

Abstract
Parkinson’s disease is an age-related degenerative disorder of the central nervous system that often impairs the sufferer’s motor skills and speech, as well as other functions. Symptoms can include tremor, stiffness, slowness of movement, and impaired balance. An estimated four million people worldwide suffer from the disease, which usually affects people over the age of 60. Presently, there is no precedent for approving any drug as having a modifying effect (i.e., slowing or delaying) for disease progression of Parkinson’s disease. Clinical trial designs such as delayed start and withdrawal are being proposed to discern symptomatic and protective effects. The current work focused on understanding the features of delayed start design using prior knowledge from published and data submitted to US Food and Drug Administration (US FDA) as part of drug approval or protocol evaluation. Clinical trial simulations were conducted to evaluate the false-positive rate, power under a new statistical analysis methodology, and various scenarios leading to patient discontinuations from clinical trials. The outcome of this work is part of the ongoing discussion between the US FDA and the pharmaceutical industry on the standards required for demonstrating disease-modifying effect using delayed start design.
Key words: delayed start, disease modification, neuroprotection, Parkinson’s disease, randomized start
INTRODUCTION
Parkinson’s disease (PD) belongs to a group of conditions called movement disorders and is principally the result of the loss of dopamine-producing brain cells in the midbrain. Pharmaceutical companies are attempting to develop drugs that can potentially slow the progression of Parkinson’s disease which is also referred to as “disease modification.” Currently, there are no US Food and Drug Administration (US FDA)-approved drugs that have a claim for Parkinson’s disease modification.
For regulatory approval of treatments that offer symptomatic benefit in early Parkinson’s disease patients, clinical trials have used a double-blind, placebo-controlled, parallel group design with fixed or flexible dosing strategy. A variety of efficacy outcome measures (one or more combinations of subscales of the Unified Parkinson’s Disease Rating Scale [UPDRS]) and need for additional symptomatic therapy such as dopaminergic agonists, levodopa, have been used to assess the effects of treatment.
However, the current criteria for approval of drugs for Parkinson’s disease based on the change in “total” UPDRS (sum of parts I, II, and III) or individual parts of “total” UPDRS at the last visit do not differentiate drug effects that could be symptomatic, disease modifying, or both (Fig. 1). A two-phase study design (e.g., randomized withdrawal, delayed start) has been proposed to discern symptomatic and disease-modifying effects (Fig. 2). However, the withdrawal design can be complicated by various challenges such as, uncertainty of the duration of the withdrawal phase, and higher likelihood of patient discontinuation during the withdrawal phase. To overcome some of these concerns, an alternate design known as a randomized start design or delayed start design (1–3) has been proposed. To the best of our knowledge, there is only one published clinical trial that utilized this design (4).
Fig. 1.

Mean change from baseline total UPDRS score in a parallel design fixed dose trial comparing placebo (1) and drug that has purely symptomatic (2), purely disease-modifying (3), or combined symptomatic plus disease-modifying treatments
Fig. 2.

Mean change in total UPDRS score in a delayed start and withdrawal design trial comparing placebo, symptomatic + disease-modifying treatment
In clinical trials utilizing a delayed start design, patients are initially randomized to placebo or study drug for a certain duration (e.g., 36 weeks). This phase is referred to as the placebo control phase. At the end of the placebo control phase, patients who were randomized to placebo are switched to the study drug. The phase on study drug post 36 weeks is referred to as the active control phase. Patients who were randomized to the study drug initially during the placebo control phase will continue to receive drug in the active control phase. The patients who received placebo in the placebo control phase are referred to as the delayed start group. Patients who received treatment in both phases are referred to as the early start group.
The evidence of a disease-modifying benefit could be potentially demonstrated by testing the following hypotheses:
In the placebo control phase, the slope of total UPDRS change over time for the study drug group is shallower than that for the placebo group.
At the end of the active control phase, the early-start group would have a lower total UPDRS change compared to the delayed start group.
In the active control phase, the slope of total UPDRS change over time for the early and delayed start groups remains parallel.
To propose a valid statistical methodology to analyze data from delay start design trials, it is important to: (1) gain insights into the characteristics of disease progression and patient discontinuation patterns, from prior clinical trials; and (2) conduct extensive simulations to gain insights into the design features. Specifically, it is important to address the following questions:
What mathematical model describes the natural progression of total UPDRS?
What are the appropriate approaches/tests to evaluate a disease-modifying effects of a drug in a delayed start design?
What are false-positive and false-negative rates of the proposed statistical tests?
METHODS
Data
Our trial database included information from nearly 1,500 patients with early Parkinson’s disease. The duration of the trials ranged from 3 to 18 months. Data from open-label extension trials (up to 3 years) were also examined. Information on demographic factors such as age, duration of Parkinson’s disease, age at onset of disease, baseline total and subscale UPDRS scores, gender, race, and concomitant medications were collected.
Disease Progression and Drug Effect Models
The models that describe the longitudinal course of total UPDRS change in placebo and treatment groups are described below.
Natural Disease Progression and Placebo Effect
The characteristics of natural disease progression were examined in patients treated with placebo in clinical trials using Eqs. 1 and 2 as shown below (5,6):For data collected from baseline and all visits
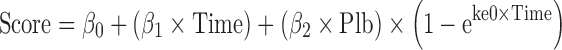 |
1 |
For data collected post mean time of 8 weeks
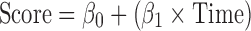 |
2 |
where Score refers to change from baseline total UPDRS score or total UPDRS score, Plb to the placebo group, β0 to the intercept, β1 to the slope of the placebo group, β2 to the symptomatic effect in the placebo group, and ke0 to the rate constant which influences the time to reach the maximum symptomatic effect.
Treatment Effect
The treatment group data are often modeled along with the placebo group data using Eq. 3 as shown below. The model is used to describe both the disease progression characteristics and symptomatic effects simultaneously.
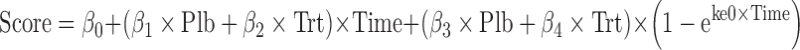 |
3 |
Where Score refers to change from baseline total UPDRS score or total UPDRS score, Plb to placebo, Trt to treatment, β0 to the intercept, β1 to the slope of the placebo group, β2 to the slope of the treatment group, β3 to the symptomatic effect in the placebo group, β4 to the symptomatic effect in the treatment group, and ke0 to the rate constant which influences the time to reach the maximum symptomatic effect. In case where a drug has no disease-modifying benefits, the difference (β1 − β2) will be zero.
Clinical Trial Simulations
The disease progression and drug effect, along with the likelihood of a patient discontinuation at each visit, were used to simulate 1,000 clinical trial replicates using SAS®. To assess the false-positive rates, we assumed the presence of only a symptomatic drug effect in which the slope of the disease progression remained the same for both treatment groups. To assess the statistical power, we simulated trials (sample size ranged from 50 to 600 per group) in which the study drug was assumed to slow disease progression by 20%, 30%, 40%, 50%, or 60%.
Clinical Trial Design
The study duration for clinical trial simulations was 72 weeks with two groups. A total of 500 virtual subjects were enrolled. The allotment was 1:1 per group (250 subjects per group). The study comprised of two phases: placebo control phase (0–36 weeks) and active control phase (37–72 weeks). Patients were assigned to the placebo group or study drug group during the placebo control phase. At the end of the placebo control phase, the patients who were randomized to the placebo group were switched to the study drug for the active control phase. Patients who received the study drug in the placebo control phase continued to receive the study drug in the active control phase. The total UPDRS score was recorded at weeks 0, 4, 12, 24, 36, 42, 48, 54, 60, 66, and 72.
Disease Progression Model
Table I lists the model parameters that were used for simulating the longitudinal time course of total UPDRS scores in the placebo and treatment groups. A rate constant of 0.693/week was used in the simulation to achieve the maximum symptomatic drug effect for the early start group within the first 12 weeks in the placebo control phase and for the delay start group in the first 12 weeks of the active control phase. No prognostic factors were included for simulating the baseline scores. It was assumed that the symptomatic effects were independent of baseline and no prognostic factors influence the rate of disease progression.
Table I.
Summary of Model Parameters Used to Simulate the Longitudinal Course of Parkinson’s Disease
| Group | Parameter | Mean | Variability (%) |
|---|---|---|---|
| Placebo | Rate of progression (slope) | 0.16 | 50 |
| Symptomatic effect | 0.8 | 50 | |
| Rate constant for time to attain maximum symptomatic effect | 0.693 | 50 | |
| Baseline UPDRS | 25 | 50 | |
| Drug | Rate of progression (slope) | 0.16 | 50 |
| Symptomatic effect | 2 | 50 | |
| Rate constant for time to attain maximum symptomatic effect | 0.693 | 50 | |
| Baseline UPDRS | 25 | 50 |
Units for rate of progression (slope)—per week; symptomatic effect—UPDRS; rate constant for time to attain maximum symptomatic effect (ke0)—per week
UPDRS Unified Parkinson’s Disease Rating Scale
Missing Data Scenarios
Missing data are commonly grouped into three missing data mechanisms based on the reasons for patient drop out (7,8).
Missing completely at random (MCAR): Data are considered to be MCAR if the reason for discontinuation is not related to the trial. For example, a patient moves to another geographical location and hence cannot continue in the trial.
Missing at random (MAR): Data are considered to be MAR if the reason for discontinuation is directly related to the observed outcome measures. For example, a patient discontinues as he/she experiences drug toxicity or the symptoms worsen. Even when toxicity develops or symptoms worsen, some patients/physicians may decide not to discontinue from the study while others may elect otherwise. These choices can be extremely subjective, and hence they are still considered random.
Missing not at random (MNAR): Data are considered to be MNAR if the discontinuation is due to unobserved outcome measures. For example, a patient’s symptoms could suddenly worsen to such an extent and have a major impact on the patient’s health that ultimately results in the patient’s death. In this case, this patient’s score on the outcome measure before death could never be collected.
The missing data are also grouped as ignorable missing and non-ignorable missing. The ignorable missingness includes the MCAR and MAR mechanisms, and non-ignorable missingness include the MNAR mechanism.
Clinical trials evaluating the effects of various treatments in patients with early Parkinson’s disease show that about approximately 30–40% of them need additional symptomatic therapy within 12 months of treatment initiation (4,9–11). Hence, in current simulations at each of these visits, it was assumed that a certain percentage of patients would discontinue from the study drug or placebo group either due to a need for additional symptomatic therapy or treatment-related adverse events (Fig. 3). The need for additional symptomatic therapy was simulated with the assumption that a patient with a higher change from baseline UPDRS is more likely to need additional therapy than others with a lower change from baseline. At each visit, patients were ranked, large positive to large negative change, based on their change from baseline UPDRS scores. A proportion of the patients based on their rank were discontinued. On the other hand, patients were randomly discontinued due to treatment-related adverse events between 12 and 20 weeks. The timing of these tolerability events was chosen such that a similar percentage of patients in the early and delay start groups would discontinue.
Fig. 3.

Percentage of patients remaining in the study under the assumption that: equal percentage of patients in early and delayed start group in the study need additional symptomatic therapy (scenario = 1), higher percentage of patients in delayed start group need additional symptomatic therapy due to initial placebo treatment(scenario = 2), and higher percentage of patients in early start group have tolerability issues with drug followed by a similar percentage of patients in delay start group when they are switched from placebo to drug (scenario = 3)
Statistical Analysis Methodology
The following hypotheses were tested at a significance level of 0.05 (two tailed) in our simulation studies evaluating a disease-modifying effect of a drug using a delayed start design. In both the placebo control and active control phases, the data collected data prior to 12 weeks of each respective phase were excluded from statistical analyses. This exclusion enabled us to test the drug effect on slope. For patients who discontinue in the placebo control phase, their data till the last visit were included in the analysis with no further imputation. The data from patients who discontinued in the placebo control phase were not included in the active control phase.
(Test any difference in slopes of the placebo and study drug groups)
In the placebo-controlled phase (using post-randomization data from 12 weeks through 36 weeks), the null hypothesis as stated below was tested based on the intent-to-treat (ITT) sample using linear mixed-effect modeling (MRM) analysis on the change from baseline scores of UPDRS.
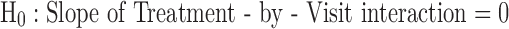 |
 |
The model included the fixed categorical effects of treatment and center, as well as the continuous fixed covariates baseline total UPDRS score, visit, and a treatment × visit interaction term. Random effects were included on slope and intercept. In the model, unstructured (UN) covariance structure was used to model the within-subject covariance of the measurements. The available data points of each subject were included in the analysis without any imputation.
-
Hypothesis 2
(Test any statistical difference in total UPDRS change from baseline between early and delay start groups at the end visit of active phase)
In the active control phase, the null hypothesis as stated below was evaluated at 72 weeks based on the available patients’ data (non-ITT sample) using mixed model repeated measure (MMRM) analysis on the change from baseline scores for total UPDRS of all available visits. In MMRM analysis, the time effect is assumed to be unstructured (i.e., time points are considered as discrete) instead of a linear effect, and this assumption allows to make a direct comparison of the endpoint mean score differences between the study drug and placebo.
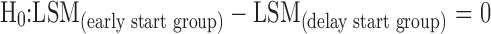 |
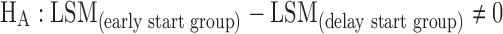 |
Where
- LSM
Least square mean.
The principal statistical analysis was a MMRM analysis on the change from baseline scores of UPDRS at the available visits (i.e., weeks 48, 54, 60, 66, and 72). The model included the fixed categorical effects of treatment, visit, center, and visit by treatment interaction, as well as the continuous fixed covariate baseline total UPDRS score. In the model, unstructured covariance structure was used to model the within-subject covariance of the measurements. The available data points of each subject in the active phase were included in the analysis without any imputation.
-
Hypothesis 3
(Non-inferiority comparison of the slopes of the two groups)
In the active control phase (using data from 48 to 72 weeks), the following non-inferiority hypothesis was evaluated based on the estimated slopes of early start group vs. delay start group of the study drug (non-ITT sample) using MRM analysis on the change from baseline scores of total UPDRS at the available visits. We used a non-inferiority margin of ≥0.15 units/week. However, it should be noted that parallelism of slopes would be evaluated if hypothesis 2 was statistically significant.
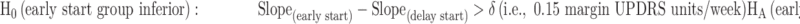 |
The principal statistical analysis was a MRM analysis on the change from baseline scores of UPDRS at the available visits (i.e., weeks 48, 54, 60, 66, and 72). The MRM model was similar to that described in hypothesis 1.
Considering that the statistical analysis of the active control phase will be based on a non-ITT sample, exploratory analyses in the active control phase data need be conducted to evaluate the impact of the dropouts on the statistical inferences.
RESULTS
What Mathematical Model Describes the Natural Disease Progression as Reflected by the Total UPDRS Scores?
Figure 4 shows that a linear model adequately describes the natural progression as reflected by the total UPDRS scores. Shown in Fig. 4 are the mean changes in the total UPDRS scores derived using MMRM analysis. The observed mean is not shown as the number of subjects at each visit decrease with time leading to inaccurate characterization of the time course of UPDRS scores. The data post-randomization were also analyzed using the model as shown in Eq. 1 to derive the parameters for simulations. The estimates of the parameters are shown in Table II. Similar or alternative models have been used in the literature to describe the progression of Parkinson’s disease (6,12). The model based on Eq. 1 could not estimate ke0 reliably due to the minimal effects in the placebo group.
Fig. 4.

Goodness of fit of natural disease progression in patients treated with placebo post 12 weeks randomization (study = 2, 3, 4) and post 9 weeks (study = 1). Shown in red line with plus symbol are the least square mean values based on MMRM analysis. The blue line is based on the fit using a linear model with time as continuous variable based on MRM analysis
Table II.
Mean (SE) and Variability (Between Patient) of Parameters Describing the Course of Progression of Parkinson’s Disease in Patients Treated with Placebo Using NLMIXED in SAS®
| Study | Parameter | Mean (SE) | Variability (%) |
|---|---|---|---|
| 1 | Rate of progression (slope) | 0.27 (0.07) | 64.15 |
| Symptomatic effect | 1.24 (3.28) | 44.90 | |
| Rate constant for time to attain maximum symptomatic effect | 0.06 (0.03) | 1,269.30 | |
| LN (baseline UPDRS) | 3.25 (0.04) | 38.85 | |
| 2 | Rate of progression (slope) | 0.13 (0.01) | 64.36 |
| Symptomatic effect | 1.47 (0.59) | 53.13 | |
| LN (rate constant for time to attain maximum symptomatic effect) | 0.35 (1.95) | 62.44 | |
| LN (baseline UPDRS) | 3.12 (0.04) | 34.64 | |
| 3 | Rate of progression (slope) | 0.11 (0.01) | 157.46 |
| Symptomatic effect | 1.59 (0.25) | 117.66 | |
| Rate constant for time to attain maximum symptomatic effect | 2 (fixed) | NE | |
| LN (baseline UPDRS) | 3.19 (0.02) | 41.23 | |
| 4 | Rate of progression (slope) | 0.14 (0.02) | 71.43 |
| Symptomatic effect | NE | NE | |
| Rate constant for time to attain maximum symptomatic effect | 0.08 (0.05) | NE | |
| LN (baseline UPDRS) | 3.11 (0.05) | 42.43 |
Units for rate of progression (slope)—per week; symptomatic effect—UPDRS; rate constant for time to attain maximum symptomatic effect (ke0)—per week
NE could not be estimated, LN natural logarithm, UPDRS Unified Parkinson’s Disease Rating Scale, SE standard error
False-Positive Rate
Placebo Control Phase
Based on the analyses of the results in the placebo control phase, the false-positive rates of concluding that a drug offers a disease-modifying benefit when it only offers symptomatic benefit are shown in Table III. These false-positive rates are approximately 6%. It is important to note that a final conclusion about whether there is a disease-modifying effect will be based upon inferences from the placebo and active control phases. Application of sequential testing, as proposed earlier, will protect the overall false-positive rate.
Table III.
False Positive Rates and Bias Under Null Model Using Linear Mixed Effects Models
| Placebo control phase (ITT) | Active control phase (non-ITT) | |||
|---|---|---|---|---|
| α (%) | Bias | α (%) | Bias | |
| Discontinuation independent of treatment outcomes (MCAR) | 5.9 | 0.0003 | 4.4 | 0.002 |
| Discontinuation dependent on observed treatment outcomes (MAR) | ||||
| Equal dropouts | 6.4 | 0.0005 | 6.0 | 0.04 |
| Unequal dropouts | ||||
| Lack of effectiveness | 6.4 | 0.0006 | 7.9 | −0.39 |
| Toxicity | 5.5 | 0.0003 | 5.8 | 0.02 |
In the placebo control and active control phase, analysis was performed using linear models with random effects and mixed model repeated measures approaches, respectively. In the placebo control phase, all patients are included in the analysis (ITT) while in active control phase patients with data post 48 weeks are only included in the analysis (no imputation for discontinued patients and hence non-ITT)
MCAR missing completely at random, MAR missing at random, ITT intent-to-treat
Active Control Phase
The initial simulations indicated that hypothesis test 2 alone with last observation carried forward (LOCF) as the imputation method inflated false-positive rates. Given the progressive nature of the disease, LOCF imputation for patients who discontinue prematurely in either phase will systematically underestimate the UPDRS score at the end of the trial. Moreover, for placebo patients who discontinue early, LOCF cannot be used to impute data during the active control phase. Consequently, LOCF imputation was not used in subsequent simulations.
Table III depicts the false-positive rates for the different missing data scenarios. Considering all the scenarios, the false-positive rate is reasonable, except for the case where more patients are assumed to discontinue in the active drug treatment arm which is based on two-sided hypothesis testing. In that case, the one-sided false-positive rate (delay start group has a lower change in total UPDRS score than early start group) is 0.8%, which implies that the statistical test is conservative (nominal is 2.5%). The mean bias in this case was estimated to be approximately −0.39 units of total UPDRS.
It is important to note that under the null hypothesis for tests 1 and 2, data were generated assuming that the drug offers only a symptomatic effect. With respect to the third null hypothesis (active control phase), data were simulated with a mean difference of δ in slopes between the two groups. We assumed δ to be 0.15 units/week which is similar to the natural disease progression slope. The simulations showed that the probability of concluding that the drug is disease-modifying using the combination of hypothesis tests 2 and 3 is zero in this case.
Power
Simulations were conducted for different sample sizes ranging from 50 to 600 patients per group under two scenarios. Approximately 80% power was achieved with 250 patients for 60% or 50% drug effects on the slope of disease progression (Fig. 5).
Fig. 5.

Power curves for a scenario where (a) equal percentage of patients in the drug and placebo groups will need additional symptomatic therapy, and (b) higher percentage of patients in the placebo group will need additional symptomatic therapy relative to the drug group. The data collected post-symptomatic therapy will not be included in the analysis although the patients might still undergo follow-up visits. Note the different lines for effects of drug on the slope of disease progression. For example, 20 refers to 20% decrease in the slope of disease progression relative to the progression in placebo group
DISCUSSION
What Mathematical Model Can Reasonably Describe the Natural Disease Progression as Reflected by the Total UPDRS Scores?
A linear model adequately describes the natural disease progression at least till 5 years in patients with early Parkinson’s disease. Various lines of evidence support this inference. First, Parkinson’s disease is a slowly progressing disease that exhibits deterioration at the rate of 8 units of total UPDRS/year (highest possible score = 124 units). During typical trial durations of up to 1.5 years for studying disease progression, a relatively limited change (∼12 units) would be expected. Hence, the change in total UPDRS scores over time are more reasonably described using a linear model.
Second, numerous literature reports suggest that disease progression follows a linear trend, beyond 12 weeks (after end of dose titration, if applicable) (9,10,13–19). Trials which followed patients up to 5 years also support a linear disease progression. There are some reports of non-linear progression of UPDRS scores after 5–8 years (12,20). However, these patients were on various study drugs.
Third, our analyses of the change in total UPDRS over time using mixed effect models provide evidence that the disease progression beyond 12 weeks is reasonably linear (Fig. 4).
Although the early time points are not included in the analysis evaluating a disease-modifying effect of a drug, it is important to collect these data for evaluating the symptomatic effect (if present). It is prudent to determine the time to peak symptomatic effect for each new molecule from the early trials. Such data can be analyzed using Eq. 3 or more complex models for designing future trials (12,21).
What Is the False-Positive Rate and the Power for Concluding That a Drug Has a Disease-Modifying Effect?
Hypothesis testing should preserve the type 1 error (or false-positive) rate at a nominal level of 5%. This approach ensures that one does not falsely conclude that a drug that provides only symptomatic benefit is modifying disease progression.
In general, the simulations suggested that the false-positive rate is acceptable. However, when more patients discontinued from the placebo group compared to active treatment due to lack of effectiveness, the false-positive rate was conservative for the active control phase analysis. It is important to note that the analysis of data from the active control phase does not agree with the regulatory requirement of the ITT principle. According to the ITT principle, all patients who were randomized must be included in the analysis. However, those patients who discontinue from the trial during the placebo control phase, especially those randomized to placebo initially, do not contribute any data on drug in the active control phase. It is not possible to impute drug effects rationally in the active control phase based on placebo effects. The only possibility is to use the data from those patients who completed the placebo control phase and who entered the active control phase. Because the active control phase analysis violates the ITT principle, analyzing the placebo control phase data (as meets the ITT principle) for comparing the slopes for the placebo and active treatment groups might be important. A case can be made that an unusually delayed symptomatic effect might give the appearance of divergent slopes for the two arms. In this case, relying on the placebo control phase will lead to erroneous conclusions. The non-inferiority margin is currently unknown (because no such drug has yet been approved for a disease-modifying claim), this margin can be determined as a result of discussion among medical experts of Parkinson’s disease. One approach would be to test whether a certain portion of the difference at the end of the placebo phase is still retained at the end of the active phase.
To achieve 80% power to conclude disease-modifying effect using the analysis methodology as proposed, a sample size of at least 600 each in drug and placebo groups with a drug effect of at least 40% on the slope of disease progression would be required.
CONCLUSIONS
We quantified the disease characteristics such as the progression and dropout rates from previous trials to explore endpoints for demonstrating disease modification effects. A set of three reasonable endpoints are proposed in the current report. Whether divergence of slopes in the placebo phase should be demonstrated in the light of testing the other two hypotheses for the active phase data needs further discussion. These endpoints can guide the individual researchers and drug developers to select the most suitable design and/or endpoints for their trials. There are two important assumptions contained in our analyses presented in the current manuscript. The first assumption is that change in the total UPDRS score is a good measure of the disease and its progression rather than a score from a individual component/subscale (e.g., motor subscale scores) of the total UPDRS. The second assumption is about the time to achieving maximum symptomatic benefit. The current analysis focuses on estimating slope for testing differences in the placebo control phase as well as parallelism in the active control phase using linear models. Early dose range finding studies can provide information about the onset and offset of drug effects with adequate measurement of total UPDRS scores.
However, because of the lack of direct prior experience, data from delayed start designs will need to be subjected to extensive explorations to accumulate substantial confidence in the inferences from these analyses and to corroborate internal consistency of various analyses. Also, it would be important to learn if disease-modifying effects can be well discerned from symptomatic effects from clinical trials with varied designs such as withdrawal and natural history-staggered design (22,23).
We believe that our model and simulations could potentially apply toward studying and assessing a disease-modifying effect of a study drug not only for any specific stage of Parkinson’s disease and for any specific efficacy outcome measure, but also to any other neurodegenerative disease (e.g., Alzheimer’s disease) and an appropriate, respective efficacy outcome measure (e.g., Alzheimer’s Disease Assessment Scale—Cognitive).
Acknowledgments
The authors wish to acknowledge Parkinson’s Study Group, NIH Exploratory Trials in Parkinson’s Disease (NET-PD) Group for providing access to clinical trial data. We are also grateful for the insightful discussions and feedback provided by numerous FDA and academic colleagues, and by professional organizations such as American Association of Pharmaceutical Scientists (AAPS) and Michael J Fox Foundation for Parkinson’s Research.
Footnotes
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official views of FDA.
References
- 1.Leber P. Slowing the progression of Alzheimer disease: methodological issues. Alzheimer Dis Assoc Disord. 1997;11(5):S10–S21. [PubMed] [Google Scholar]
- 2.Mani RB. The evaluation of disease modifying therapies in Alzheimer's disease: a regulatory viewpoint. Stat Med. 2004;23(2):305–314. doi: 10.1002/sim.1718. [DOI] [PubMed] [Google Scholar]
- 3.Clarke CE. A “cure” for Parkinson's disease: can neuroprotection be proven with current trial designs? Mov Disord. 2004;19(5):491–498. doi: 10.1002/mds.20057. [DOI] [PubMed] [Google Scholar]
- 4.Parkinson Study Group A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol. 2004;61(4):561–566. doi: 10.1001/archneur.61.4.561. [DOI] [PubMed] [Google Scholar]
- 5.Chan PL, Holford NH. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol. 2001;41:625–659. doi: 10.1146/annurev.pharmtox.41.1.625. [DOI] [PubMed] [Google Scholar]
- 6.Guimaraes P, Kieburtz K, Goetz CG, et al. Non-linearity of Parkinson's disease progression: implications for sample size calculations in clinical trials. Clin Trials. 2005;2(6):509–518. doi: 10.1191/1740774505cn125oa. [DOI] [PubMed] [Google Scholar]
- 7.Rubin DB. Inference and missing data. Biometrika. 1976;63:581–592. doi: 10.1093/biomet/63.3.581. [DOI] [Google Scholar]
- 8.Little RJA, Rubin DB. Statistical analysis with missing data. New York: Wiley; 1987. [Google Scholar]
- 9.The NINDS NET-PD Investigators A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66(5):664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- 10.Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59(10):1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 11.The NINDS NET-PD Investigators A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68(1):20–28. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- 12.Holford NH, Chan PL, Nutt JG, et al. Disease progression and pharmacodynamics in Parkinson disease—evidence for functional protection with levodopa and other treatments. J Pharmacokinet Pharmacodyn. 2006;33:281–311. doi: 10.1007/s10928-006-9012-6. [DOI] [PubMed] [Google Scholar]
- 13.Watts RL, Jankovic J, Waters C, et al. Randomized, blind, controlled trial of transdermal rotigotine in early Parkinson disease. Neurology. 2007;68(4):272–276. doi: 10.1212/01.wnl.0000252355.79284.22. [DOI] [PubMed] [Google Scholar]
- 14.Korczyn AD, Brunt ER, Larsen JP, et al. A 3-year randomized trial of ropinirole and bromocriptine in early Parkinson's disease. The 053 Study Group. Neurology. 1999;53(2):364–370. doi: 10.1212/wnl.53.2.364. [DOI] [PubMed] [Google Scholar]
- 15.Olanow CW, Kieburtz K, Stern M, et al. Double-blind, placebo-controlled study of entacapone in levodopa-treated patients with stable Parkinson disease. Arch Neurol. 2004;61(10):1563–1568. doi: 10.1001/archneur.61.10.1563. [DOI] [PubMed] [Google Scholar]
- 16.Larsen JP, Boas J, Erdal JE. Does selegiline modify the progression of early Parkinson's disease? Results from a five-year study. The Norwegian-Danish Study Group. Eur J Neurol. 1999;6(5):539–547. doi: 10.1046/j.1468-1331.1999.650539.x. [DOI] [PubMed] [Google Scholar]
- 17.Parkinson Study Group A controlled trial of rasagiline in early Parkinson disease: the TEMPO Study. Arch Neurol. 2002;59(12):1937–1943. doi: 10.1001/archneur.59.12.1937. [DOI] [PubMed] [Google Scholar]
- 18.Parkinson Study Group Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA. 2000;284(15):1931–1938. doi: 10.1001/jama.284.15.1931. [DOI] [PubMed] [Google Scholar]
- 19.Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson's disease. N Engl J Med. 2004;351(24):2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 20.Palhagen S, Heinonen E, Hagglund J, et al. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology. 2006;66(8):1200–1206. doi: 10.1212/01.wnl.0000204007.46190.54. [DOI] [PubMed] [Google Scholar]
- 21.Chan PL, Nutt JG, Holford NH. Modeling the short- and long-duration responses to exogenous levodopa and to endogenous levodopa production in Parkinson's disease. J Pharmacokinet Pharmacodyn. 2004;31(3):243–268. doi: 10.1023/B:JOPA.0000039566.75368.59. [DOI] [PubMed] [Google Scholar]
- 22.Ploeger BA, Holford NH. Washout and delayed start designs for identifying disease modifying effects in slowly progressive diseases using disease progression analysis. Pharm Stat 2008, in press. [DOI] [PubMed]
- 23.Hendrix SB. AAPS workshop on demonstrating disease-modifying effects for the treatment of Parkinson’s disease: drug development and regulatory issues, Arlington, VA; 2008