

Abstract
The review summarizes the most recent achievements in structure–activity relationship (SAR) studies of tariquidar and its analogs. Tariquidar is one of the most promising representatives of the third generation of multidrug resistance (MDR) modulators created so far. This fact determines the strong interest of different research groups in the development of tariquidar-like structures as selective inhibitors of MDR transporters in resistant human cancer cells. After the discovery of tariquidar, a number of analogs have been synthesized and pharmacologically tested, thus supplying good data for comprehensive analyses of their structure–activity relationships. In the review, the structural and pharmacological data of newly synthesized tariquidar-like compounds are first presented. Next, the main achievements in the SAR studies are described focusing on two main transport proteins: P-glycoprotein and breast cancer resistance protein. The reported results are discussed from the point of view of their significance and importance for future directions in the rational design of effective MDR modulators.
Key words: BCRP, modeling, multidrug resistance, pharmacophore, P-glycoprotein, QSAR
INTRODUCTION
Multidrug resistance (MDR) in tumor cells has a significant impact on the efficacy of cancer chemotherapy and appears as a major obstacle in the modern cancer treatment. MDR is mainly related to the expression of the ATP-binding cassette (ABC) transporters. These proteins actively transport a wide variety of structurally different substrates out of the tumor cells, thereby decreasing their intracellular concentrations. So far, 49 members of the ABC superfamily in humans have been identified and classified into seven subfamilies (coded by the letters A to G) based on phylogenetic similarity (1). Among them, three proteins from the B, C, and G subfamilies have been primarily associated with the MDR phenomenon: P-glycoprotein, P-gp (ABCB1); the multidrug resistance-associated protein, MRP1 (ABCC1); and the breast cancer resistance protein, BCRP (ABCG2) (2,3). These transporters can simultaneously be overexpressed in tumor cells, thus outlining MDR as a multifactorial problem for the treatment of cancer.
P-gp is perhaps the best-known MDR protein. It is the first ABC efflux transporter that has been initially discovered in resistant CHO cells (4) and found later in various resistant tumor cell lines of animal and human origin (5). The protein is also naturally expressed in many tissues with barrier functions such as liver, BBB, kidney, and intestine. A large number of compounds have been reported to be its substrates and/or inhibitors, including many cytotoxic anticancer drugs such as anthracylines, Vinca alkaloids, taxanes, and epipodophyllotoxins (6). After P-gp, MRP1 is known as a second major MDR protein. It has been discovered in human small cell lung cancer cells NCI-H69 (7). MRP1 is present in almost all mammalian cells in small quantities, and it is expressed in the sinusoidal membrane of liver hepatocytes. The protein functions as a multispecific organic anion transporter and transports also neutral or weakly basic organic compounds (8). Both P-gp and MRP1 confer resistance to a similar but not identical spectrum of cytotoxic drugs (9,10). BCRP was first identified in the MCF-7/AdrVp cell line that does not express P-gp and MRP1 (11–13). The protein has also been found in cell lines selected for resistance to mitoxantrone—a poor substrate of P-gp and MRP1 (14). BCRP is expressed in the intestine, the bile canalicular membrane, and the placenta, particularly in the synctiotrophoblastic cells (15). More recently, high levels of BCRP have been detected in cancer stem cells (16). Similarly to P-gp and MRP1, this transporter shows wide substrate recognition properties, including neutral, positively, and negatively charged compounds. The common and most striking feature of these MDR proteins is the diversity of the recognized and transported substrates. They belong to various chemical classes and generally do not share structural homology (13).
Since the discovery of the first P-gp inhibitor, verapamil (17), a lot of studies have been performed to understand the protein efflux function and to create specific and effective MDR inhibitors, called also MDR modulators. Currently, the known MDR modulators are classified into three generations. To the first generation belong compounds already used clinically for other therapeutic applications (like verapamil, cyclosporin A, and quinidine). They showed high toxicity when applied in doses required for MDR reversal. The intensive search for more specific and less toxic compounds led to the development of next generations of MDR inhibitors. Nowadays, the third generation of MDR modulators are in the focus of interest (18). They represent novel molecules composed of structural features preselected on structure–activity relationships and then submitted to pharmacological screening (19). In contrast to the second-generation MDR modulators, these inhibitors are not cytochrome P450 3A4 substrates, and do not influence significantly the pharmacokinetic profile of co-administered drugs (18,20).
Prominent members among the third-generation MDR modulators are elacridar (GF120918) and tariquidar (XR9576), both containing a dimethoxytetrahydroisoquinoline–ethyl–phenylamine partial structure. Tariquidar belongs to a series of compounds, called XR compounds, which have been developed by Xenova Group Ltd. (21,22). A number of new tariquidar analogs have been synthesized and pharmacologically tested (23–32), thus supplying good data for a profound structure–activity investigation of this promising class of MDR modulators.
In this review, the recent achievements in structure–activity relationship investigations of tariquidar analogs are summarized. First, the main groups of the inhibitors studied are described. Next, the results from the structure–activity relationship analyses are presented for the particular protein studied. Finally, some conclusions are drawn about the main structural features related to the anti-MDR effects and interactions of the modulators with the MDR transporters.
PHARMACOLOGICAL AND STRUCTURAL DATA OF TARIQUIDAR AND ITS ANALOGS
Tariquidar and Other XR Compounds
Tariquidar is one of the most potent MDR inhibitors created so far. The compound has been reported to achieve a complete reversal of resistance at very low concentrations (25–80 nM) and to hold a long duration of activity in a panel of murine and human cell lines with different degrees of P-gp expression. The potency of XR9576 is between tenfold and 30-fold greater than that of the second-generation MDR modulator PSC833, and it is several logs more potent than the first-generation modulators cyclosporin A and verapamil (33,34). Tariquidar is shown to be about 50-fold more potent than verapamil in inhibiting rhodamine uptake, while for calcein AM uptake, a nearly 1,000-fold difference has been reported in MDR human plasma membrane vesicles from a human lymphoblastoid cell line (CEM Col1000) (35).
XR9576 has been assumed to specifically inhibit the P-gp transport function and to have no effect on the MDR-associated protein MRP1 (36). Recently, it has been shown to be a BCRP inhibitor, although less potent compared to P-gp (37). Thus, tariquidar is still considered as an appropriate compound for testing the role of P-gp in resistant cancer cells (38).
The XR derivatives have been synthesized and pharmacologically studied for their ability to potentiate the toxicity of doxorubicin in the mouse mammary carcinoma cell line EMT6/AR1.0 overexpressing P-gp. The effect of compounds on the accumulation of [3H]-daunorubicin has been measured as IC50 calculated relative to a dose of 100 μM verapamil, which restores the level to that of the parental EMT6/P cells. Figure 1 shows the structure of tariquidar. Under the code XR9576, it has been selected from 178 compounds developed from the MDR program of Xenova Group Ltd. (21) Table I summarizes the structural and activity data of selected XR inhibitors. All compounds possess a 6,7-dimethoxytetrahydroisoquinoline substructure and most have a second amide group in position X. The main structural variations relate to the linker Y between the substructures and to the substituents R1, R2, and R.
Fig. 1.

Structure of tariquidar (XR9576) (22)
Table I.
Structural and Activity Data of Selected XR Compounds
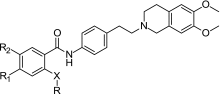
| |||||
|---|---|---|---|---|---|
| No. | X | R | R1 | R2 | IC50 (nM)a daunorubicin accumulation |
| 1 | – | Phenyl | H | H | 1,700 (n = 1) |
| 2 | O | Phenyl | H | H | 2,700 ± 350 |
| 3 | S | Phenyl | H | H | 2,300 (n = 1) |
| 4 | CO | Phenyl | H | H | 1,000 (n = 1) |
| 5 (XR9456) | NHCO | Phenyl | H | H | 250 ± 25 |
| 6 | NHCO | 4-i-Propylphenyl | H | H | 850 ± 370 |
| 7 | NHCO | 3-Thiophenyl | H | H | 730 ± 270 |
| 8 | NHCO | 2-Furanyl | H | H | 3,300 ± 800 |
| 9 | NHCO | 3-Furanyl | H | H | 920 ± 520 |
| 10 | NHCO | 2-Pyrazinyl | H | H | 204 ± 10 |
| 11 | NHCO | 2-(5-Methylpyrazinyl) | H | H | 67 ± 17 |
| 12 | NHCO | 2-Pyridyl | H | H | 680 ± 110 |
| 13 | NHCO | 3-Pyridyl | H | H | 880 ± 380 |
| 14 | NHCO | 3-(6-Methylpyridyl) | H | H | 67 ± 25 |
| 15 | NHCO | 2-Quinoxalinyl | H | H | 92 ± 13 |
| 16 | NHCO | 2-Quinolinyl | H | H | 790 ± 15 |
| 17 (XR9544) | NHCO | 3-Quinolinyl | H | H | 87 ± 13 |
| 18 | NHCO | 3-Isoquinolinyl | H | H | 4,900 (n = 1) |
| 19 | NHCO | 3-Quinolinyl | H | F | 60 ± 16 |
| 20 | NHCO | 3-Quinolinyl | F | H | 38 ± 13 |
| 21 | NHCO | 3-Quinolinyl | H | Cl | 149 ± 26 |
| 22 | NHCO | 3-Quinolinyl | Cl | H | 141 ± 19 |
| 23 | NHCO | 3-Quinolinyl | H | Me | 220 ± 66 |
| 24 | NHCO | 3-Quinolinyl | H | NMe2 | 293 ± 75 |
| 25 | NHCO | 3-Quinolinyl | NO2 | H | 45 ± 16 |
| 26 (Tariquidar) | NHCO | 3-Quinolinyl | OCH3 | OCH3 | 38 ± 18 |
Other Tariquidar Analogs
Tariquidar has been used as a template to develop new MDR modulators by different research groups. Modifications in different parts of the XR9576 structure (Fig. 1) have been explored. In all series, variations in the linker that connect the tetrahydroisoquinoline substructure to the rest of the molecule have been investigated. In some analogs, the anthranilamide nucleus is kept; in others, it has been modified. Different substituents at the anthranilamide-like and tetrahydroisoquinoline substructure have been introduced. Intensively studied is also the hydrophobic part connected to the anthranilamide. The structural and activity data of tariquidar analogs proposed by various research groups are described below.
A number of tariquidar analogs have been synthesized and tested by Wiese and coworkers (23–27). Recently, a small molecule library has been reported (28) with 39 compounds that possess a tetrahydroisoquinoline–ethyl–phenylamine substructure as in XR9576. However, in contrast to the XR derivatives, this substructure is connected only to one (hetero)aromatic residue and lacks the second (hetero)aromatic ring system (R substituent, Table I). This results in a smaller hydrophobic part and subsequently in molecules of lower molecular weight. Table II presents the structures and activity data of the compounds. R codes for the structural variations in the hydrophobic part and R3 for the substituents at the tetrahydroisoquinoline (H or –OCH3). Four different types of linkers X connect the tetrahydroisoquinoline–ethyl–phenylamine substructure to the hydrophobic part: “–”, direct bond (amide linker), –CH=CH– (amide–styryl linker), –O–CH2– (amide–ether linker), –NH– (urea linker) (28). The P-gp inhibitory effect of these compounds has been estimated using two functional assays: the calcein AM assay and a new assay with the substrate Hoechst 33342 (26). Several well-recognized P-gp substrates and inhibitors have also been measured to help in evaluating the inhibitory effects of the new modulators, among them representatives of the first generation of MDR modulators (verapamil, diltiazem, and cyclosporin A) and tariquidar. Table II summarizes some of the activity data determined.
Table II.
Inhibitory Effect of Tariquidar-Related MDR Inhibitors Determined by Calcein AM Accumulation (28)
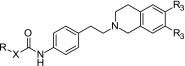
| ||||
|---|---|---|---|---|
| No. | X | R | R3 | IC50 ± SD (μM) calcein AM accumulation |
| 27 | – | Phenyl | OCH3 | 4.1 ± 1.2 |
| 28 | – | 2-Nitrophenyl | OCH3 | 5.4 ± 0.7 |
| 29 | – | 2-Nitrophenyl | H | 14 ± 3 |
| 30 | – | 2-Aminophenyl | OCH3 | 8.5 ± 1.6 |
| 31 | – | 2-Aminophenyl | H | 9.9 ± 4.7 |
| 32 | – | 4-Nitrophenyl | OCH3 | 1.4 ± 0.5 |
| 33 | – | 4-Nitrophenyl | H | 5.6 ± 2.7 |
| 34 | – | 4-Aminophenyl | OCH3 | 4.4 ± 2.8 |
| 35 | – | 4-Aminophenyl | H | 12 ± 3 |
| 36 | – | 3-Quinolinyl | OCH3 | 0.57 ± 0.18 |
| 37 | – | 3-Quinolinyl | H | 0.43 ± 0.11 |
| 38 | – | 2-Quinolinyl | OCH3 | 0.85 ± 0.36 |
| 39 | – | 4-Quinolinyl | OCH3 | 4.7 ± 0.7 |
| 40 | – | 6-Quinolinyl | OCH3 | 0.65 ± 0.11 |
| 41 | – | 2-Quinoxalinyl | OCH3 | 0.47 ± 0.05 |
| 42 | – | 1-Naphthyl | OCH3 | 1.4 ± 0.4 |
| 43 | – | 2-Naphthyl | OCH3 | 0.63 ± 0.18 |
| 44 | – | 3-Pyridyl | OCH3 | 4.8 ± 1.5 |
| 45 | – | 2-Bromophenyl | OCH3 | 3.3 ± 2.3 |
| 46 | – | 3-Bromophenyl | OCH3 | 1.8 ± 0.2 |
| 47 | – | 4-Bromophenyl | OCH3 | 2.4 ± 1.1 |
| 48 | – | 3,4-Dimethoxyphenyl | OCH3 | 4.2 ± 2.1 |
| 49 | – | 4,5-Dimethoxy–2-nitrophenyl | OCH3 | 13 ± 2 |
| 50 | – | 3,4-Methylendioxyphenyl | OCH3 | 2.1 ± 0.5 |
| 51 | –CH=CH– | Phenyl | OCH3 | 1.4 ± 0.4 |
| 52 | –CH=CH– | 2-Nitrophenyl | OCH3 | 1.1 ± 0.1 |
| 53 | –CH=CH– | 4-Chlorophenyl | OCH3 | 0.67 ± 0.09 |
| 54 | –CH=CH– | 4,5-Dimethoxy-2-nitrophenyl | OCH3 | 1.5 ± 0.4 |
| 55 | –CH2–O– | Phenyl | OCH3 | 3.1 ± 0.1 |
| 56 | –CH2–O– | 2-Nitrophenyl | OCH3 | 0.22 ± 0.03 |
| 57 | –CH2–O– | 2-Aminophenyl | OCH3 | 3.4 ± 1.0 |
| 58 | –NH– | 2-Nitrophenyl | OCH3 | 0.33 ± 0.07 |
| 59 | –NH– | 2-Nitrophenyl | H | 0.77 ± 0.15 |
| 60 | –NH– | 3-Nitrophenyl | OCH3 | 0.67 ± 0.20 |
| 61 | –NH– | 4-Nitrophenyl | OCH3 | 1.5 ± 0.2 |
| 62 | –NH– | 2-Aminophenyl | OCH3 | 21 ± 3 |
| 63 | –NH– | 2-Aminophenyl | H | 19 ± 8 |
| 64 | –NH– | 3-Aminophenyl | OCH3 | 14 ± 3 |
| 65 | –NH– | 4-Aminophenyl | OCH3 | 31 ± 17 |
| 26 (Tariquidar) | 0.078 ± 0.013 | |||
| Diltiazem | 49 ± 20 | |||
| Verapamil | 5.2 ± 2.0 | |||
| Cyclosporin A | 1.4 ± 0.3 | |||
Reproduced with permission from Elsevier
A number of compounds in the series show P-gp inhibitory potency at submicromolar concentrations. The best compounds are ten to 20 times more active than verapamil and three to six times more active than cyclosporin A. Among the derivatives synthesized, compound 56 possesses the strongest inhibitory potency against P-gp (Table II). It contains an elongated amide linker between the tetrahydroisoquinoline–phenylethylamine substructure and the hydrophobic 2-nitrophenyl group. This substance inhibits P-gp more than 20 times greater than verapamil and three times less than tariquidar (IC50 = 0.078 μM) and can be considered as a promising lead structure.
Most of the compounds possessing an amide linker have been additionally tested in the Hoechst 33342 assay using the same P-gp overexpressing adriamycin-resistant A2780adr cell line. Assays using two substrates, which are supposed to bind to different sites of P-gp (see the pharmacophore section below for more detailed explanations), have been applied in order to get a deeper insight into the binding sites of the compounds and the mechanism of their interaction with the transporter. A high correlation between the pIC50 values in both assays was observed, and it was shown that at the low substrate concentrations used in the assays, the type of interaction of the compounds with P-gp (competitive or non-competitive) could not be distinguished from the IC50 values only. When additional experiments were performed with different substrate concentrations to appraise the type of inhibition, non-competitive inhibition was found with calcein AM and competitive inhibition was observed for Hoechst 33342 (27).
The same compounds have also been tested for their potency to inhibit BCRP. They showed moderate activity with IC50 values of 5 μM or more, while some XR compounds were found to be more potent BCRP inhibitors with IC50 values in the range of 1 to 5 μM (Table III) (40). The comparison of pIC50 values shows that all compounds are approximately tenfold stronger inhibitors of P-gp as compared to BCRP using the same substrate Hoechst 33342. However, there is no correlation between the inhibition data against the two proteins, and compound 66 (XR9577) is the best BCRP inhibitor, being only three times less potent than against P-gp.
Table III.
pIC50 Values of Selected XR Derivatives and a Related Compound for Inhibition of BCRP and P-gp Determined with the Hoechst 33342 Assay (40)
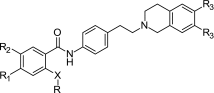
| |||||
|---|---|---|---|---|---|
| Compound no. | R | R1,2 | R3 | BCRP | P-gp |
| pIC50 ± SD Hoechst 33342 | pIC50 ± SD Hoechst 33342 | ||||
| 5 (XR9456) | Phenyl | H | OCH3 | 5.40 ± 0.13 | 6.29 ± 0.06 |
| 17 (XR9544) | 3-Quinolinyl | H | OCH3 | 5.30 ± 0.19 | 6.78 ± 0.09 |
| 26 (Tariquidar) | 3-Quinolinyl | OCH3 | OCH3 | 5.84 ± 0.04 | 7.14 ± 0.12 |
| 66 (XR9577) | 3-Quinolinyl | H | H | 6.00 ± 0.23 | 6.45 ± 0.07 |
| 67 (XR9504) | 4-Methylphenyl | H | OCH3 | 5.41 ± 0.25 | 6.28 ± 0.23 |
| 68 | 3,4-Dimethoxyphenyl | H | OCH3 | 5.68 ± 0.12 | 6.48 ± 0.15 |
Reproduced with permission from Elsevier
To assess the importance of the spacer connecting the anthranilamide nucleus and the tetrahydroisoquinoline substructure of tariquidar (Fig. 1), Labrie et al. (29) synthesized new anthranilamide MDR modulators in which the ethylphenyl linker between the nitrogen atoms of the amide group and the tetrahydroisoquinoline substructure was replaced by a flexible alkyl chain of two to six carbon atoms. In another series, the dimethoxytetrahydroisoquinoline group was replaced by a N-methyl-3,4-dimethoxyphenethylamine substructure and the same variations in the linker were explored. The P-gp inhibition potency and the cytotoxic activity of selected compounds were assessed using human CEM/VLB500 leukemia cells. To compare the P-gp inhibition effects of the modulators, the EC50 values of the compounds were determined in the presence of a fixed concentration of vinblastine or daunorubicin. All synthesized derivatives proved to be much less potent than XR9576, tested under the same conditions. Interestingly, there were only small activity differences for analogs with intact or “opened” tetrahydroisoquinoline substructure. The derivatives with two (and three) carbon linkers were found to be the most potent ones. In a continuing study (30), more rigid spacers were investigated resulting in active P-gp inhibitors. Figure 2 illustrates the structures of the two most potent representatives in the series. The ethyl–tetrahydroisoquinoline moiety containing a basic nitrogen was replaced by a N-benzylpiperazinyl group. In the CEM/VLB500 cells, the inhibitory potency EC50 were 124 ± 76 nM (R1,2 = H) and 59 ± 35 nM (R1,2 = OCH3). In the same assay, tariquidar had an EC50 = 68 ± 40 nM. Additionally, the authors performed in vitro biotransformation analyses using human CYP-450 isoforms and found that conversely to XR9576, the two compounds inhibited CYP3A4 that could limit their potential clinical use.
Fig. 2.

Structures of two tariquidar analogs with a rigidified piperazinyl instead of an ethyl linker acting as highly potent P-glycoprotein inhibitors (R1,2 = H, OCH3) (30)
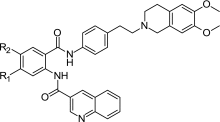
Egger et al. (31) performed synthesis of some tariquidar derivatives with different substituents at the central anthranilamide ring. The structures and inhibition data reported in their study are summarized in Table IV. The more lipophilic bromine derivative is even slightly more effective than tariquidar in inhibiting the P-gp-mediated calcein AM transport. Interestingly, large substituents at the anthranilamide are also tolerated as seen by the 2-ethoxyethoxy derivative (compound 72, Table IV) that is equipotent to tariquidar, while the large and hydrophilic morpholino substituent leads only to less than threefold decrease in activity. For this small series of five compounds, a linear dependence between calculated logD values and pEC50 is obtained with R2 = 0.93.
Table IV.
Structures and P-gp Inhibition by Tariquidar Derivatives Determined in the Calcein AM Accumulation Assay with Kb-V1 cells
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | EC50 (nM) | LogD 7.4 a |
| 26 (Tariquidar) | OCH3 | OCH3 | 223 ± 8 | 5.94 |
| 69 | H | Br | 145 ± 12 | 6.80 |
| 70 | H | NH–(CH2)2–O–(CH2)2–OCH3 | 1,575 ± 98 | 4.10 |
| 71 | H | Morpholino | 593 ± 21 | 4.80 |
| 72 | H | O–(CH2)2–O–C2H5 | 181 ± 6 | 5.87 |
Activity data are taken from (31)
STRUCTURE–ACTIVITY RELATIONSHIPS
P-glycoprotein Binding Site and Ligand Pharmacophore of Tariquidar
Tariquidar is assumed to bind to the same binding site of P-gp as the P-gp substrate Hoechst 33342 (41,42). This site has initially been proposed by Shapiro and Ling (43,44) who named it H-site to differentiate it from the binding site of rhodamine 123 (the so-called R-site) (43). According to Martin et al. (42), the H-site is in some way unique as it combines both transport and regulatory functions. Using the fluorescence resonance energy transfer analysis, Qu and Sharom (45) localized the H-site within the inner leaflet of the membrane at 10.5 to 14.5 Ǻ apart from the membrane surface in both the resting and transition state of P-gp. Litman et al. (46) also suggested that XR9576 inhibits P-gp pumping from the cytoplasmic face of the membrane as vinblastine and cyclosporin A presumably do. A number of small molecules called QB compounds, which are structurally related to pifithrin, are assumed to bind to the same site as Hoechst 33342 (47). These compounds can strongly affect the substrate specificity of P-gp, making the protein pumping much more active against some and less active against other substrates. Cyclic pifithrin-α (QB 102; Fig. 3b) has been shown to increase the efflux of substrates like doxorubicin, daunorubicin, etoposide, and rhodamine 123 and to decrease the efflux of taxol, vinblastine, vincristine, and Hoechst 33342. Thus, the compound’s behavior resembles the R–H classification of the P-gp binding sites of Shapiro and Ling by interacting potentially with the H-site.
Fig. 3.

a Structures of Hoechst 33342 and b cyclic pifithrin-α (QB 102). In gray circles indicated are the common pharmacophore points. All pharmacophore points lie in the same plane: H 1, H 2, H 3—hydrophobic centers; A, D—hydrogen bond acceptor and donor points, respectively
Pajeva et al. (48) have used these facts to develop a minimal ligand pharmacophore for the H-binding site of P-gp. Figure 3a, b shows the structure of Hoechst 33342 and QB 102 together with the pharmacophoric points identified from overlays of Hoechst 33342 and QB compounds. This pharmacophore has been taken into account to decide on the potential functional groups and atoms in the structures of tariquidar and its analogs in the subsequent 3D QSAR studies. In a similar fashion, the common pharmacophoric points may be identified for Hoechst 33342 and XR9576, as shown in Fig. 4a, b, respectively. Clearly seen is the excellent correspondence between the hydrophobic and hydrogen bond (HB) donor and acceptor points in both structures as well as the deviating quinolinyl group having no correspondence to any Hoechst 33342 substructure. Considering the flexibility of the tariquidar analogs, the authors investigated also flexible alignments between Hoechst 3342 and different XR analogs. Figure 5 illustrates possible overlays generated. In Fig. 5a, the anthranilamide nucleus of compound XR9456 is aligned on the ethoxy-phenyl ring of Hoechst 33342, and a very good correspondence between the pharmacophore points (Fig. 3) and groups of the same functionality in XR9456 is observed similar to XR9576 (Fig. 4). As the quinolinyl substructure in tariquidar, the terminal benzamide substituent deviates from the common overlay (pointing “down”, Fig. 5a). An opposite orientation of this group has also been observed (pointing “up”, overlay not shown). Figure 5b illustrates another alignment produced on compound XR9544. Compared to Fig. 5a, a mirror-like overlay of the whole structure is generated that could potentially correspond to an inverse binding mode. The tetrahydroisoquinoline part is overlaid on the ethoxy-phenyl ring of Hoechst 33342 and the quinolinyl substituent again deviates from the common overlap, this time at the opposite side. Based on the above overlays and considering the high inhibitory activities of the XR derivatives, it has been presumed that the deviating substructure in the XR compounds interacts with an additional site on the protein, and this interaction is facilitated by the ligand flexibility. Results of the flexible and pharmacophore overlays of the XR inhibitors suggest that the H-site could be a potential binding site for these compounds, in agreement with the experimental observations (42). Furthermore, the possibility for binding to an additional site of the protein could help in the understanding of their remarkably high inhibitory effect on the P-gp function.
Fig. 4.

a Structure of Hoechst 33342 and b XR9576 with atoms and groups potentially involved in common interactions: green, hydrophobic centers; cyan, HB acceptors; magenta, HB donors; red, O-atoms; blue, N-atoms; cyan, H-atoms; gray, C-atoms. Reprinted from (48) with permission from Elsevier
Fig. 5.

Alignments of a XR9456 (no. 1, Table I), b XR9544 (no. 17, Table I), c compound 36 (structures displayed as sticks) on the lowest energy conformer of Hoechst 33342 (line display); red, O-atoms; blue, N-atoms; cyan, H-atoms; gray, C-atoms. Reprinted from (27) with permission from Elsevier
Quantitative structure–activity relationships (QSARs) of XR analogs have extensively been studied by two research groups (39,49). Globisch et al. (39) performed combined classical QSAR, pharmacophore, and 3D QSAR analyses of the XR derivatives reported by Roe et al. (22) The QSAR (Free-Wilson) analysis of 31 XR derivatives outlined the tetrahydroisoquinoline substructure, bonded to the anthranilamide core through a phenyl moiety, and a bulky aromatic ring system with a heteroatom in position 3 in the R-part (see the template structure in Table I) to have the most significant impact on the MDR reversal activity of the compounds. The subsequent 3D QSAR analysis applying the comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) approaches confirmed this observation. The highest contribution to activity variance related mostly to the R-part of the structures. Using the neutral forms of all compounds, it has been demonstrated that the HB acceptor indices yield the best single field model with a cross-validated correlation coefficient, q2cv, of 0.747. In contrast, the HB donor field model has shown the lowest internal predictivity (q2cv = 0.240). Using combinations of fields, the best results have been obtained by combining the hydrophobic and acceptor fields (q2cv = 0.794) and the steric with the hydrophobic and acceptor fields simultaneously (q2cv = 0.778). When splitting the data into training and test sets (nine compounds have been randomly selected to form the test set and the validation procedure has been repeated 20 times), these models yielded also high external predictivity (0.66–0.75). Thus, the HB acceptor, steric and hydrophobic properties of the compounds have been shown to be most important for the inhibitory effect of the XR inhibitors towards P-gp.
Considering that the compounds have been aligned based on the pharmacophore of the Hoechst 33342 structure, it has been suggested that the anthranilamide core and the tetrahydroisoquinoline substructure bonded to the core through the phenyl moiety drive the compounds’ binding to the H-site. The anthranilamide nucleus provides HB interaction points; the phenyl moiety is an essential hydrophobic point. At the same time, the R-substituent that has no corresponding part in the structure of Hoechst 33342 additionally contributes to the modulating effect in accordance with the results of the pharmacophore and flexible alignments. However, it remains to be established how the XR compounds interact with the H-site of P-gp. As mentioned in the pharmacophore section, alternative binding modes could also be possible.
Labrie et al. (49) have also performed 3D QSAR analyses of XR derivatives using a larger number of 62 selected out of 178 compounds of Xenova Ltd. (21). The data set was split into a training set of 49 and a test set of 13 compounds. Three different conformations and two different alignment rules were explored. Additionally, several CoMFA and CoMSIA parameters were varied in order to optimize the q2 value taken as a measure of the model performance. In the CoMFA models, the best q2 values varied between 0.496 and 0.645 depending on the chosen template and alignment rule. In four out of six cases, the best models were obtained with the electrostatic field only. Using CoMSIA, models with similar q2 values (0.502–0.646) were generated, but the included fields varied greatly. Comparing the important regions, the authors noticed several discrepancies where the CoMFA and CoMSIA models had no commonalities. Due to these differences and the various field combinations yielding the best CoMSIA models, the contour maps of the different models were discussed separately. Despite the variety in the field combinations, the authors concluded on the steric, electrostatic, and hydrogen bond HB acceptor fields as the most important properties. They outlined the role of a nitrogen (as an acceptor group) in position 3 in a condensed heteroaromatic ring system in the R-part (Table I), a HB acceptor group like a methoxy group or an electronegative atom like fluorine or chlorine in the anthranilamide part (substituents R1 and R2, Table I), and a HB acceptor group in the tetrahydroisoquinoline part to have the most significant impact on pharmacological activity of the anthranilamide P-gp inhibitors. In general, these results confirm those reported by Globisch et al. (39).
Similar to the XR compounds, the tariquidar analogs synthesized and tested by Wiese and co-workers were subjected to 3D QSAR analyses. To decide on the most appropriate overlay of the compounds, the authors performed flexible and pharmacophore alignments of their compounds and Hoechst 33342, taking into account two main facts: (1) the result of Martin et al. (42) about interactions of tariquidar with the Hoechst 33324 binding site of P-gp and (2) their own results from competition experiments between Hoechst 33342 and inhibitors (27). Twenty-four tariquidar analogs and four XR compounds (9456, 9544, 9577, and 9504, Table III) were modeled. The flexible and pharmacophore alignments of the 3D structures of different representatives in the series showed that the compounds meet the structural and functional requirements for binding to the H-site of the protein.
Figure 5c illustrates one of the possible alignments of compound 36 (Table II). Similar to the XR compounds (Fig. 5a, b), overlays with different orientations of the quinoline N-atom (“up” and “down”) and an alternative (mirror-like) alignment have been produced. In the series, the tetrahydroisoquinoline substructure appears as either unsubstituted or 6,7-dimethoxysubstituted (Table II). The differences in the inhibitory effects of the methoxy-substituted and unsubstituted compounds suggest that this substructure could play a role for the inhibitory effect. A Free-Wilson QSAR analysis outlined a statistically significant contribution of these groups to the inhibitory activities of the compounds in both, the calcein AM and Hoechst 33342 assays (50).
The 3D QSAR modeling revealed the important role of more than one field for the inhibitory potency of the compounds in the Hoechst 33342 assay. The models were derived based on the data set of small tariquidar analogs and validated on an external test set of XR compounds taken from (22). A very good correspondence between the experimental and predicted inhibitory effects of the XR compounds was observed, thus confirming the model validity. Mostly contributing were the CoMSIA electrostatic, steric, hydrophobic, and HB acceptor indices. The best model combined electrostatic and HB acceptor indices and had high internal (q2cv = 0.834) and external predictivity (0.58–0.75) (27).
The contribution of the hydrophobic properties to the inhibitory effect of the tariquidar-like compounds serves a special attention. No correlation has been outlined with the logP values for the XR compounds (39) as well as for the smaller tariquidar analogs (27). These results show that logP, although an important lipophilicity characteristic, is not linearly related to the activities in these two series of compounds. In contrast to logP, the hydrophobic indices, generated by CoMSIA, showed high correlations; the hydrophobic field alone produced models with q2 = 0.718 (27). This observation is in agreement with our previous studies of different classes of first-generation MDR modulators that pointed to the role of hydrophobicity as a space-distributed molecular property for the MDR modulating effect (51,52). It should be noticed that, in contrast to the previous generations of modulators, the predictive ability of the hydrophobic field alone remains relatively low, and the same has been detected in the 3D QSAR study of anthranilamide analogs (39). These results confirm that, although an important structural property, hydrophobicity is not the only determinant for the inhibitory potency of the third-generation MDR modulators.
BCRP Inhibition
A structure–activity relationship study of five XR derivatives and 25 structurally related compounds has been reported using data obtained in BCRP-overexpressing tumor cells (40). Three-dimensional QSAR analyses were performed using CoMFA and CoMSIA approaches. The best models yielded q2 values of more than 0.8 with leave-one-out cross-validation and proved to be stable when leave-many-out cross-validation was applied. The developed models were compared to those derived for P-gp inhibition by the same set of compounds. The CoMSIA approach yielded better 3D QSAR models for both MDR transporters. In contrast to P-gp, for inhibition of BCRP, the field combinations involving the HB acceptor similarity index gave much worse q2 values. Instead, the combination of electrostatic and steric plus HB donor similarity index yielded the best CoMSIA models. Another difference is related to the effect of the two methoxy groups on the tetrahydroisoquinoline substructure. In the case of P-gp, as discussed above, they generally increase inhibitory potency, while for inhibition of BCRP, the opposite is true.
Very recently, activity data of tariquidar analogs were reported, in which small structural changes at the benzamide core resulted in large shifts in activity and selectivity from P-gp towards BCRP (32). By moving the amide-attached quinoline substituent of tariquidar to the meta position resulting in a meta-benzamide core (Fig. 6), the inhibitory activity of the compounds against P-gp was greatly diminished, while it was retained against BCRP. The removal of the methoxy group in position R1 and its replacement by a methylester in position R2 further increased activity against BCRP. Keeping this feature, different quinoline substituents, 2-quinoxalinyl, 2-pyrazinyl, and 3-pyridyl in position R, were investigated. The most active and selective BCRP inhibitor (R = 2-quinolinyl, R1 = COOCH3, R2 = H) was about 15 times more active than tariquidar in inhibiting BCRP and about four times more active than the most potent BCRP inhibitor known so far, Ko143. However, the new inhibitors did not reach the maximum inhibition obtained with Ko143, or fumitremorgin C, instead leveled off at about 50% of the maximum effect reached by fumitremorgin C. This fact was attributed to the low water solubility of the compounds.
Fig. 6.

General structure of new tariquidar-related derivatives acting as selective BCRP inhibitors (32). The compounds contain a meta-amino benzamide instead of an anthranilamide core
These results suggest that although sharing some general similarity, the structural requirements for binding of tariquidar analogs to P-gp and BCRP differ, and this is probably related to differences in the topology and physicochemical properties of the protein binding sites.
CONCLUSION
Developed as products of a rational design, the third-generation MDR modulators, including tariquidar and its analogs, are suggested to be more potent and more specific than their precursors. However, they are still far from being perfect MDR modulators able to effectively and safely overcome resistance in cancer cells. Thus, elucidating their structure–activity relationships is a necessary task which is expected to contribute to both better understanding of the interactions of this promising class of inhibitors with the MDR transporters and directing the synthesis of new and improved MDR modulators for clinical use. Many efforts are still necessary in this direction. The structure–activity studies of the tariquidar analogs performed so far revealed no single property as explicitly important; vice versa, they illustrated the complex role of more than one molecular field (steric, electrostatic, hydrophobic, and hydrogen bonding) for the inhibitory potency of these compounds. Some new substances have been synthesized that appear as promising lead structures for use in further studies.
Acknowledgments
We thank our colleagues and especially Christoph Globisch, Werner Klinkammer, and Henrik Müller whose efforts contributed to our results and knowledge on the topic. The financial support from the Alexander von Humboldt Foundation and Deutsche Forschungsgemeinschaft is also highly recognized.
References
- 1.Klein I, Sarkadi B, Varadi A. An inventory of the human ABC proteins. Biochim Biophys Acta. 1999;1461:237–262. doi: 10.1016/S0005-2736(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 2.Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene. 2003;22:7512–7523. doi: 10.1038/sj.onc.1206951. [DOI] [PubMed] [Google Scholar]
- 3.Bodo A, Bakos E, Szeri F, Varadi A, Sarkadi B. The role of multidrug transporters in drug availability, metabolism and toxicity. Toxicol Lett. 2003;140:133–143. doi: 10.1016/S0378-4274(02)00497-6. [DOI] [PubMed] [Google Scholar]
- 4.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 5.Gottesmann MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 6.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 7.Mirski SEL, Gerlach JH, Cole SPC. Multidrug resistance in a human small cell lung cancer cell line selected in adriamycin. Cancer Res. 1987;47:2594–2598. [PubMed] [Google Scholar]
- 8.Boumendjel A, Baubichon-Cortay H, Trompier D, Perrotton T, Di Pietro A. Anticancer multidrug resistance mediated by MRP1: recent advances in the discovery of reversal agents. Med Res Rev. 2005;25:453–472. doi: 10.1002/med.20032. [DOI] [PubMed] [Google Scholar]
- 9.Hipfner DR, Deeley RG, Cole SPC. Structural, mechanistic and clinical aspects of MRP1. Biochim Biophys Acta. 1999;1461:359–376. doi: 10.1016/S0005-2736(99)00168-6. [DOI] [PubMed] [Google Scholar]
- 10.Leslie EM, Deeley RG, Cole SPC. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167:3–23. doi: 10.1016/S0300-483X(01)00454-1. [DOI] [PubMed] [Google Scholar]
- 11.Kusuhara H, Sugiyama Y. ATP-binding cassette, subfamily G (ABCG family) Pflugers Arch. 2007;453:735–744. doi: 10.1007/s00424-006-0134-x. [DOI] [PubMed] [Google Scholar]
- 12.Doyle LA, Yang WD, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Litman T, Brangi M, Hudson E, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113:2011–2021. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 14.Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 15.Maliepaard M, van Gastelen MA, Tohgo A, et al. Circumvention of breast cancer resistance protein (BCRP)-mediated resistance to camptothecins in vitro using non-substrate drugs or the BCRP inhibitor GF120918. Clin Cancer Res. 2001;7:935–941. [PubMed] [Google Scholar]
- 16.Abbott BL. ABCG2 (BCRP): a cytoprotectant in normal and malignant stem cells. Clin Adv Hematol Oncol. 2006;4:63–72. [PubMed] [Google Scholar]
- 17.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Enhancement of vincristine- and adriamycin-induced cytotoxicity by verapamil in P388 leukemia and its sublines resistant to vincristine and adriamycin. Biochem Pharmacol. 1982;31:3138–3140. doi: 10.1016/0006-2952(82)90097-1. [DOI] [PubMed] [Google Scholar]
- 18.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 19.Robert J, Jarry C. Multidrug resistance reversal agents. J Med Chem. 2003;46:4805–4817. doi: 10.1021/jm030183a. [DOI] [PubMed] [Google Scholar]
- 20.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/S0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 21.Ryder, H, Ashworth PA, Roe MJ, et al. Anthranilic acid derivatives as multi drug resistance modulators. WO98/17648, April 30, 1998.
- 22.Roe M, Folkes A, Ashworth P, et al. Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg Med Chem Lett. 1999;9:595–600. doi: 10.1016/S0960-894X(99)00030-X. [DOI] [PubMed] [Google Scholar]
- 23.Klinkhammer W. Design, Synthese und 3D-QSAR neuartiger P-gp-Modulatoren. [Ph D thesis]. Bonn: University of Bonn; 2006. URN: urn:nbn:de:hbz:5 N-08459. http://deposit.ddb.de/cgi-bin/dokserv?idn=981124488&dok_var=d1&dok_ext=pdf&filename=981124488.pdf.
- 24.Jekerle V, Klinkhammer W, Reilly RM, Piquette-Miller M, Wiese M. Novel tetrahydroisoquinolin–ethyl–phenylamine based multidrug resistance inhibitors with broad-spectrum modulating properties. Cancer Chemother Pharmacol. 2007;59:61–69. doi: 10.1007/s00280-006-0244-3. [DOI] [PubMed] [Google Scholar]
- 25.Jekerle V, Klinkhammer W, Scollard DA, Breitbach K, Reilly RM, Piquette-Miller M, Wiese M. In vitro and in vivo evaluation of WK-X-34, a novel inhibitor of P-glycoprotein and BCRP, using radio imaging techniques. Int J Cancer. 2006;119:414–422. doi: 10.1002/ijc.21827. [DOI] [PubMed] [Google Scholar]
- 26.Müller H, Klinkhammer W, Globisch C, Kassack MU, Pajeva IK, Wiese M. New functional assay of P-glycoprotein activity using Hoechst 33342. Bioorg Med Chem. 2007;15:7470–7479. doi: 10.1016/j.bmc.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 27.Müller H, Pajeva IK, Globisch C, Wiese M. Functional assay and structure–activity relationships of new third-generation P-glycoprotein inhibitors. Bioorg Med Chem. 2008;16:2448–2462. doi: 10.1016/j.bmc.2007.11.057. [DOI] [PubMed] [Google Scholar]
- 28.Klinkhammer W, Müller H, Globisch C, Pajeva IK, Wiese M. Synthesis and biological evaluation of a small molecule library of 3rd generation multidrug resistance modulators. Bioorg Med Chem. 2009;17:2524–2535. doi: 10.1016/j.bmc.2009.01.072. [DOI] [PubMed] [Google Scholar]
- 29.Labrie P, Maddaford SP, Lacroix J, et al. In vitro activity of novel dual action MDR anthranilamide modulators with inhibitory activity at CYP-450. Bioorg Med Chem. 2006;14:7972–7987. doi: 10.1016/j.bmc.2006.07.055. [DOI] [PubMed] [Google Scholar]
- 30.Labrie P, Maddaford SP, Lacroix J, et al. In vitro activity of novel dual action MDR anthranilamide modulators with inhibitory activity on CYP-450 (Part 2) Bioorg Med Chem. 2007;15:3854–3868. doi: 10.1016/j.bmc.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 31.Egger M, Li X, Müller C, Bernhardt G, Buschauer A, König B. Tariquidar analogues: synthesis by CuI-catalysed N/O–aryl coupling and inhibitory activity against the ABCB1 transporter. Eur J Org Chem. 2007;2643–49.
- 32.Kühnle M, Egger M, Müller C, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the P-glycoprotein (ABCB1) modulator tariquidar. J Med Chem. 2009;52:1190–1197. doi: 10.1021/jm8013822. [DOI] [PubMed] [Google Scholar]
- 33.Mistry P, Stewart AJ, Dangerfield W, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61:749–758. [PubMed] [Google Scholar]
- 34.Walker J, Martin C, Callaghan R. Inhibition of P-glycoprotein function by XR9576 in a solid tumour model can restore anticancer drug efficacy. Eur J Cancer. 2004;40:594–605. doi: 10.1016/j.ejca.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 35.Kohler S, Stein WD. Optimizing chemotherapy by measuring reversal of P-glycoprotein activity in plasma membrane vesicles. Biotechnol Bioeng. 2003;81:507–517. doi: 10.1002/bit.10488. [DOI] [PubMed] [Google Scholar]
- 36.Leyers S, Wiese M. Tariquidar at a concentration of 10 μM has no effect on MRP1 activity in 2008MRP1 cells. Unpublished results.
- 37.Robey RW, Steadman K, Polgar O, et al. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.CAN-03-3298. [DOI] [PubMed] [Google Scholar]
- 38.Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- 39.Globisch C, Pajeva IK, Wiese M. Structure–activity relationships of a series of tariquidar analogs as multidrug resistance modulators. Bioorg Med Chem. 2006;14:1588–1598. doi: 10.1016/j.bmc.2005.10.058. [DOI] [PubMed] [Google Scholar]
- 40.Pick A, Müller H, Wiese M. Structure–activity relationships of new inhibitors of breast cancer resistance protein (ABCG2) Bioorg Med Chem. 2008;16:8224–8236. doi: 10.1016/j.bmc.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 41.Martin C, Berridge G, Mistry P, Higgins C, Charlton P, Callaghan R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br J Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 43.Shapiro B, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 44.Shapiro AB, Fox K, Lam P, Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- 45.Qu Q, Sharom F. Proximity of bound hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmatic membrane leaflet. Biochemistry. 2002;41:4744–4752. doi: 10.1021/bi0120897. [DOI] [PubMed] [Google Scholar]
- 46.Litman T, Skovsgaard T, Stein WDJ. Pumping of drugs by P-glycoprotein: a two-step process? Pharmacol Exp Ther. 2003;307:846–853. doi: 10.1124/jpet.103.056960. [DOI] [PubMed] [Google Scholar]
- 47.Kondratov RV, Komarov PG, Becker Y, Ewenson A, Gudkov AV. Small molecules that dramatically alter multidrug resistance phenotype by modulating the substrate specificity of P-glycoprotein. Proc Natl Acad Sci U S A. 2001;98:14078–14083. doi: 10.1073/pnas.241314798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pajeva IK, Globisch C, Wiese M. Structure–function relationships of multidrug resistance P-glycoprotein. J Med Chem. 2004;47:2523–2533. doi: 10.1021/jm031009p. [DOI] [PubMed] [Google Scholar]
- 49.Labrie P, Maddaford SP, Fortin S, et al. A comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) of anthranilamide derivatives that are multidrug resistance modulators. J Med Chem. 2006;49:7646–7660. doi: 10.1021/jm060239b. [DOI] [PubMed] [Google Scholar]
- 50.Müller, H. Funktionelle Untersuchungen des ABC-transporters P-glykoprotein. PhD thesis, University of Bonn, Bonn; 2007. urn:nbn:de:hbz:5N-12814. http://hss.ulb.uni-bonn.de/diss_online/math_nat_fak/2007/mueller_henrik.
- 51.Pajeva I, Globisch C, Fleischer R, Tsakovska I, Wiese M. Molecular modeling of P-glycoprotein and related drugs. Med Chem Res. 2005;14:106–117. doi: 10.1007/s00044-005-0127-x. [DOI] [Google Scholar]
- 52.Pajeva IK, Wiese M. Molecular modeling of phenothiazines and related drugs as multidrug resistance modifiers: a comparative molecular field analysis study. J Med Chem. 1998;41:1815–1826. doi: 10.1021/jm970786k. [DOI] [PubMed] [Google Scholar]