about 8% of a population of cells are constitutive for SOS (SOSC) expression. This can be increased to 100% by the recAo1403 operator mutation which increases the basal rate of transcription 2–3 fold (Long et al., 2008; Wertman and Mount, 1985). In this report, all strains with recA4142 also have recAo1403 as well and for simplicity will be referred to simply as recA4142. In summary, the results are consistent with a model whereby RecA4142 is loaded by RecBCD at double strand ends that exist in log phase cells and that RecA730 binds to other ssDNA in the cell, possibly at replication forks, in a manner yet to be determined.
Figure 1.
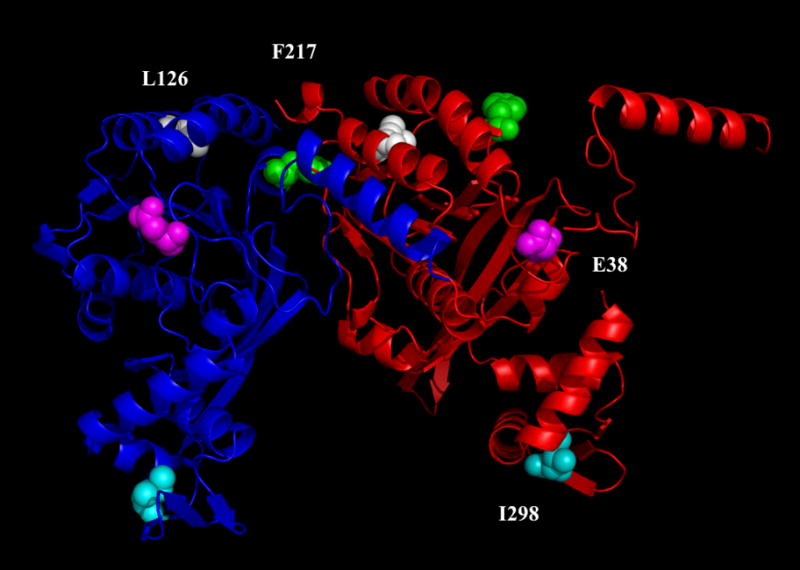
This figure shows a depiction of two adjacent RecA molecules (red and blue) as they may appear in a helical filament based on the crystal structure without DNA (Story and Steitz, 1992; Story et al., 1992). The green residues represent the positions of F217Y, the magenta residues show the positions of E38K, the white residues are L126V and the cyan residues are I298V.
Historically, two intragenic suppressor mutations for the SOSC phenotype of recA730 were found. The suppressor mutations are named herein recA4162 (I298V) and recA4164 (L126V) so that the paper is clear when it discusses these suppressor mutations in the presence of other recAC alleles or by themselves. The recA4162 suppressor was originally identified as one of the mutations in the recA allele first called tif-1 for temperature-inducible filamentation (Kirby et al., 1967). This allele was later shown to contain two mutations (E38K and I298V) and re-named recA441 (Knight et al., 1984). This allele showed the SOSC phenotype in a temperature dependent manner (at 42°C, not at 30°C). We named this allele recA730,4162. The other suppressor, recA4164, was isolated as one of the mutations in an allele originally called recA718 (contains both recA730 and recA4164) (McCall et al., 1987; Witkin et al., 1982). Unlike recA730,4162, SOSC expression is suppressed in recA730,4164 cells at all temperatures. The positions of the codons mutated in recA4162 and recA4164 (as well as recA730 and recA4142) are different from one another (Figure 1). The codon position mutated in recA4162 is in the carboxy-terminal domain and the codon position mutated in recA4164 is close to the face of the protein thought to be on the inside of the RecA-DNA helical filament near the DNA (Chen et al., 2008).
Even though these two suppressor mutations have been known for many years, the mechanism by which they function is not yet clear. Intragenic suppression is sometimes found to be allele specific. When it is allele specific, the suppression mechanism often proposed is that the first mutation locally distorts the structure of the protein and the second mutation restores it. A priori, in the case of recA730,4162 or recA730,4164, this mechanism of suppression is unlikely to explain these situations since the two suppressors mutations are not in proximity of the “offending” recA730 mutation even if one considers the location of the mutations in adjacent monomers in a filament (Figure 1). Nonetheless, it is possible that the suppressor mutations restore local structure by some indirect effect. If the suppressors do not restore the structure in an allele specific manner, then it is possible that these mutations affect some general property of the RecA protein such that they no longer support SOSC expression. It is also possible that the suppression does not occur at the level of individual monomers, but at the level of a filament.
It is shown that suppression of the SOSC phenotype by recA4162 and recA4164 is not allele specific since they suppress another recAC allele, recA4142, in cis. It is then shown that recA4162 or recA4164 are able to suppress (and are dominant to) to both recA730 and recA4142 for the ability to produce SOSC expression. This inability to allow SOSC expression is not due to an inability to properly interact with the DNA or properly induce the SOS response since all strains with either of the two suppressor mutations individually or in combination with recA730 and recA4142 are able to induce the SOS Response after UV treatment like wild type and are Rec+ and UVR. Lastly, it is shown that both RecX and UvrD are required for suppression in recA730,4164 and recA142,4162 mutants, but not in recA730,4162 and recA4142,4164 mutants. Several UvrD missense mutants are tested for their ability to aid in the suppression of SOSC expression and differential abilities are seen. It is suggested that these two suppressor mutations inhibit SOSC expression by mimicking the structure of the RecA filament that occurs at the end of the SOS Response. This structure is better at recruiting and or responding to the proteins that destabilize or dissolve the RecA filament.
RESULTS
recA4162 and recA4164 suppress the SOSC expression of recA4142
The suppression provided by recA4162 and recA4164 to recA730 could be due to specific interactions (allele specific) within the recA730 protein. If the suppression is not due to some special interactions, then the mutations should be able to suppress the SOSC expression of other recAC allele such as recA4142. As indicated above, recA4142 was picked for this study because the mutation causing the recAC phenotype is in a different part of RecA and its requirements for SOSC expression is different as compared to recA730 (Figure 1 and see above). To test this, recA4142 was combined with each of the suppressor mutations on the chromosome and measured for its ability to produce SOSC expression in log phase cells. The level of SOS expression was measured using the sulAp-gfp transcriptional reporter system as has been described previously (Long et al., 2008; McCool et al., 2004). All measurements were done with log phase cells grown in minimal medium.
The average Relative Fluorescence Intensity (RFI) and the percentage of cells in a population expressing SOSC six-fold above wild type for the recA730 single mutant and recA730 with the two suppressor mutations are shown in Table 1. recA730 mutants show that nearly all their cells have a RFI greater than six-fold above wild type and that the average RFI of the population is about 40-fold above wild type. As expected, the addition of recA4162 and recA4164 to the recA730 mutant decreased both the percentage of cells in the population that had high levels of SOSC expression and the average RFI across the population to just 2-fold above wild type (or a 20 fold decrease from recA730). Table 1 shows that the series of recA4142 strains with and without the suppressors mutations reveal a pattern very similar to the recA730 series. It is notable that recA4142,4162 displayed no SOSC expression at 42°C unlike its recA730 counterpart (data not shown). It is possible that the low levels of SOSC expression in the recA4142,4162 and recA4142,4164 were due the inactivation of the RecA protein. Table 2 shows that these double mutants are all active as wild type for conjugal recombination, survival after UV irradiation (DNA repair) and induction of the SOS response after UV treatment. Therefore, we conclude that the suppression provided by recA4162 and recA4164 is not allele specific since they can suppress the SOSC expression of at least two different recAC alleles that produce SOSC expression by very different mechanisms and that the suppression is not due to inactivation of the RecA protein for its normal functions.
Table 1.
Suppression of constitutive SOS expression by recA4162 and recA4164 in cis and in trans with recA730 and recA4142
| Strains | Chr.recAo | Chr.recA | Plasmid recA | RFI ratioa | % of cells expressing SOSC ≥ 6-fold |
|---|---|---|---|---|---|
| SS996 | + | + | 1.0 ± 0.0 | 0.2 ± 0.1 | |
| SS4629 | + | 730 | 40.4 ± 7.1 | 99.6 ± 0.2 | |
| SS6013 | + | 4142 | 3.6 ± 0.7 | 7.6 ± 2.8 | |
| SS6052 | + | 4162 | 1.8 ± 0.1 | 3.6 ± 2.9 | |
| SS6027 | + | 4164 | 2.0 ± 0.1 | 2.0 ± 1.3 | |
| SS4630 | + | 730,4162 b | 1.0 ± 0.6 | 0.3 ± 0.2 | |
| SS5292 | + | 730,4164 | 0.8 ± 0.2 | 1.0 ± 0.1 | |
| SS4976 | o1403 | 4142 | 34.5 ± 1.5 | 100 ± 0.0 | |
| SS6089 | o1403 | 4142,4162 | 1.0 ± 0.2 | 0.5 ± 0.4 | |
| SS6062 | o1403 | 4142,4164 | 2.2 ± 0.2 | 1.9 ± 1.0 | |
| SS5376 | + | 730 | pDPT429 | 56.1 ± 4.3 | 98.9 ± 1.7 |
| SS6074 | + | 730 | recA | 48.0 ± 8.3 | 99.8 ± 0.2 |
| SS6077 | + | 730 | recA4162 | 7.8 ± 1.9 | 13.3 ± 1.5 |
| SS6079 | + | 730 | recA4164 | 7.1 ± 2.0 | 18.9 ± 7.3 |
| SS5377 | o1403 | 4142 | pDPT429 | 34.0 ± 8.9 | 89.3 ± 18.5 |
| SS6075 | o1403 | 4142 | recA | 14.9 ± 2.8 | 81.8 ± 13.1 |
| SS6076 | o1403 | 4142 | recA4162 | 7.0 ± 2.6 | 15.5 ± 6.7 |
| SS6078 | o1403 | 4142 | recA4164 | 6.6 ± 1.3 | 10.1 ± 2.0 |
| SS6118 | + | del | 730 | 33.6 ± 2.7 | 99.2 ± 1.0 |
| SS6086 | + | recA4162 | 730 | 1.6 ± 0.5 | 1.64 ± 1.4 |
| SS6085 | + | recA4164 | 730 | 1.0 ± 0.6 | 1.38 ±0.5 |
| SS6100 | + | del | 4142 | 28.2 ± 1.9 | 81.5 ± 7.0 |
| SS6081 | + | recA4162 | 4142 | 1.7 ± 0.4 | 3.95 ± 2.0 |
| SS6082 | + | recA4164 | 4142 | 2.3 ± 0.7 | 2.66 ± 2.9 |
See Materials and Methods for specific protocols. All Strains were grown in minimal medium at 37°C in log phase before sampling. All numbers are the average of three independent measurements. For each strain approximately 1000–2000 cells were counted. All strains having recA4142 on the chromosome also have recAo1403. Plasmid versions of recA4142 are recAo+. RFI stand for Relative Fluorescence Intensity and is a measure of the SOSC expression.
recA730,4162 (recA441) are sampled at 30°C.
Table 2.
Summary of phenotypic analysis of recA mutants used in this studya
| Strain | recAo | recA | Relative Recombinationb | % Survival at 5J/m of UV | Average RFI after 5J of UV |
|---|---|---|---|---|---|
| SS996 | + | + | 1.09 ± 0.26 | 80.0 ± 3.77 | 8.7 ± 2.8 |
| SS391 | + | 938::cat | 0.0006 ± 0.0002 | <0.001 | ND d |
| SS4629 | + | 730 | 1.50 ± 0.14 | 78.0 ± 2.00 | ND |
| SS6013 | + | 4142 | 1.79 ± 0.34 | 87.8 ± 6.27 | 11.1 ± 1.8 |
| SS6052 | + | 4162 | 1.38 ± 0.36 | 76.4 ± 4.31 | 8.1 ± 2.2 |
| SS6027 | + | 4164 | 1.15 ± 0.16 | 78.0 ± 2.64 | 6.7 ± 0.9 |
| SS4630 | + | 730,4162 | 0.82 ± 0.18c | 82.2 ± 6.29 | 8.3 ± 0.81 |
| SS5292 | + | 730,4164 | 0.79 ± 0.30 | 83.3 ± 4.32 | 12.3 ± 3.1 |
| SS4976 | 1403 | 4142 | 1.11 ± 0.28 | 83.1 ± 5.61 | ND |
| SS6089 | 1403 | 4142,4162 | 1.32 ± 0.35 | 87.1 ± 9.89 | 10.0 ± 1.1 |
| SS6062 | 1403 | 4142,4164 | 0.97 ± 0.25 | 83.6 ± 1.59 | 11.4 ± 2.0 |
See Materials and Methods for specific protocols. All numbers are the average of at least three independent measurements.
Relative to JC13509.
The Hfr mating for this Recombination test done at 37°C. The same test done at 30°C is 0.08±0.01.
ND is Not Determined.
recA4162 or recA4164 suppress SOSC expression in trans
RecA polymerizes on ssDNA in a head to tail fashion to create a RecA-DNA filament. Given this type of arrangement, it is possible that the mechanism of suppression of SOSC expression by RecA4162 and RecA4164 is not due to suppression within individual RecA monomers but is manifested at the level of the filament; possibly through interaction of adjacent monomers or some conformation of the filament.
To test if these suppressors can act in trans, a 4.5 kb Bam HI fragment carrying the recA4162 and recA4164 genes and their surrounding regions (this includes ygaD1::kan upstream of recA, the wild type recA promoter before the recA gene and the recX gene that follows immediately) were cloned into a low-copy vector pDPT429 (Taylor and Cohen, 1979). These plasmids were used to transform cells containing recA730 or recA4142. Table 1 shows that when recA4162 or recA4164 is expressed in trans in a recA730 mutant, an eight-fold decrease in RFI is seen. This suggests that both suppressor mutations are dominant to recA730. It is possible that the decrease is not due to a specific property of the suppressor mutation and that a copy of recA+ on a plasmid would also decrease SOSC expression. To test this, recA+ was expressed in trans on a plasmid with recA730 on the chromosome. Table 1 shows that no significant decrease in RFI is seen. This suggests that the decrease is specific to the suppressor mutations and that recA730 is dominant to wild type for this phenotype.
These series of experiments were then repeated for cells containing recA4142. Similar results are seen when the recA4162 or recA4164 plasmids were placed in the recA4142 strain: an eight-fold decrease in RFI (Table 1). A difference, however, was seen when recA+ is expressed in trans with recA4142. Here a two-fold decrease in the RFI is seen when compared to the recA4142 strain with vector alone. These results suggest that the two suppressor mutations are also dominant to recA4142 and that although recA+ can achieve some degree of suppression of the SOSC phenotype when expressed in trans, full suppression requires the suppressor mutation.
It is possible that the decrease seen occurs because the suppressor genes on the plasmids are expressed at a higher level than that of the chromosomal recAC alleles and thus their phenotype dominates. Therefore the situations were reversed; recA730 or recA4142 were cloned and expressed from the low copy vector and recA4162 and recA4164 were expressed from the chromosome. With one minor exception, similar patterns of SOSC expression and suppression were seen (Table 1). The minor exception is that the plasmid containing recA4142 in a recA deletion strain showed a population of cells that were only 28-fold above background instead of 34-fold and the percentage of cells having six-fold above background RFI was only 81% instead of 100%. Although the cause of this small decrease is not known, it is possible that this is due to the fact that the plasmid version of recA4142 contains recAo+ instead of recAo1403. Nonetheless, when this plasmid was placed in strains with recA4162 and recA4164 on the chromosome, SOSC expression was found to be similar to background levels (Table 1).
It is possible that the level of SOSC expression seen in the in trans experiment is due to the fact that the suppressor alleles by themselves are inactive or that when the two alleles are mixed (suppressor and recAC), they inactivate one another. These ideas were tested. Table 2 shows that two mutants harboring the suppressor allele are as active for recombination, DNA repair and induction of SOS after UV treatment as wild type. Surprisingly, these strains show a low level of SOSC expression by themselves (Table 1). Additionally, the recombination proficiency and UV-induced SOS expression was tested for all four in trans combinations of suppressor and recAC allele. They were found to be Rec+ and have UV-induced SOS expression like wild type (data not shown).
These results show that when recA4162 or recA4164 are expressed in trans with either recA730 or recA4142, suppression of constitutive SOS expression is seen suggesting that the mechanism of suppression occurs at the level of the filament.
The combination of RecX and UvrD are required for suppression of SOSC expression by recA4162 and recA4164
The in trans experiments above suggested that suppression should be occurring at the level of the RecA filament and not at the level of the RecA monomer. It is known that at least two proteins interact with RecA filaments and destabilize the filaments or remove RecA from the DNA. These are RecX and UvrD respectively. To test if RecX and or UvrD are required to suppress the SOSC expression produced by recA730 and recA4142, the four double mutants (recA730,4162, recA730,4264, recA4142,4162 and recA4142,4164) were combined singly with either a del(recX) or a del(uvrD) mutation. If these gene products are required to suppress the SOSC expression, then their removal should result in an increase in SOSC expression. Table 3 shows that individually, deletion of just recX had little effect. Removal of uvrD, however, revealed a modest 2–4 fold increase in RFI in recA730,4162, recA4142,4162 and recA4142,4164 over a recA+ strain. Removal of both uvrD and recX had a much larger, about an 8-fold increase in recA730,4164 and recA4142,4162 strains over a recA+ strain. Table 3 shows that the recA730,4162 and recA4142,4164 mutants were not additionally affected. From this we can tentatively conclude that at least two of the suppressor mutations require the action of the UvrD and RecX proteins to inhibit the SOSC expression of RecA730 and RecA4142. There may, however, be other proteins and or mechanisms that are also required for inhibition since two of the mutants showed only small, if any, increases in the absence of RecX and UvrD and none of the four mutants showed full levels of SOSC expression (e.g., equal to that of a recA730 mutant).
Table 3.
The effect of recX and uvrD deletion mutations on the ability of recA constitutive allele with suppressor to produce SOSC expression a
| Strain | recA | recX | uvrD | RFI ratioa | % of cells expressing SOSC ≥ 6-fold |
|---|---|---|---|---|---|
| SS996 | + | + | + | 1.0 | 0.3 |
| SS4630 | 730,4162b | + | + | 1.0 ± 0.6 | 0.3 ± 0.2 |
| SS5292 | 730,4164 | + | + | 0.8 ± 0.2 | 1.0 ±0.1 |
| SS6089 | 4142,4162 | + | + | 1.0 ± 0.2 | 0.5 ± 0.4 |
| SS6062 | 4142,4164 | + | + | 2.2 ± 0.2 | 1.9 ±1.0 |
| SS6080 | + | del | + | 1.7 ± 0.2 | 1.5 ± 0.6 |
| SS7474 | 730,4162 | del | + | 1.5 ± 0.5 | 2.5 ± 1.2 |
| SS7475 | 730,4164 | del | + | 1.0 ± 0.3 | 1.1 ± 1.0 |
| SS7449 | 4142,4162 | del | + | 2.3 ± 0.4 | 5.3 ± 0.6 |
| SS7476 | 4142,4164 | del | + | 1.5 ± 0.4 | 4.2 ± 1.8 |
| SS7139 | + | + | del | 1.8 ± 0.6 | 4.8 ± 1.0 |
| SS7464 | 730,4162 | + | kan | 3.3 ± 0.5 | 8.6 ±2.5 |
| SS7463 | 730,4164 | + | del | 1.6 ± 0.2 | 3.4 ± 0.7 |
| SS7434 | 4142,4162 | + | del | 6.2 ± 1.1 | 25.8 ± 6.8 |
| SS7465 | 4142,4164 | + | del | 7.7 ± 1.5 | 39.1 ± 10.6 |
| SS7467 | + | del | del | 2.0 ± 0.4 | 7.2 ± 1.5 |
| SS7477 | 730,4162 | del | del | 2.6 ± 1.1 | 5.1 ± 1.9 |
| SS7478 | 730,4164 | del | del | 18.3 ± 2.6 | 56.0 ± 6.2 |
| SS7450 | 4142,4162 | del | del | 16.5 ± 1.2 | 94.5 ± 1.3 |
| SS7479 | 4142,4164 | del | del | 5.9 ± 1.6 | 29.0 ± 13.6 |
Same as Table 1. All strains having recA4142 also have recAo1403.
All recA730,4162 strains were measured at 30°C.
Differential ability of uvrD missense mutant to aid in suppression
UvrD deletion mutant are UVS, hyper-recombinogenic and mutators. Biochemically, UvrD has been shown to have ATPase activity, helicase/translocase activity, participate in Nucleotide Excision Repair (NER) reactions, Methyl-Directed Mismatch (MMR) repair reactions and remove RecA from ssDNA (Atkinson et al., 2009; Matson and Robertson, 2006; Veaute et al., 2005). uvrD3 (E387K), uvrD252 (G30D), uvrD701 (deletion of the C-terminal 40 aa) and uvrD307 (R284A) were combined with either recA730,4162 or recA4142,4164 to test if they can aid the suppression of SOSC expression in the absence of recX. Figure 2 shows that uvrD307 and uvrD252 revealed the highest levels of SOSC expression of the four uvrD alleles tested. SOSC expression in the uvrD3 and uvrD701 strains were very low like uvrD+ for both recA mutants, whereas uvrD307 and uvrD252 showed higher levels. In general, higher levels were seen with recA4142,4164 than with recA730,4162. Interestingly, when compared to the null mutant, only the uvrD252 allele showed nearly the same average RFI, but that the percentage of cells in the population producing SOS expression six-fold above background was much lower (57% compared to 95%). From these we conclude that different uvrD alleles have differential ability to aid in the suppression.
Figure 2.
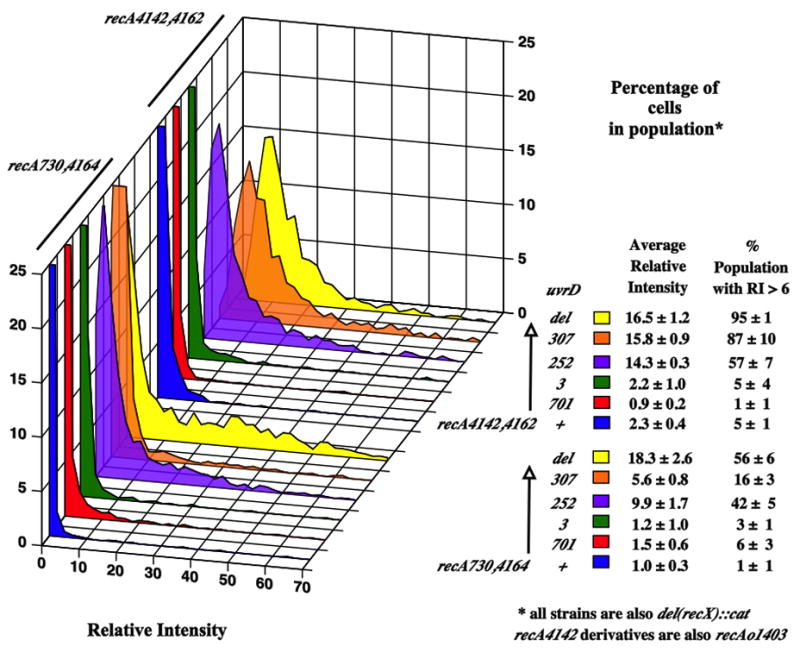
This figure shows the distributions of cells with different levels of constitutive SOS expression (detected as GFP fluorescence) expressed as the percentage of cells in the population. The graphs truncate the percentage of cells at 25%. The strains are in order from top of the graph to the bottom. Unless otherwise indicated, all strains were grown in minimal medium at 37°C with aeration. The strains are: SS7450 {recA4142,4162 del(recX)::cat del(uvrD)}, SS7457 (recA4142,4162 del(recX)::cat uvrD307), SS7459 (recA4142,4162 del(recX)::cat uvrD252), SS7489 (recA4142,4162 del(recX)::cat uvrD3), SS7451 (recA4142,4162 del(recX)::cat uvrD701), SS7449 (recA4142,4162 del(recX)::cat), SS7478{recA730,4164 del(recX)::cat del(uvrD)}, SS7488 (recA730,4164 del(recX)::cat uvrD307), SS7487 (recA730,4164 del(recX)::cat uvrD252), SS7485 (recA730,4164 del(recX)::cat uvrD3), SS7486 (recA730,4164 del(recX)::cat uvrD701), SS7475 (recA730,4164 del(recX)::cat). The SOS expression ratio of the recA+ del(recX)::cat versions of these uvrD mutants are on the order of 1–2 RFI depending on the particular set of mutants (data not shown).
Other mutations in genes required for NER and MMR do not increase SOSC expression in the recA recX double mutants like del(uvrD)
Since it is known that UvrD participates in two DNA repair processes in the cell: NER and MMR and that cells deficient in these processes presumably have higher backgrounds of DNA damage, this in turn could be responsible for the increase in SOSC expression we observed in the recA730,4164 del(recX) del(uvrD) and recA4142,4164 del(recX) del(uvrD) mutants. To test this, we combined null mutations in uvrA, uvrB, mutH, mutL and mutS with recA730,4164 del(recX) and recA4142,4164 del(recX) and measure the level of SOSC expression. Table 4 shows that, relative to the recA+ recX+ strains, there are not any significant increases in SOSC expression when any of the five mutations are added to the two mutants above with the exception of uvrA mutation in the recA730,4164 del(recX) strain. Here, about a two-fold increase is seen. In summary, across the 10 strains (the five pairs of mutants), there is much less SOSC expression than when del(uvrD) is added to these two combinations of mutations (Table 3 and Figure 2). This supports the idea that the increase in SOSC expression seen in recA730,4164 del(recX) del(uvrD) and recA4142,4164 del(recX) del(uvrD) mutants is due to absence of UvrD’s activity in removing RecA from the DNA and not from an increase in background levels of DNA damage.
Table 4.
The effect of NER and MMR mutations on the ability of recA constitutive allele with suppressor to produce SOSC expressiona
| Strain | recA | recX | other | SOSC expression ratioa | % of cells expressing SOSC ≥ 6-fold |
|---|---|---|---|---|---|
| SS5752 | + | + | uvrA | 1.2 ± 0.2 | 2.9 ± 0.5 |
| SS5767 | + | + | uvrB | 0.9 ± 0.1 | 0.5 ± 0.4 |
| SS5949 | + | + | mutL | 1.2 ± 0.3 | 3.4 ± 1.6 |
| SS7604 | + | + | mutH | 1.2 ± 0.3 | 4.3 ± 2.2 |
| SS7605 | + | + | mutS | 1.4 ± 0.5 | 3.0 ± 1.7 |
| SS7615 | 4142,4162 | cat | uvrA | 1.2 ± 0.6 | 4.4 ± 1.8 |
| SS7612 | 4142,4162 | cat | uvrB | 0.8 ± 0.2 | 1.5 ± 1.1 |
| SS7611 | 4142,4162 | cat | mutL | 1.2 ± 0.2 | 4.5 ± 1.4 |
| SS7619 | 4142,4162 | cat | mutH | 1.6 ± 0.6 | 3.1 ± 2.2 |
| SS7617 | 4142,4162 | cat | mutS | 1.4 ± 0.4 | 2.6 ± 0.9 |
| SS7616 | 730,4164 | cat | uvrA | 1.8 ± 0.6 | 6.2 ± 0.5 |
| SS7614 | 730,4164 | cat | uvrB | 1.3 ± 0.2 | 2.1 ± 0.8 |
| SS7613 | 730,4164 | cat | mutL | 1.5 ± 0.4 | 2.4 ± 0.2 |
| SS7620 | 730,4164 | cat | mutH | 1.1 ± 0.3 | 3.7 ± 1.2 |
| SS7618 | 730,4164 | cat | mutS | 1.0 ± 0.1 | 2.1 ± 1.6 |
Same as Table 1. All strains having recA4142 also have recAo1403.
DISCUSSION
Intragenic suppressors of the SOSC phenotype of recA730 have been known for many years, but very little was known about how these mutations suppressed SOSC expression. The amino acid changes of recA4162 (I298V) and recA4164 (L126V) mapped to different parts of RecA and it was not clear if they worked by similar or different mechanisms. This paper shows that the ability of these suppressor mutations to inhibit SOSC is not allele specific, they can suppress in cis or in trans and that they are likely to operate at the level of the filament possibly to better attract and or respond to UvrD and RecX. The results also showed, however, that all alleles were not equally affected by the absence of UvrD and RecX. recA730,4164 and recA4142,4162 revealed little or no increase when either UvrD or RecX were removed, but when both were removed, a fairly large increase in SOSC expression is seen suggesting that the two gene products act independently to suppress SOSC expression. This was not seen, however, with recA4142,4164. Here, recA4142,4164 showed an increase in SOSC expression when UvrD was removed, but did not show an additional increase when RecX was also removed. Lastly, recA730,4162 showed very little increase when either or both UvrD and RecX were removed. Thus, it is very difficult to unambiguously and systematically categorize all these changes since all four recA mutants did not respond equally to the absence of either RecX or UvrD alone or both at once. One reason why all four recA alleles may not have behaved the same with both the uvrD and recX mutations is that some other mechanism may also be operating to inhibit full SOSC expression. This idea is supported by the observations that all four recA mutants did have SOSC expression at the level of a recA730 or recA4142 mutant.
The study of suppressors, whether intragenic or extragenic, has long been a potent tool to investigate molecular mechanisms. The suppressors here serve to suppress the ability of two recAC mutants to turn on the SOS Response when they should not. The suppressor mutations by themselves, or in conjunction with the recAC mutations, do not inhibit RecA’s normal ability to function in any way that we have tried to measure (Table 2 and data not shown). Thus, the only function of the suppressor is to inhibit the ability of the recAC mutation to turn on the SOS Response when it should not. These suppressors, however, are very selective in their ability to repress SOSC expression, they only do so in log phase cells in the absence of external DNA damage. When the cells are treated with UV light, SOS is normally induced in the suppressor strains, whether they are single mutants, in cis or in trans with the recAC allele. Their ability to discriminate between the two situations suggests that recAC mutants binding to DNA in vivo is somehow different from that when normal SOS is induced. Perhaps the DNA substrate, its location or context in the cell is different.
How might the suppressors function? Evidence supports the notion that not all RecA filaments in the cell induce the SOS Response. It is known that about 15% of cells in a log phase population have RecA filaments as measured by RecA-GFP formation or the effects of RecA-mediated of recombination, but less than about 0.5% of the population are induced for SOS ((McCool et al., 2004; Pennington and Rosenberg, 2007; Renzette et al., 2005; Steiner and Kuempel, 1998) and summarized in (Long et al., 2008)). Thus, there must be some difference between the filaments that induce the SOS response and those that form in log phase cells presumably to fix DNA damage caused by normal metabolic functions. While the specific differences are not known, it has been hypothesized that the SOS inducing filaments could be longer or have a slightly different conformation. Here, it is further suggested that RecA filaments may have slightly different forms as the cell progresses through the stages of the SOS response. In particular, recAC mutants may resemble those filaments that induce the SOS Response (after UV treatment) and this is the reason why when they are loaded onto the DNA, they promote LexA auto-cleavage. As the damage is repaired, the SOS Response should decrease. A necessary part of this is the disassembly of the RecA filaments. It is possible that at this point, the RecA filaments adopt a slightly different structure making it more susceptible to the destabilizing and dismantling effects of RecX and UvrD. The fact that uvrD and recX are induced during the SOS response and would be in higher quantities at the end of the SOS response may also aid in this process. Since the suppressor mutations (recA4162 and recA4164) are dominant to the recAC mutations (recA730 and recA4142) in cis or in trans, it suggests that turning off SOS is dominant to turning it on. This seems like an appropriate way to prevent SOS expression unless it is absolutely necessary.
There were significant differences between the abilities of the different alleles of uvrD to aid in the suppression. Each of the uvrD mutants tested has been characterized for standard uvrD phenotypes (the uvrD deletion mutant is UVS, hyper-rec and a mutator; see above). uvrD701 is missing the C-terminal forty amino acids and behaves as a monomer instead of a dimer. It is has wild type activity both in vitro and in vivo (Centore et al., 2009; Mechanic et al., 1999). uvrD307 (R284A) is mutant in a highly conserved helicase motif close to the P-loop motif. Genetic studies show that this mutant is UVS, hyper-rec and a mutator like a uvrD deletion mutant (Hall and Matson, 1997; Zhang et al., 1997). In vitro, it has ability to bind DNA like wild type, but has a greatly decreased ability to bind ATP and almost no helicase activity. The crystal structure of UvrD (Lee and Yang, 2006) shows that uvrD252 (G30D) (also called recL152 (Rothman and Fried, 1984)) is located very close to the uvrD307 (R284A) mutation and the P-loop motif. In vivo, uvrD252 mutants are UVS. The literature is, however, controversial on the mutator and hyper-rec phenotypes of uvrD252. It has reported that uvrD252 strains have the ability to act with wild type (Washburn and Kushner, 1991), low (Arthur and Lloyd, 1980) or high (Marinus, 1980) mutator activity. Similarly, the hyper-rec phenotype has been reported to be either low ((Zieg et al., 1978) and our unpublished results) or high (Arthur and Lloyd, 1980). Although here, the methods for measuring the hyper-rec phenotype were quite different. In the former case, recombinants were measured and in the latter case, transcription from recombinational intermediates was measured. This mutant also maintains the ability to ameliorate the negative effects of recombination proteins at certain types of stopped replication forks (Lestini and Michel, 2007, 2008). In vitro, it has reduced ATP binding and helicase activity (Washburn and Kushner, 1993). uvrD3 (E387K) is located in the 2B domain thought to be important in the regulation of the helicase activity (Brendza et al., 2005). uvrD3 (E387K) mutants are dominant, UVS and do not display a mutator, but have a hyper-rec phenotype (Maples and Kushner, 1982; Marinus, 1980; Ogawa et al., 1968; Zieg et al., 1978). Thus the data is consistent with the interpretation that uvrD3 is a partial activity mutant that can still undo SOSC RecA filaments, but cannot undo RecA filaments that lead to the hyper-rec phenotype. UvrD252 is then a mutant of the opposite type; it can still undo recombinational intermediates (this assumes that uvrD252 is not hyper-rec and that Arthur and Lloyd measured some other phenotype of uvrD252 with their assay), but not SOSC RecA filaments. Lastly, UvrD307 has lost both abilities. Biochemically, the inability to aid recA4162 or recA4164 in suppression of SOSC expression correlates with the low levels of helicase activity, the inability to bind ATP and the region of the protein that binds ATP. It is therefore possible that UvrD exerts its destabilizing effect through its helicase activity or through a special conformation it might adopt when bound with a nucleotide.
Lastly, it was shown that an inability to do NER or MMR through mutations in uvrA, uvrB, mutH, mutL and mutS does not increase the ability of recA730,4164 del(recX) and recA4142,4164 del(recX) to produce SOSC expression like a del(uvrD) mutation. This supports the idea that UvrD has a specific role in repression of the SOSC expression and this increase does not correspond to a lack of DNA repair capacity in cells lacking the NER and MMR pathways. It is also known that UvrAB and MutL can aid UvrD in loading onto certain substrates and augment its activity (reviewed in (Matson and Robertson, 2006) and (Atkinson et al., 2009)). Since uvrA, uvrB and mutL mutants do not display high levels of SOSC like del(uvrD) mutants, one can speculate that either UvrD has an UvrAB-MutL-independent method to load onto the DNA or it does not need to load onto the DNA to repress SOSC expression. Further experiments will be necessary to test these ideas.
EXPERIMENTAL PROCEEDURES
Bacterial strains
All bacterial strains used in this work are derivatives of E. coli K-12 and are described in Table 5. The protocol for P1 transduction has been described elsewhere (Willetts et al., 1969). All P1 transductions were selected on 2% agar plates containing either minimal or rich media. Where appropriate plates also contained the following antibiotics at these final concentrations: tetracycline 10 μg ml−1, chloramphenicol 25 μg ml−1 or kanamycin 50 μg ml−1. All transductants were purified on the same type of media on which they were selected. When necessary, the recA alleles were placed on the chromosome in the place of recA+ as previously described (see below). Table 2 shows the characterization of these mutants for their survival to UV irradiation, ability to inherit markers during conjugation and the ability to induce the SOS response. Specific protocols for these tests have been previously described (Sandler et al., 1996).
Table 5.
Strains used in this work
| Strain | ygaD | recAo | recA | RecX | uvrD | attλ | Other relevant genotype | Origin of reference |
|---|---|---|---|---|---|---|---|---|
| CAG18491 | + | + | + | + | + | + | metE3079::Tn10 | E.coli Stock Center |
| DM1187 | + | + | 730,4162 (recA441) | + | + | + | D. Mount | |
| GY8382 | + | + | 938::cat | + | + | + | B. Michel | |
| JC13509a | + | + | + | + | + | + | Lab Stock | |
| JJC7643 | + | + | + | + | 307 | + | yigE::cat | B. Michel |
| K253 | + | + | 730,4164 (recA718) | + | + | + | srlC300::Tn10 | M. Volkert |
| KL880 | + | + | + | + | 252 | + | B. Low | |
| N14–4 | + | + | + | + | 3 | + | B. Low | |
| N3055 | + | + | + | + | + | + | uvrA277::Tn10 | R.G. Lloyd |
| SS391 | + | + | 938::cat | + | + | + | GY8382→JC13509c | |
| SS996 | + | + | + | + | + | Ωgfp | (McCool et al., 2004) | |
| SS1054 | + | + | + | + | + | + | metE3079::Tn10 | (Centore et al., 2009) |
| SS1436 | + | + | 938::cat | + | + | Ωgfp | (McCool et al., 2004) | |
| SS4070 | + | + | + | + | + | + | del(mutL)100::kan | (Baba et al., 2006) |
| SS4073 | + | + | + | + | + | + | del(mutH)100::kan | (Baba et al., 2006) |
| SS4539 | + | + | + | + | 252 | + | KL880→SS1054g | |
| SS4540 | + | + | + | + | 3 | + | N14–4→SS1054g | |
| SS4558 | + | + | + | + | 252 | Ωgfp | gal-76::Tn10 | SS1465→SS4539c |
| SS4559 | + | + | + | + | 3 | Ωgfp | gal-76::Tn10 | SS1465→SS4540c |
| SS4570 | + | + | + | + | kan | + | (Baba et al., 2006) | |
| SS4626 | + | + | + | + | + | Ωgfp | zfj-3131::Tn10 alaS5 | (Long et al., 2008) |
| SS4629 | + | + | 730 | + | + | Ωgfp | (Long et al., 2008) | |
| SS4630 | + | + | 730,4162 | + | + | Ωgfp | DM1187→SS4626e | |
| SS4976 | kan | o1403 | 4142 | + | + | Ωgfp | (Long et al., 2008) | |
| SS5130 | + | + | + | + | + | + | del(uvrB)100::kan | (Baba et al., 2006) |
| SS5802 | + | + | + | + | + | Ωgfp | metE3079::Tn10 | CAG18491→SS996c |
| SS5292 | + | + | 730,4164 | + | + | Ωgfp | srlC300::Tn10 | K253→SS996c |
| SS5296 | kan | + | 803,4162 | + | + | Ωgfp | Lab Stock | |
| SS5376 | + | + | 730 | + | + | Ωgfp | pDPT429→SS4629d | |
| SS5377 | kan | o1403 | 4142 | + | + | Ωgfp | pDPT429→SS4976d | |
| SS5737 | + | + | + | + | 701 | Ωgfp | gal-76::Tn10 | (Centore et al., 2009) |
| SS5752 | + | + | + | + | + | Ωgfp | uvrA277::Tn10 | N3055→SS996c |
| SS5753 | + | + | + | + | 307 | Ωgfp | yigE::cat | JJC2643→SS5802g |
| SS5767 | + | + | + | + | + | Ωgfp | del(uvrB)100::kan | SS5130→SS996b |
| SS5949 | + | + | + | + | + | Ωgfp | del(mutL)100::kan | SS4070→SS996b |
| SS5958 | + | + | del(recA)100::kan | + | + | Ωgfp | SS4952→SS996b | |
| SS6013 | kan | + | 4142 | + | + | Ωgfp | SS6009→SS996h | |
| SS6089 | kan | o1403 | 4142,4162 | + | + | Ωgfp | SS5323→SS996b, h | |
| SS6027 | kan | + | 4164 | + | + | Ωgfp | SS6026→SS996b, h | |
| SS6052 | kan | + | 4162 | + | + | Ωgfp | SS6430→SS4630b, h | |
| SS6062 | kan | o1403 | 4142,4164 | + | + | Ωgfp | SS6444→SS996b, h | |
| SS6074 | + | + | 730 | + | + | Ωgfp | pNR122→SS4629d | |
| SS6075 | kan | o1403 | 4142,4164 | + | + | Ωgfp | pNR122→SS4976d | |
| SS6076 | kan | o1403 | 4142,4164 | + | + | Ωgfp | pNR123→SS4976d | |
| SS6077 | + | + | 730 | + | + | Ωgfp | pNR123→SS4629d | |
| SS6078 | kan | o1403 | 4142,4164 | + | + | Ωgfp | pNR124→SS4976d | |
| SS6079 | + | + | 730 | + | + | Ωgfp | pNR124→SS4629d | |
| SS6080 | + | + | + | cat | + | Ωgfp | (Long et al., 2008) | |
| SS6081 | kan | + | 4162 | + | + | Ωgfp | pEL16→SS6052d | |
| SS6082 | kan | + | 4164 | + | + | Ωgfp | pEL16→SS6027d | |
| SS6085 | kan | + | 4164 | + | + | Ωgfp | pNR127→SS6027d | |
| SS6086 | kan | + | 4162 | + | + | Ωgfp | pNR127→SS6052d | |
| SS6118 | + | + | del | + | + | Ωgfp | pNR127→SS5958d | |
| SS6100 | + | + | del | + | + | Ωgfp | pEL16→SS5958d | |
| SS7125 | + | + | + | + | kan | Ωgfp | SS4570→SS996b | |
| SS7139 | + | + | + | + | del | Ωgfp | SS7134i | |
| SS7434 | kan | 1403 | 4142,4162 | + | del | Ωgfp | SS5323→SS7139b | |
| SS7449 | kan | 1403 | 4142,4162 | cat | + | Ωgfp | SS7444→SS996f | |
| SS7450 | kan | 1403 | 4142,4162 | cat | del | Ωgfp | SS7444→SS7139f | |
| SS7451 | kan | 1403 | 4142,4162 | cat | 701 | Ωgfp | gal-76::Tn10 | SS7444→SS5737f |
| SS7457 | kan | 1403 | 4142,4162 | cat | 307 | Ωgfp | yigE::cat | SS7444→SS5753f |
| SS7459 | kan | 1403 | 4142,4162 | cat | 252 | Ωgfp | SS7444→SS4558f | |
| SS7463 | + | + | 730,4164 | + | del | Ωgfp | srlC300::Tn10 | K253→SS7139c |
| SS7464 | + | + | 730,4162 | + | kan | Ωgfp | SS4570→SS4630b | |
| SS7465 | kan | 1403 | 4142,4164 | + | del | Ωgfp | SS6444→SS7139b | |
| SS7467 | + | + | + | cat | del | Ωgfp | SS6080→SS7139f | |
| SS7468 | + | + | + | cat | 252 | Ωgfp | SS6080→SS4558f | |
| SS7470 | + | + | + | cat | 701 | Ωgfp | SS6080→SS7139f | |
| SS7474 | + | + | 730,4162 | cat | + | Ωgfp | SS7471→SS996j, f | |
| SS7475 | + | + | 730,4164 | cat | + | Ωgfp | srlC300::Tn10 | SS7472→SS996j, f |
| SS7476 | kan | 1403 | 4142,4164 | cat | + | Ωgfp | SS7473→SS996j, f | |
| SS7477 | + | + | 730,4162 | cat | del | Ωgfp | SS7471→SS7139f | |
| SS7478 | + | + | 730,4164 | cat | del | Ωgfp | srlC300::Tn10 | SS7472→SS7139f |
| SS7479 | kan | 1403 | 4142,4164 | cat | del | Ωgfp | SS7473→SS7139f | |
| SS7480 | + | + | + | cat | + | Ωgfp | metE3079::Tn10 | CAG18491→SS6080c |
| SS7481 | + | + | + | cat | 307 | Ωgfp | yigE::cat | JJC7643→SS7480g |
| SS7485 | + | + | 730,4164 | cat | 3 | Ωgfp | SS7472→SS4559f | |
| SS7486 | + | + | 730,4164 | cat | 701 | Ωgfp | SS7472→SS5737f | |
| SS7487 | + | + | 730,4164 | cat | 252 | Ωgfp | SS7472→SS4558f | |
| SS7488 | + | + | 730,4164 | cat | 307 | Ωgfp | yigE::cat srlC300::Tn10 | SS7472→SS5753c |
| SS7489 | kan | 1403 | 4142,4162 | cat | 3 | Ωgfp | SS7444→SS4559f | |
| SS7604 | + | + | + | + | + | Ωgfp | del(mutH)100::kan | SS4073→SS996b |
| SS7605 | + | + | + | + | + | Ωgfp | del(mutS)100::kan | SS4882→SS996b |
| SS7611 | kan | o1403 | 4142,4162 | cat | + | Ωgfp | del(mutL)100::kan | SS7444→SS5949j, f |
| SS7612 | kan | o1403 | 4142,4162 | cat | + | Ωgfp | del(uvrB)100::kan | SS7444→SS5767f |
| SS7613 | + | + | 730,4164 | cat | + | Ωgfp | del(mutL)100::kan srlC300::Tn10 | SS7472→SS5949j, f |
| SS7614 | + | + | 730,4164 | cat | + | Ωgfp | del(uvrB)100::kan srlC300::Tn10 | SS7472→SS5767f |
| SS7615 | kan | 1403 | 4142,4162 | cat | + | Ωgfp | del(uvrA)100::kan | SS7444→SS5752f |
| SS7616 | + | + | 730,4164 | cat | + | Ωgfp | del(uvrA)100::kan srlC300::Tn10 | SS7472→SS5752f |
| SS7617 | kan | 1403 | 4142,4162 | cat | + | Ωgfp | del(mutS)100::kan | SS7444→SS7605f |
| SS7618 | + | + | 730,4164 | cat | + | Ωgfp | del(mutS)100::kan srlC300::Tn10 | SS7472→SS7605f |
| SS7619 | kan | 1403 | 4142,4162 | cat | + | Ωgfp | del(mutH)100::kan | SS7444→SS7604f |
| SS7620 | + | + | 730,4164 | cat | + | Ωgfp | del(mutH)100::kan srlC300::Tn10 | SS7472→SS7604f |
JC13509 has the following genotype: sulB103 lacMS286 φ 80dIIlacBK1 argE3 hi-4 thi-1 xyl-5 mtl-1 rpsL31 tsx. The lacMS286φ80dIIlacBK1 code for two partial non-overlapping deletions of the lac operon (Konrad, 1977; Zieg and Kushner, 1977). Full notation for ygaD mutation is ygaD1::kan. Full notation for Ωgfp mutation is Δattλ::sulApΩgfp-mut2.
Select for KanR and then screen for other marker phenotypically or by PCR.
Select for TetR and then screen for other marker phenotypically or by PCR.
Select for AmpR
Select for AlaS+
Select for CatR
Select for Met+
These recAo or recA mutations were first constructed on a plasmid as described in the Materials and Methods. They were then transferred to the chromosome using the method of Datsenko and Wanner (Datsenko and Wanner, 2000) using a strain that was lexA3 malE::Tn10 in a JC13509 background with pKD46 encoding exo and bet. This original combination of mutants were named and saved as the strain indicated as the donor in this cross.
This deletion allele was created by first transducing the kan resistant derivative from the Keio collection into the strain as indicated in the reference column. pLH29, carrying the flp gene, was then introduced and Kan sensitive derivatives were screened.
recX::cat was amplified with prSJS748,749 using pACYC184 (New England Biolabs) as a template. recX::cat was transferred to the chromosome using the exo-bet method next to the recA allele indicated. This original combination of mutants were named and saved as the strain indicated as the donor in this cross.
Construction of plasmids
All plasmids used in this work are derivatives of a low copy number vector called pDPT429 (Taylor and Cohen, 1979). All plasmids have a 4.5 kb BamHI fragment cloned into the BamHI site of pDPT429 in the same orientation. The BamHI fragment contain ygaD1::kan {kan gene inserted at BsiWI site (Renzette et al., 2005)}, recA (some contain different alleles as indicated) and recX genes. In all cases where the construction called for PCR amplification, these sequences were confirmed by DNA sequence analysis.
To create the plasmid pNR122 (containing recA+), pNR117 and pDPT429 was restricted with BamHI. pNR117 is a derivative of pBR322 with the 4.5 kb BamHI fragment containing ygaD1::kan recAo+ recA+ recX+. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA+. This plasmid is called pNR122.
To create the plasmid pNR123 (containing recA4162), pRecAN99 (gift from Steve Kowalczykowski) (Mirshad and Kowalczykowski, 2003) and pSJS1373 (like pNR117 but with recA803) were first restricted with RsrII and PmeI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA803, N99. This plasmid is called pNR64. The recAI298V fragment was then amplified using prSJS453 (5′ GAAATCTACGGACCGGAATCTTCCGG3′) and prSJS472 (5′ TCTTCTCCTTTACTGATGCTCCCAAAATCTTCGTTAGTTTCTGC3′) with SS5296 as the template DNA. The resulting fragment was then restricted with RsrII and KpnI and ligated into the same sites of pNR64 to produce a plasmid containing ygaD1::kan recA803,4162. This plasmid is called pNR102. pNR102 and pNR115 was then restricted with RsrII and KpnI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA4162. This plasmid is called pNR118. pNR118 and pDPT429 were then restricted with BamHI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA4162. This plasmid is called pNR123.
To create the plasmid pNR124 (containing recA4164), pSJS1337 (Renzette and Sandler, 2008) and pJC869 (Madiraju et al., 1988) was first restricted with HindIII and SphI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing recA803,2201. This plasmid is called pNR53. pNR53 and pSJS1373 was then restricted with RsrII and PmeI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA803,2201. This plasmid is called pEL12. The recA4164 fragment was then amplified using pSJS453 and prSJS472 with SS5292 as the template DNA. The resulting fragment was then restricted with RsrII and BlpI and ligated into the same sites of pEL12 to produce a plasmid containing ygaD1::kan recA803,4164. This plasmid is called pNR106. pNR106 and pNR115 were then restricted with RsrII and KpnI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA4164. This plasmid is called pNR119. pNR119 and pDPT429 were then restricted with BamHI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA4164. This plasmid is called pNR124.
To create pEL16 (containing recA4142), pSJS1373 and pDPT429 were restricted with BamHI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA803. This plasmid is called pNR105. pNR105 and pNR115 (Long et al., 2008) was then restricted with RsrII and KpnI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA recX. This plasmid is called pNR117. pNR115 and pNR117 was then restricted with BpII and RsrII. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA4142 recX. This plasmid is called pEL15. pEL15 and pDPT429 were then restricted with BamHI. The appropriate fragments were isolated, mixed and treated to produce a plasmid containing ygaD1::kan recA4142 recX. This plasmid is called pEL16.
To create the plasmid pNR127 (containing recA730), pEAW305 (gift from Mike Cox) and pSJS1373 were restricted with NcoI and PmeI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA730. This plasmid is called pNR59. pNR59 and pDPT429 were then restricted with BamHI. The appropriate fragments were isolated, mixed and treated with DNA ligase to produce a plasmid containing ygaD1::kan recA730. This plasmid is called pNR127.
Acknowledgments
This work was supported by AI059027 from the National Institutes of Health. We would like to thank Brooks Low, Mike Volkert, Mike Cox and Steve Kowalczykowski for strains and plasmids and Benedicte Michel and Kunal Baxi for reading the manuscript before publication and offering helpful suggestions.
References
- Arthur HM, Lloyd RG. Hyper-recombination in uvrD mutants of Escherichia coli K-12. Mol Gen Genet. 1980;180:185–191. doi: 10.1007/BF00267368. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Guy CP, Cadman CJ, Moolenaar GF, Goosen N, McGlynn P. Stimulation of UvrD Helicase by UvrAB. J Biol Chem. 2009;284:9612–9623. doi: 10.1074/jbc.M808030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitin DM, Gruenig MC, Cox MM. SSB antagonizes RecX-RecA interaction. J Biol Chem. 2008;283:14198–14204. doi: 10.1074/jbc.M801511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork JM, Cox MM, Inman RB. RecA protein filaments disassemble in the 5′ to 3′ direction on single- stranded DNA. J Biol Chem. 2001;276:45740–45743. doi: 10.1074/jbc.M109247200. [DOI] [PubMed] [Google Scholar]
- Brendza KM, Cheng W, Fischer CJ, Chesnik MA, Niedziela-Majka A, Lohman TM. Autoinhibition of Escherichia coli Rep monomer helicase activity by its 2B subdomain. Proc Natl Acad Sci U S A. 2005;102:10076–10081. doi: 10.1073/pnas.0502886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore RC, Sandler SJ. UvrD limits the number and intensities of RecA-green fluorescent protein structures in Escherichia coli K-12. J Bacteriol. 2007;189:2915–2920. doi: 10.1128/JB.01777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore RC, Leeson MC, Sandler SJ. UvrD303, a hyper-helicase mutant that antagonizes RecA-dependent SOS expression by a mechanism that depends on its C-terminus. J Bacteriol. 2009;191:1429–1438. doi: 10.1128/JB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–484. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ. Homologous genetic recombination: the pieces begin to fall into place. Crit Rev Microbiol. 1994;20:125–142. doi: 10.3109/10408419409113552. [DOI] [PubMed] [Google Scholar]
- Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees JC, Lusetti SL, Chitteni-Pattu S, Inman RB, Cox MM. A RecA filament capping mechanism for RecX protein. Mol Cell. 2004a;15:789–798. doi: 10.1016/j.molcel.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Drees JC, Lusetti SL, Cox MM. Inhibition of RecA protein by the Escherichia coli RecX protein: modulation by the RecA C terminus and filament functional state. J Biol Chem. 2004b;279:52991–52997. doi: 10.1074/jbc.M409050200. [DOI] [PubMed] [Google Scholar]
- Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- Flores MJ, Sanchez N, Michel B. A fork-clearing role for UvrD. Mol Microbiol. 2005;57:1664–1675. doi: 10.1111/j.1365-2958.2005.04753.x. [DOI] [PubMed] [Google Scholar]
- Hall MC, Matson SW. Mutation of a highly conserved arginine in motif IV of Escherichia coli DNA helicase II results in an ATP-binding defect. J Biol Chem. 1997;272:18614–18620. doi: 10.1074/jbc.272.30.18614. [DOI] [PubMed] [Google Scholar]
- Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, Richard GF. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Biol. 2009;16:159–167. doi: 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby EP, Jacob F, Goldthwait DA. Prophage induction and filament formation in a mutant strain of Escherichia coli. Proc Natl Acad Sci U S A. 1967;58:1903–1910. doi: 10.1073/pnas.58.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight KL, Aoki KH, Ujita EL, McEntee K. Identification of the amino acid substitutions in two mutant forms of the recA protein from Escherichia coli: recA441 and recA629. J Biol Chem. 1984;259:11279–11283. [PubMed] [Google Scholar]
- Konrad EB. Method for the isolation of Escherichia coli mutants with enhanced recombination between chromosomal duplications. J Bacteriol. 1977;130:167–172. doi: 10.1128/jb.130.1.167-172.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestini R, Michel B. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007;26:3804–3814. doi: 10.1038/sj.emboj.7601804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestini R, Michel B. UvrD and UvrD252 counteract RecQ, RecJ, and RecFOR in a rep mutant of Escherichia coli. J Bacteriol. 2008;190:5995–6001. doi: 10.1128/JB.00620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW. Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie. 1991;73:411–421. doi: 10.1016/0300-9084(91)90108-d. [DOI] [PubMed] [Google Scholar]
- Long JE, Renzette N, Centore RC, Sandler SJ. Differential requirements of two recA mutants for constitutive SOS expression in Escherichia coli K-12. PLoS ONE. 2008;3:e4100. doi: 10.1371/journal.pone.0004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- Lusetti SL, Hobbs MD, Stohl EA, Chitteni-Pattu S, Inman RB, Seifert HS, Cox MM. The RecF protein antagonizes RecX function via direct interaction. Mol Cell. 2006;21:41–50. doi: 10.1016/j.molcel.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macris MA, Sung P. Multifaceted role of the Saccharomyces cerevisiae Srs2 helicase in homologous recombination regulation. Biochem Soc Trans. 2005;33:1447–1450. doi: 10.1042/BST0331447. [DOI] [PubMed] [Google Scholar]
- Madiraju MVVS, Templin A, Clark AJ. Properties of a mutant recA-encoded protein which reveal a possible role for Escherichia coli recF-encoded protein in genetic recombination. Proc Natl Acad Sci USA. 1988;85:6592–6569. doi: 10.1073/pnas.85.18.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maples VF, Kushner SR. DNA repair in Escherichia coli: identification of the uvrD gene product. Proc Natl Acad Sci U S A. 1982;79:5616–5620. doi: 10.1073/pnas.79.18.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinus MG. Influence of uvrD3, uvrE502, and recL152 mutations on the phenotypes of Escherichia coli K-12 dam mutants. J Bacteriol. 1980;141:223–226. doi: 10.1128/jb.141.1.223-226.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson SW, Robertson AB. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res. 2006;34:4089–4097. doi: 10.1093/nar/gkl450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall JO, Witkin EM, Kogoma T, Roegner-Maniscalco V. Constitutive expression of the SOS response in recA718 mutants of Escherichia coli requires amplification of RecA718 protein. J Bacteriol. 1987;169:728–734. doi: 10.1128/jb.169.2.728-734.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool JD, Long E, Petrosino JF, Sandler HA, Rosenberg SM, Sandler SJ. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol Microbiol. 2004;53:1343–1357. doi: 10.1111/j.1365-2958.2004.04225.x. [DOI] [PubMed] [Google Scholar]
- McGrew DA, Knight KL. Molecular design and functional organization of the RecA protein. Crit Rev Biochem Mol Biol. 2003;38:385–432. doi: 10.1080/10409230390242489. [DOI] [PubMed] [Google Scholar]
- Mechanic LE, Hall MC, Matson SW. Escherichia coli DNA helicase II is active as a monomer. J Biol Chem. 1999;274:12488–12498. doi: 10.1074/jbc.274.18.12488. [DOI] [PubMed] [Google Scholar]
- Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Mirshad JK, Kowalczykowski SC. Biochemical characterization of a mutant RecA protein altered in DNA-binding loop 1. Biochemistry. 2003;42:5945–5954. doi: 10.1021/bi027233i. [DOI] [PubMed] [Google Scholar]
- Mishra S, Mazumdar PA, Dey J, Das AK. Molecular modeling of RecX reveals its mode of interaction with RecA. Biochem Biophys Res Commun. 2003;312:615–622. doi: 10.1016/j.bbrc.2003.10.164. [DOI] [PubMed] [Google Scholar]
- Nastri HG, Guzzo A, Lange CS, Walker GC, Knight KL. Mutational analysis of the RecA protein L1 region identifies this area as a probable part of the co-protease substrate binding site. Mol Microbiol. 1997;25:967–978. doi: 10.1111/j.1365-2958.1997.mmi533.x. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Shimada K, Tomizawa J. Studies on radiation-sensitive mutants of E. coli. I. Mutants defective in the repair synthesis. Mol Gen Genet. 1968;101:227–244. doi: 10.1007/BF00271625. [DOI] [PubMed] [Google Scholar]
- Pages V, Koffel-Schwartz N, Fuchs RP. recX, a new SOS gene that is co-transcribed with the recA gene in Escherichia coli. DNA Repair (Amst) 2003;2:273–284. doi: 10.1016/s1568-7864(02)00217-3. [DOI] [PubMed] [Google Scholar]
- Pennington JM, Rosenberg SM. Spontaneous DNA breakage in single living Escherichia coli cells. Nature Genetics. 2007;39:797–802. doi: 10.1038/ng2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragone S, Maman JD, Furnham N, Pellegrini L. Structural basis for inhibition of homologous recombination by the RecX protein. EMBO J. 2008;27:2259–2269. doi: 10.1038/emboj.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, Sandler SJ. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol Microbiol. 2005;57:1074–1085. doi: 10.1111/j.1365-2958.2005.04755.x. [DOI] [PubMed] [Google Scholar]
- Renzette N, Sandler SJ. Requirements for ATP binding and hydrolysis in RecA function in Escherichia coli. Mol Microbiol. 2008;67:1347–1359. doi: 10.1111/j.1365-2958.2008.06130.x. [DOI] [PubMed] [Google Scholar]
- Rothman RH, Fried B. Long repair replication patches are produced by the short-patch pathway in a uvrD252 (recL152) mutant of Escherichia coli K-12. J Bacteriol. 1984;158:749–753. doi: 10.1128/jb.158.2.749-753.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Samra HS, Clark AJ. Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics. 1996;143:5–13. doi: 10.1093/genetics/143.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Bork JM, Webb BL, Inman RB, Cox MM. RecA Protein Filaments: End-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol. 1997;265:519–540. doi: 10.1006/jmbi.1996.0748. [DOI] [PubMed] [Google Scholar]
- Skiba MC, Knight KL. Functionally important residues at a subunit interface site in the RecA protein from Escherichia coli. J Biol Chem. 1994;269:3823–3828. [PubMed] [Google Scholar]
- Steiner WW, Kuempel PL. Sister chromatid exchange frequencies in Escherichia coli analyzed by recombination at the dif resolvase site. Journal of Bacteriology. 1998;180:6269–6275. doi: 10.1128/jb.180.23.6269-6275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Brockman JP, Burkle KL, Morimatsu K, Kowalczykowski SC, Seifert HS. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem. 2003;278:2278–2285. doi: 10.1074/jbc.M210496200. [DOI] [PubMed] [Google Scholar]
- Story RM, Steitz TA. Structure of the RecA protein-ADP complex. Nature. 1992;355:374–376. doi: 10.1038/355374a0. [DOI] [PubMed] [Google Scholar]
- Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monmer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- Taylor DP, Cohen SN. Structural and functional analysis of cloned DNA segments containing the replication and incompatibility regions of a miniplasmid derived from a copy number mutant of NR1. J Bacteriol. 1979;137:92–104. doi: 10.1128/jb.137.1.92-104.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessman ES, Peterson P. Plaque color method for rapid isolation of novel recA mutants of Escherichia coli K-12: new classes of protease-constitutive recA mutants. J Bacteriol. 1985;163:677–687. doi: 10.1128/jb.163.2.677-687.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanLoock MS, Yu X, Yang S, Galkin VE, Huang H, Rajan SS, Anderson WF, Stohl EA, Seifert HS, Egelman EH. Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J Mol Biol. 2003;333:345–354. doi: 10.1016/j.jmb.2003.08.053. [DOI] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, Fabre F, Petit MA. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–189. doi: 10.1038/sj.emboj.7600485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker G. In: The SOS Response of Escherichia coli Escherichia coli and Salmonella. Neidhardt FC, editor. Vol. 1. Washington, D. C: American Society of Microbiology; 1996. pp. 1400–1416. [Google Scholar]
- Washburn BK, Kushner SR. Construction and analysis of deletions in the structural gene (uvrD) for DNA helicase II of Escherichia coli. J Bacteriol. 1991;173:2569–2575. doi: 10.1128/jb.173.8.2569-2575.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn BK, Kushner SR. Characterization of DNA helicase II from a uvrD252 mutant of Escherichia coli. J Bacteriol. 1993;175:341–350. doi: 10.1128/jb.175.2.341-350.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertman KF, Mount DW. Nucleotide sequence binding specificity of the LexA repressor of Escherichia coli K-12. J Bacteriol. 1985;163:376–384. doi: 10.1128/jb.163.1.376-384.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts NS, Clark AJ, Low B. Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. J Bacteriol. 1969;97:244–249. doi: 10.1128/jb.97.1.244-249.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin EM, McCall JO, Volkert MR, Wermundsen IE. Constitutive expression of SOS functions and modulation of mutagenesis resulting from resolution of genetic instability at or near the recA locus of Escherichia coli. Mol Gen Genet. 1982;185:43–50. doi: 10.1007/BF00333788. [DOI] [PubMed] [Google Scholar]
- Zhang G, Deng E, Baugh LR, Hamilton CM, Maples VF, Kushner SR. Conserved motifs II to VI of DNA helicase II from Escherichia coli are all required for biological activity. J Bacteriol. 1997;179:7544–7550. doi: 10.1128/jb.179.23.7544-7550.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieg J, Kushner SR. Analysis of genetic recombination between two partially deleted lactose operons of Escherichia coli K-12. J Bacteriol. 1977;131:123–132. doi: 10.1128/jb.131.1.123-132.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieg J, Maples VF, Kushner SR. Recombinant levels of Escherichia coli K-12 mutants deficient in various replication, recombination, or repair genes. J Bacteriol. 1978;134:958–966. doi: 10.1128/jb.134.3.958-966.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]