

Abstract
Accurate predictions of human pharmacokinetic and pharmacodynamic (PK/PD) profiles are critical in early drug development, as safe, efficacious, and “developable” dosing regimens of promising compounds have to be identified. While advantages of successful integration of preclinical PK/PD data in the “anticipation” of human doses (AHD) have been recognized, pharmaceutical scientists have faced difficulties with practical implementation, especially for PK/PD profile projections of compounds with challenging absorption, distribution, metabolism, excretion and formulation properties. In this article, practical projection approaches for formulation-dependent human PK/PD parameters and profiles of Biopharmaceutics Classification System classes I-IV drugs based on preclinical data are described. Case examples for “AHD” demonstrate the utility of preclinical and clinical PK/PD modeling for formulation risk identification, lead candidate differentiation, and prediction of clinical outcome. The application of allometric scaling methods and physiologically based pharmacokinetic approaches for clearance or volume of distribution projections is described using GastroPlus™. Methods to enhance prediction confidence such as in vitro–in vivo extrapolations in clearance predictions using in vitro microsomal data are discussed. Examples for integration of clinical PK/PD and formulation data from frontrunner compounds via “reverse pharmacology strategies” that minimize uncertainty with PK/PD predictions are included. The use of integrated softwares such as GastroPlus™ in combination with established PK projection methods allow the projection of formulation-dependent preclinical and human PK/PD profiles required for compound differentiation and development risk assessments.
Key words: formulation, human dose prediction, modeling, PBPK, PK/PD
INTRODUCTION
The successful development of new drug candidates with both efficacious and safe systemic exposures in humans with “developable” formulations based on preclinical data remains a major challenge in the pharmaceutical industry (1–4). One reason is that systemic exposure after oral dosing is determined by a variety of physicochemical, biopharmaceutical, and pharmacokinetic (PK) factors including drug solubility, dissolution, dosage form, permeability, efflux, and first-pass effects (5–8). Moreover, desirable molecular properties for orally delivered drugs fall into a narrow range for most marketed drugs (8,9). Another reason is that both human PK and pharmacodynamic (PD) parameters have to be predicted based on preclinical PK/PD relationships to accurately assess efficacious human dosing regimen (3,10).
Most published literature describe single PK parameter projection methods based on interspecies scaling for systemic clearance (CL) (11–17), volume of distribution at steady state (Vss) (14–18), which includes physiologically based PK (PBPK) models (19–22), and bioavailability (F) (17,23,24,25). Human oral PK or PK/PD plasma profile predictions are not frequently reported. Successful profile predictions for a few compounds were performed by Wajima and others (19,23,26). Formulation-dependent human PK/PD profile projections (3) following oral dosing are rare. Often human predictions rely solely on an unchanging absorption rate constant (ka) and a bioavailability estimate obtained from the average of preclinical species data as absorption input parameters (23). As a result, these PK/PD models can have limited flexibility to evaluate the impact of formulation parameters or modified dosage forms on oral PK/PD profiles, which can be important especially for compounds with solubility or dissolution rate limited absorption.
We report examples for the practical projection of human PK and PK/PD concentration and effect time profiles based on preclinical PK/PD and absorption models (27,28). We illustrate modeling approaches which can be readily implemented by pharmaceutical scientists utilizing common tools such as allometric scaling (11–14,29,30), the Wajima et al. method (23,26), and GastroPlus™ including its PBPK, PK/PD, and absorption, distribution, metabolism, excretion, and toxicity (ADMET) Predictor™ software modules (31). Case examples will demonstrate the use of “reverse pharmacology approaches” and in vitro–in vivo extrapolation (IVIVE) strategies for selections of clinical backup candidates and risk identification based on preclinical PK/PD and formulation modeling with inclusion of data from frontrunner compounds. The cases discussed here have challenging absorption, distribution, metabolism, and excretion (ADME) properties, such as low gastrointestinal solubility, high or unpredictable first pass effects, or unknown potency/efficacy in humans, for which human PK/PD profiles had to be established for decision making.
GENERAL PROCESS FOR ANTICIPATION OF HUMAN DOSES AND PK/PD PROFILES IN DRUG DEVELOPMENT
The prediction of efficacious human doses is a complex process as biopharmaceutical, pharmacokinetic, and pharmacodynamic properties must be projected from preclinical data (1). While the maximum recommended starting dose for first in human (FIH) trials is covered by an Food and Drug Administration guidance (32), the prediction of efficacious doses is performed at early and late development stages for several reasons, including (a) selection of clinical candidate back-up compounds with improved ADMET properties, (b) identification of safe and efficacious doses for FIH trials, (c) identification of formulation development risks, and (d) estimation of drug supply needed for toxicokinetic and FIH trials.
Lowe et al. (1) had described a general basic four-step projection process for the anticipation of human doses (AHD), schematically shown in Fig. 1a, which includes (step 1) characterization of preclinical exposure–response or effect relationships, (step 2) “humanization” or correction for interspecies differences, such as protein binding, or differences in potency, (step 3) compound ADME properties evaluation and prediction of human pharmacokinetic parameters, and (step 4) prediction of human dose–responses for dose selection in phase I protocols. Steps 3 to 4 include the integration and scaling of preclinical PK parameters to the human (e.g., CL, Vss, and F) and PD parameters such as 50% inhibitory concentration (IC50), i.e., the plasma drug concentration that is required to maintain 50% inhibition against a target. Clinical PK/PD data from frontrunner candidates can then be used to validate preclinical PK/PD strategies. The original AHD paradigm (1) did not include biopharmaceutics and formulation strategies, which are especially critical for Biopharmaceutics Classification System (BCS) classes II to IV compounds. The incorporation of formulation and biopharmaceutical parameters for human PK/PD predictions is shown schematically in Fig. 1b. Human PK and PK/PD profiles can be predicted for varying dosage forms and varied drug properties, such as particle size, solubility, and dissolution, among others, for compounds with solubility-/dissolution-dependent bioavailability (BCS classes II and IV), or for BCS classes I and III compounds where modified release dosage forms are being sought to modify systemic exposure, such as to reduce peak to trough plasma concentrations.
Fig. 1.

a Anticipation of human dose (AHD), PK/PD, and projections of efficacious dosing regimen often involves multiple steps (see text). The predictions of human PK parameters such as CL, V ss, F, and a targeted C ssavg are usually performed once preclinical PK data are available for at least one species. After incorporating PD data from preclinical efficacy models, human PK/PD projections can be performed. Formulation strategies are needed during all steps and are especially critical prior to first in human (FIH) studies. With FIH data, the AHD process can be validated with the frontrunner data and the models can be refined. “Reverse pharmacology” strategies utilize clinical PK/PD data to establish a link and validate preclinical PK/PD models and can aid in the selection of back-up candidates. b Formulation-dependent PK/PD requires the integration of parameters, such as solubility and dosage form with PK and PD parameters. Systemic exposure or bioavailability (F) is controlled by fraction of drug absorbed (F a), the fraction that escapes gut wall metabolism (F g), which is often negligible, and the systemic clearance (CL) (2,41). F a is determined by permeability (P app), solubility, intestinal efflux, and formulation parameters. CL can be estimated via several methods including allometry, the rule of exponents (13), IVIVE (14,15), or the Wajima et al. method (26). Formulation-dependent PK concentration–time profiles can be determined after input of V ss, which can be predicted using allometry or mechanistic methods (17,20). PD parameters, e.g., IC50 values can be obtained from preclinical efficacy studies using direct and indirect PD models (46) to generate a formulation-dependent response time-course profile (effect vs. time; PD profiles after Meibohm and Derendorf (40))
The impact of formulation variables on drug absorption and resulting PK profiles can be evaluated with simulation tools, such as GastroPlus™ and PDPlus™ (Simulations Plus, Inc., CA, USA) or Simcyp™ (33) (Simcyp Ltd, Sheffield, UK). GastroPlus™, which evolved from Yu’s compartmental and absorption and transit (CAT) model (34), now allows oral absorption and systemic PK/PD simulation using a nine compartmental pharmacokinetic advanced CAT model (31). Since recently, PK/PD profiles based on direct and indirect PD models (35) can be simulated with GastroPlus™ and its integrated PDPlus™. Case examples of PK profile projection and AHD demonstrate the utility of preclinical PK/PD (cases 1–3); incorporation of species differences (cases 1 and 2); practical projections of human CL and Vss parameter (cases 1-4); F predictions using GastroPlus™ (cases 1–4); and a PBPK approach (case 3).
Case Study 1: Formulation-Dependent PK/PD Simulations via a “Reverse Pharmacology Approach” Using Metabolism IVIVE from Multiple Species
Purpose
Backup B is a follow-on candidate to frontrunner A which has known comparable efficacy in humans and dogs. Human PK/PD modeling was performed for backup B vs. frontrunner A at lead optimization stage for differentiation assessment. The following goals were set for FIH projections: (a) establish if backup B has higher potency and percent target inhibition over time, (b) determine PK properties, including Cmin and Cmax, (c) determine if backup B could be administered at lower doses than frontrunner A to maintain levels above the 80% inhibitory concentration (IC80), and (d) evaluate if Cmax could be reduced with a controlled release (CR) formulation to minimize levels between Cmin and Cmax.
Background
Backup B is a strong base (pKa = 8.4) with a molecular weight (MW) < 500 and an equilibrium solubility of 2 to 10 mg/mL at intestinal pH in biorelevant media. Given the log P of 2.5 and a high permeability in a Caco-2 assay (Papp apical to basolateral, 13 × 10−6 cm/s), backup B was classified as a likely BCS class I drug. Backup B showed bioavailabilities with ∼20%, 42%, and 60% in the monkey, rat, and dog, after solution dosing, respectively. Low systemic clearances were observed across species when compared to their respective liver blood flows (36) (Table I). Human CL was estimated using the following “proven” methods which had predicted observed human CL of clinical frontrunner A (<1.5-fold error). The CL projection methods and obtained estimates for backup B were (a) microsomal data (1,14,15), 7.7 mL/min/kg; (b) Wajima et al. method (26), 6.5 mL/min/kg; (c) multispecies allometry (17), 6.0 mL/min/kg; and (d) fraction unbound corrected intercept method (FCIM) (13), 5.9 mL/min/kg suggesting concordance between methods. The CL values generated from microsomes were consistent with in vivo results, suggesting useful metabolism IVIVE (Table I). Overall low to moderate Vss was observed across species (1.3, 5.8, and 6.9 L/kg for rat, dog, and monkey, respectively), and elimination half-life was moderate (3 to 7 h). The Vss was estimated using (a) Oie–Tozer (2.8 L/kg) (1,15), (b) multispecies allometry (3.6 L/kg), and (c) ADMET Predictor™ using in silico predictions with a PBPK model for a default human (3.5 L/kg) (37–39). These methods had predicted Vss of frontrunner A within 2-fold (not shown). Frontrunner A’s IC50 was compared to that of backup B in the dog efficacy model following oral dosing to determine their relative potencies.
Table I.
Case 1: Human CL Estimates Can Be Obtained via Several Methods
| Compound | Species | CLint (mL/min/mg protein) | Predicted in vivo CL from microsomes (mL/min/kg BW) | Observed in vivo CL (mL/min/kg BW) |
|---|---|---|---|---|
| Backup B | Rat | 0.026 | 25 | 27 |
| Backup B | Dog | 0.019 | 14 | 14 |
| Backup B | Monkey | 0.049 | 25 | 22 |
| Backup B | Human | 0.015 | 7.7 | NDb |
| Other CL predicteda—5.9, 6.0, 6.5 (FCIM, allometry, Wajima et al. methods) | ||||
| Frontrunner A | Human | ND | ND | 5.2b |
| Other CL predicteda—5.7, 5.6, 7.5 (FCIM, allometry, Wajima et al. methods) |
For backup B, an IVIVE with in vitro metabolism data can be used to predict human in vivo CL with confidence, as the predicted in vivo CL from microsomal data is in agreement with the observed in vivo CL
BW body weight, ND not determined, FCIM fraction unbound corrected intercept method, CL clearance
aPrediction methods chosen, as described
bHuman measured clinical data for CL was only available for frontrunner A
Method
Since both preclinical and clinical PK/PD data were available for the frontrunner A, a “reverse pharmacology strategy” was used to validate and predict human PK/PD profiles of backup B:
Establishment of PK/PD link between human and preclinical PK/PD with frontrunner A: A dog preclinical PK/PD model was established by using measured dog PK and PD data from three animals at 1 mg/kg. PK data were described by a two-compartment model. PD data were modeled using a direct Imax model within the GastroPlus PDPlus™ module, as the PD response and PK concentration occurred at the same time, without delay (40). For frontrunner A, the IC50 value was then used in combination with clinical PK data to predict the known human PK/PD profile (step 5 below).
Determination of the potency for backup B in the dog PD model and comparison to frontrunner A: The IC50 values were determined. The protein binding interspecies correction factors were near unity, as both drugs have low protein binding in dog and in human (<40%). However, protein binding corrections can be important especially for highly bound drugs (10).
Prediction of CL and Vss parameters for backup B: As described, for CL, multiple methods were used (Table I). For Vss the Oie–Tozer methods and the PBPK model from Lukacova et al. were used (37–39), but other methods could have been employed (17).
Prediction of human oral PK profiles following immediate release (IR) or CR formulations for backup B using GastroPlus™: The default absorption model for a 70-kg human with a fasted gastrointestinal physiology was used.
Prediction of human PD profiles following oral IR or CR formulations for backup B and comparison to known human PK/PD profile of clinical frontrunner A: Here, the determined IC50 values were used with the GastroPlus PDPlus™ module to simulate human plasma PK/PD profiles after single and multiple oral daily dosing regimens.
Dose and formulation selection scenarios: To assess differentiation and to identify risks, the following human model scenarios were used: (a) 50 and 100 mg IR doses of frontrunner A, (b) 50 and 100 mg IR doses of backup B, and (c) 100 mg CR of backup B. PK and PD were compared for both dose strengths.
Results and Discussion
The dog PK/PD data following a 1 mg/kg oral dose of frontrunner A and backup B are shown in Fig. 2a, b, respectively. Both figures show double-Y plots with plasma levels (left axis) and the measured PD response (right axis). The dog PK/PD model predicted an IC50 and IC80 of 3.4 and 20 ng/mL, respectively, for backup B. For frontrunner A, the IC50 and IC80 were predicted to be 15 and 89 ng/mL, respectively, suggesting lower potency than backup B.
Fig. 2.

a, b Preclinical dog PK/PD of clinical frontrunner A (a) compared to the backup B (b). Both compounds were administered orally at 1 mg/kg. Measured plasma concentrations (CP) are shown on the left axis, while the PD response is shown on the right. The data were fitted to a direct I max model using GastroPlus PKPDPlus™. The dog IC50 for backup B was 3.4 ng/mL and was 15 ng/mL for frontrunner A
Using human estimates for CL and Vss and using the same IC50 values for humans, the human PK and PD profiles were simulated with GastroPlus™. Figure 3a shows an overall acceptable agreement for the frontrunner A predicted vs. the clinically observed pharmacokinetic profile following a daily dose of 100 mg IR (dashed line and solid circles, respectively), even though Cmax values were overpredicted. Figure 3b shows frontrunner A’s simulated human PD response after a 100 mg (dashed line) or a 50 mg (dashed-dotted line) daily oral dose administered as an IR formulation. Frontrunner A is simulated to yield approximately 80% of inhibition for 24 h in human with a 100 mg daily IR dose at steady state. These results are in line with demonstrated clinical efficacy at this dose (data not shown). However, a 50 mg IR dose of frontrunner A is simulated to provide 80% inhibition for only approximately 10 h (Table II; Fig. 3b), indicating a 50 mg dose is too low to maintain the desired PD response over the dosing interval. These PK/PD results support frontrunner A’s recommended efficacious daily dose of 100 mg. For backup B, Fig. 3b demonstrates that a lower dose, 50 mg, is sufficient to maintain a desired >80% inhibition PD response within the 24-h dosing interval. This is mainly attributable to the higher potency of backup B, as simulated oral PK profiles are similar for both compounds at 50 mg (Fig. 3a). As expected, both compounds provide sufficient PD effect at 100 mg daily doses (Table II). A 100 mg CR formulation of backup B lowers Cmax and thus may reduce systemic side effects when compared to 100 mg IR formulation (Table II).
Fig. 3.

a, b Human PK/PD of frontrunner A and the backup candidate, backup B using GastroPlus™ for 50 and 100 mg IR doses. Predicted PK plasma concentrations are shown on the left (a). The solid symbols represent observed clinical PK data after a single dose of 100 mg of frontrunner A. The corresponding simulated PD responses using a direct I max model are shown on the right (b). The solid vertical line represents the targeted inhibition profile with a desired PD response of 80% inhibition. Fifty milligrams IR of backup B was simulated to yield a PD response of 80% for 24 h, while frontrunner A requires a 100 mg daily IR dose
Table II.
Case 1: Human PK/PD Predictions Following IR or CR Formulations for Frontrunner A Compared to Backup B
| Compound | Observed dog IC50 (ng/mL) | Predicted human IC50 (ng/mL) | C min (ng/mL) | C max (ng/mL) | PD effect min | PD effect max | Time (h) above IC80 | |
|---|---|---|---|---|---|---|---|---|
| Backup B | 50 mg IR | 3.4 | 3.4 | 16 | 195 | 80 | 98 | 24 |
| Backup B | 100 mg IR | 3.4 | 3.4 | 33 | 363 | 88 | 99.7 | 24 |
| Backup B | 100 mg CR | 3.4 | 3.4 | 27 | 192 | 85 | 99 | 24 |
| Frontrunner A | 50 mg IR | 15 | 15 | 20 | 162 | 59 | 95 | 10 |
| Frontrunner A | 100 mg IR | 15 | 15 | 45 | 329 | 84 | 100 | 24 |
To maintain efficacy, the time above IC80 has to be approximately 24 h. A 50 mg IR dose of backup B is sufficient to maintain IC80 for 24 h, while for frontrunner A, 100 mg IR dose is required due to the lower potency
IC 50 50% inhibitory concentration, IC 80 80% inhibitory concentration, PD pharmacodynamic
For successful PK predictions, multiple methods should generally be attempted for CL and Vss to evaluate concordance between methods (set arbitrarily to within less than 2-fold) (17). When metabolism IVIVE is observed (Table I), this can increase the confidence in projections, and this approach is useful for compounds which are mainly metabolized by the liver. “Species-connects”, where projected parameters are within less than 2-fold (Table I), can increase confidence in predictions. For the PD response, prediction confidence is enhanced as human data were available for frontrunner A, which allows a “reverse pharmacology” approach, where clinical PK/PD is linked to an established preclinical PK/PD model.
Conclusion
A reverse pharmacology approach was used to establish a link between clinical PK/PD data and a preclinical PK/PD dog model. Simulations showed that backup B may provide differentiation against frontrunner A, by requiring a lower human efficacious dose. For backup B, a 50 mg IR capsule provides sufficient PD effects over the dosing interval of 24 h, while for frontrunner A, 100 mg is required to achieve a similar effect level. Simulations also showed that a CR formulation could reduce Cmax while maintaining the PD response throughout the dosing interval. The lower dose for backup B is more desirable and can be attributed to its higher potency, as both compounds showed similar PK profiles.
Case Study 2: Formulation-Dependent PK/PD Assessment for Poorly Aqueous Soluble Compounds with Varying Systemic CL and Bioavailability
Purpose
Compound K and compound P are novel lipid lowering agents for the treatment of cardiovascular diseases and both have low aqueous solubility. PK/PD projections were to be performed at lead optimization stage (1) to choose one of the compounds for further development and to evaluate formulation development risks.
Background
Compound K is a lipophilic (clogP > 5.5, ADMET Predictor™) zwitterion (calculated basic pKa = 7.9, acidic pKa = 4.4) which exhibits low solubility at physiological pH (0.001 mg/mL at pH 6.8) and medium permeability in Caco-2 (Papp apical to basolateral, 2.7 × 10−6 cm/s). Similarly, compound P is a zwitterion (calculated basic pKa = 4.21 and acidic pKa = 4.8) with a solubility at physiological pH (0.02 mg/mL at pH 6.8) and low to medium permeability in Caco-2 (Papp apical to basolateral, 0.68 × 10−6 cm/s). The Caco-2 permeability was converted to a human permeability using an internally developed correlation equation. Both compounds were classified as potential BCS IV drugs (5,6).
Pharmacokinetic studies were performed in several species. Compound K’s systemic clearance was low (hamster, monkey), moderate (rabbit), and high (rat) when compared to their respective liver blood flows (36) with a range from 2.5 to 52 mL/min/kg. Moderate to high volume of distribution across species (4.4 to 10.7 L/kg), along with poor to moderate oral bioavailability (4% to 53%), were observed. For compound P, pharmacokinetics were studied in rat, hamster, and monkey, and low total systemic clearance were consistently observed (from 0.43 to 14 mL/min/kg) when compared to their respective liver blood flows (36), indicating “species agreement”. Moreover, low to moderate volume of distribution (0.57 to 1.52 L/kg) and consistent moderate oral bioavailability (50% to 53%) were observed in all species. Thus, compound K showed differences in preclinical PK parameters, while compound P showed moderate oral bioavailability and low clearance across species.
To establish metabolism IVIVE, in vitro data were obtained from rat, dog, monkey, and human liver microsomes for both compounds. For compound K, there was lack of IVIVE. While the rat showed high clearance both in vitro and in vivo, the monkey had high in vitro clearance but low in vivo total systemic clearance (not shown). Hence, human liver microsomes could not be used to predict human in vivo clearance with high confidence. On the contrary, for compound P, IVIVE was observed as all animal species had low to moderate clearance in liver microsomes (data not shown), which was consistent with the low systemic in vivo clearance. Thus, human liver microsomes could predict human in vivo clearance with confidence (17). Various allometric methods were also used to project human clearance in vivo (17). For compound K, rat and rabbit predicted moderate CL (7.3 mL/min/kg), while monkey and hamster predicted low CL (0.25 mL/min/kg). Using all four species, the predicted CL was low (0.83 mL/min/kg). In contrast, for compound P, the predicted human CL from various methods was consistently low (0.1 to 0.4 mL/min/kg). Compound K had demonstrated an IC50 of 83 ng/mL in the preclinical efficacy model, while compound P had demonstrated efficacy with an IC50 of 110 ng/mL.
Method
For compound K, various risk scenarios for 20–500 mg doses for human PK/PD profile prediction were conducted assuming that solubility could be improved by formulation optimization (Table III): (a) low CL and high solubility, (b) low CL and low solubility, (c) moderate CL and high solubility, and (d) moderate CL and low solubility. As dose depends on CL and F, a higher CL translates into a higher dose, while a lower solubility can lead to lower F by reducing the fraction absorbed, Fa (Fig. 1b) (41). Similar analyses were conducted for compound P (details not shown).
Table III.
Case 2: Predicted Human PK/PD Parameters of Compound K Using Different Modeling Scenarios for Risk Assessments
| Compound K model scenarioa | CL (mL/min/kg) | Vp (L/kg) | T 1/2 (h) | QD dose (mg) | Solubility (mg/mL) | C max (ng/mL) | F a (%) | F (%) | PD effect (%) |
|---|---|---|---|---|---|---|---|---|---|
| “Best” low CL, high solubility | 0.83 (4-species) | 3.3 | 56 | 20 | 0.010 | 315 | 91 | 87 | 84 |
| Low CL, low solubility | 0.83 (4-species) | 3.3 | 56 | 40 | 0.002 | 284 | 48 | 46 | 84 |
| Low CL, high solubility | 0.83 (4-species) | 3.3 | 56 | 40 | 0.010 | 585 | 89 | 81 | 92 |
| Low CL, low solubility | 0.83 (4-species) | 3.3 | 56 | 100 | 0.002 | 629 | 44 | 42 | 94 |
| Moderate CL, high solubility | 7.3 (2-species, rat, rabbit) | 2.5 | 9 | 100 | 0.010 | 129 | 81 | 51 | 56 |
| “Worst” moderate CL, low solubility | 7.3 (2-species, rat, rabbit) | 2.5 | 9 | 500 | 0.002 | 133 | 9.1 | 5.8 | 56 |
CL clearance, QD quaque die, PD pharmacodynamic
aHuman projections are based on in vivo IV data from four species, rat, rabbit, hamster, and monkey, using two-compartment model allometry. Since exposure is apparently solubility rate-limited, solubility scenarios chosen were (a) 0.002 mg/mL, which represents minimal formulation efforts and (b) 0.01 mg/mL assuming a 10-fold increase in solubility due to formulation optimization (28). An IR capsule and a default average particle size of 25 μm was assumed. V p was assumed to be approximately equal to V ss. The first-pass extraction in GastroPlus was calculated as described by Jones (55)
Results and Discussion
For compound K, the dose range to achieve more than 50% PD effect was wide, 20–500 mg, using an IR capsule (Table III; Fig. 4a, b) due to a wide range in CL estimates (0.83–7.3 mL/min/kg), caused by species dependent metabolism, and solubility dependent bioavailability (5.8–87%). Notably, even a 500 mg dose could not achieve the upper targeted PD effect (>80%), under the “worst” scenario with low solubility and moderate CL. In contrast, for compound P, the projected required dose range was narrow (10-40 mg, not shown) imparted by moderate to high oral bioavailability (∼40–70%, data not shown), higher solubility compared to compound K, and a narrow and consistently low predicted human CL from preclinical species (0.1 to 0.4 mL/min/kg, data not shown) indicating “species agreement”.
Fig. 4.

a, b Predicted human PK/PD relationship shown as a double-Y plot for compound K after IR dosing. PK plasma concentrations are shown on the left. The PD response is shown on the right. a The PK profile after a 20 mg oral QD dose with low systemic CL (0.83 mL/min/kg) and a solubility of 0.010 mg/mL (“best case scenario”). b PK and PD following a 500 mg oral dose for a moderate systemic CL (7.3 mL/min/kg) and low solubility (worst case scenario)
PK/PD projections for poorly soluble compounds can be challenging, especially for compounds with moderate or high CL where “first-pass” can reduce systemic exposure and oral bioavailability. Although species disconnects for in vivo CL were observed (approximately 9-fold difference; Table III) and IVIVE with microsomal data could not be established, compound K human CL was predicted to be low to moderate with confidence. For development risk identification, a range of predictions with varying scenarios can be included (Table III). Since formulations can often be optimized, the model scenarios should focus on the prediction of CL and F. Using the model scenarios, compound K and compound P could be differentiated based on required doses to maintain a desired PD effect with high confidence, and only compound P was chosen for further development.
Conclusion
Formulation parameters were integrated with preclinical PK and PD data to project the human dosing regimen for two compounds with varying CL and solubility/dissolution rate-limited bioavailability. Clear differentiation based on the projected required human dosing regimen was possible, thus allowing science-driven candidate selection at lead optimization. For BCS II or BCS IV drugs (5,6), softwares such as GastroPlus™ offer advantages as solubility, dissolution, or formulation impacts on PK plasma profiles and PD response can be readily evaluated.
Case Study 3: Prediction of Human IV and PO Profiles for Different Formulations Using PBPK Modeling for Vss and Allometry for CL
Purpose
Compound R is a novel antiviral drug. At the lead optimization phase, its primary targeted use is intravenous dosing in a hospital setting. However, for community use, oral dosage forms were also to be developed. Human PK modeling was performed with the following goals: (a) to predict its intravenous PK profile following 75 mg infusions for FIH projections, (b) determine if compound R can be dosed orally and achieve a bioavailability greater than 50%, and (c) predict food effects on PK parameters.
Background
Compound R is dibasic (pKa = 10.7, 3.1) with a MW < 500 and an aqueous solubility near 0.003 mg/mL in its free form. The predicted solubility using the ADMET Predictor™ was 0.007 mg/mL at pH 6.6. Using a self-emulsifying drug delivery systems (SEDDS) formulation (42) of its salt, the measured solubility was increased to >1 mg/mL in a simulated intestinal fluid dispersion. Its permeability in the Caco-2 assay was moderate (Papp apical to basolateral, 4.5 × 10−6 cm/s). Based on these data, compound R was classified as a BCS class II drug. Preclinical studies were conducted in the rat, dog, and monkey. In the rat, compound R showed a moderate plasma clearance of 17 mL/min/kg, a high Vss of 5.3 L/kg, and a moderate to high bioavailability (60% to 100%). In the dog, compound R showed a low plasma clearance of 5 mL/min/kg and a moderate Vss of 1.8 L/kg. In the monkey, clearance was moderate, 18 mL/min/kg, and Vss was moderate (2.1 L/kg). Thus, low to moderate systemic clearances and first-pass extraction ratio were observed across species when compared to their respective liver blood flows (36).
Formulation studies in the dog revealed that a lipid-based SEDDS (42) showed the highest bioavailability due to greatly enhanced solubility. Human CL was estimated using (a) allometry with in vitro microsomal metabolism correction suitable for compounds with overall low CL (43), (b) the “FCIM” (11,13), and (c) Wajima et al. method (26). For method (a), the log of the measured in vivo CL was plotted against the log of the body weight for rat, monkey, and dog. With the general allometric formula, 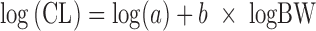 , and including intrinsic metabolism correction, one obtains
, and including intrinsic metabolism correction, one obtains 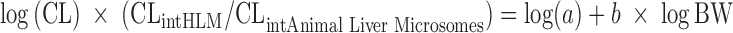 (43). The human Vss was estimated using (a) Oie–Tozer (2.4 L/kg) (1,15) and (b) ADMET Predictor™ using in silico predictions and PBPK for a 85-kg human (2.1 L/kg) and employing the Lukacova et al. method derived from the method from Rodgers et al. (37–39) (Table IV).
(43). The human Vss was estimated using (a) Oie–Tozer (2.4 L/kg) (1,15) and (b) ADMET Predictor™ using in silico predictions and PBPK for a 85-kg human (2.1 L/kg) and employing the Lukacova et al. method derived from the method from Rodgers et al. (37–39) (Table IV).
Table IV.
Case 3: Human Predicted vs. Observed PK Parameters for Compound R
| Parameter compound R | Method(s), parameters | Comment | Predicted point estimate | Observed (± SD) | Absolute predictive error (%) AUC, C max |
|---|---|---|---|---|---|
| V ss (L/kg) | GastroPlus™ PBPK modulea | PBPK 14 compartments | 2.1, 2.4 | 2.0 ± 0.4 | NA |
| Oie–Tozer (17,18) | |||||
| CL (mL/min/kg) | Allometry with in vitro metabolism correction | References (14,43) | 2.1. 3.6, 3.1 | 0.95 ± 0.21 | NA |
| FCIM | |||||
| Wajima et al. | |||||
| IV AUC (ngh/mL) (fasted) | Dose = 75 mg | 0.4 h IV infusion | 15,900 | 19,500 ± 4,800 | 18.4 |
| IV C max (ng/mL) | Dose = 75 mg | 1,830 | 1,920 ± 470 | 4.69 | |
| Oral AUC (ngh/mL) (fasted) | Dose = 75 mg | Salt SEDDS solution fasted human C s = 1.4 mg/mL | 14,800 | 17,500 ± 4,600 | 15.4 |
| Oral C max (ng/mL) | Dose = 75 mg | 764 | 930 ± 240 | 17.8 | |
| %F | Dose = 75 mg | 93 | 90 | ||
| Oral AUC (ngh/mL) (fasted) | Dose = 75 mg fasted human | Free base suspension, C s = 0.007 mg/mL | 2,180 | 3,230 ± 1,360 | 32.5 |
| Oral C max (ng/mL) | Dose = 75 mg fasted human | 55.2 | 64.9 ± 26.6 | 14.9 | |
| %F | Dose = 75 mg, fasted human | 21 | 14 | ||
| Oral AUC (ngh/mL) | Dose = 450 mg, fasted human | Free base suspension, C s = 0.007 mg/mL | 5,140 | 7,320 ± 3,590 | 29.8 |
| Oral C max (ng/mL) | Dose = 450 mg, fasted human | 135 | 121 ± 59.3 | 10.3 | |
| %F | 5.8 | 6.2 | |||
| Oral AUCc (ngh/mL) | Dose = 450 mg, fed human | Free base suspension, C s = 0.07 mg/mL | 52,300 | 19,200 ± 7,240 | 172 |
| Oral C max (ng/mL) | Dose = 450 mg, fed human | 1,520 | 741 ± 281 | 105 | |
| %F | 57 | 16 |
Predictions were performed using GastroPlus™ modules and employing interspecies scaling methods for CL and V ss (14)
NA not applicable, C s equilibrium solubility, SD standard deviation, AUC area under the concentration curve, SEDDS self-emulsifying drug delivery systems, PBPK physiologically based PK, FCIM fraction unbound corrected intercept method
aAll simulation were done with GastroPlus. V ss was 2.1 L/kg using the Lukacova et al. PBPK method (37–39); CL used was 0.95 mL/min/kg. A human fasted default model was used. P app was derived from Caco-2 using an internally developed model; 0.007 mg/mL was estimated from ADMET Predictor™; 0.07 mg was measured at intestinal pH
Method
To establish a link from preclinical PBPK models to the human situation, the following steps were taken:
Modeling of rat IV profile: Since the drug was targeted for intravenous dosing, rat intravenous data were modeled using GastroPlus™, with measured pKa and log P as input parameters. The default rat PBPK model in GastroPlus™ was chosen. Figure 5 shows the simulated and measured bolus IV profile in the rat after a 2 mg/kg dose, with r2 = 0.82.
Modeling of human IV profiles: Since IV data in the rat could be described with the Lukacova et al. method (37–39) for Vss, IV profiles were simulated for the human situation using the same method. The targeted dosing regimen was 75 mg via infusion over 25 min using compound R salt SEDDS formulation. The predicted vs. observed human IV profile is shown in Fig. 6a, demonstrating a good fit with r2 = 0.76. Selected predicted and observed PK parameters are listed in Table IV.
Modeling of oral profiles in fasted humans: The targeted dose of interest was 75 mg given daily as a SEDDS suspension. A GastroPlus™ default physiology model of a fasted human was selected. The predicted vs. the observed fasted human oral profile is shown in Fig. 6b, demonstrating a good fit with r2 = 0.76.
Simulation of oral profiles were performed for a 450-mg dose in fasted and fed humans (Table IV): The impact of food on oral absorption was modeled, since compound R solubility is approximately 9- to 10-fold higher in the presence of bile acids (data not shown).
Fig. 5.

Rat observed (solid circles) and predicted (solid line) IV profile after a 2-mg/kg bolus dose for compound R. The r 2 was 0.82
Fig. 6.

a Human observed (solid circles) vs. predicted (solid line) IV plasma concentration–time profile following a single 75 mg infusion over 25 min for compound R. The r 2 value was 0.76. b Human observed and predicted oral plasma concentration–time profiles after a single 75 mg oral SEDDS solution administration of compound R (right). The r 2 value was also 0.76
Results and Discussion
Accurate dose projections rely on accurate CL estimates, as higher projected clearance translates directly into higher doses. Here, all methods overpredicted the low observed human CL (0.95 mL/min/kg). Compound R estimates for CL were (a) 2.1, (b) 3.6, and (c) 3.1 mL/min/kg (Table IV). The best results were obtained using the microsomal correction method by Lave (43), and predicted CL values were within approximately 2-fold of the in vivo CL (Table IV). While this confirms that in vitro metabolism correction is suitable for compounds with low CL, it highlights the difficulty to project CL when overall low CL is observed in preclinical species. While all methods did correctly project low systemic CL in humans (<5 mL/min/kg; Table IV) compared to the human liver blood flow (36), this examples shows the importance of accurately predicting and multiple CL selection scenarios should be considered for projections.
The PBPK in silico method showed good agreement with measured Vss in humans. While no corrections for tissue partitioning were needed for compound R (log P < 3), compounds with high log P values should utilize the Vss method derived by Lukacova et al. and based on the method from Rodgers et al. (37–39), as conventional calculation methods can overpredict Vss often by several orders. Rat IV data can be used to identify Vss overprediction errors, as in vivo Vss can be readily compared with the in silico Vss. If available, additional corrections can be made with measured rat tissue distribution data, but these are often not available, as radiolabeled compound is required.
Formulation- and solubility-dependent PK parameters were observed in human (Table IV). While the free base suspension had a low bioavailability of 6.2%, an SEDDS-optimized solution of a salt enhanced the bioavailability to 90%. The presence of food also enhanced bioavailability of a 450-mg free base from 6.2% to 16%, likely due to enhanced solubility in presence of bile salts (Table IV). Overall, the in vivo variability (%CV) was high, larger than 35% (data not shown), which is not uncommon for poorly bioavailable compounds (44). The PK parameters (area under the concentration curve (AUC) and Cmax) could be predicted with predictive errors less than 33% after a 75-mg oral dose suspension or solution using in vitro solubility data or the ADMET Predictor™ estimate (Table IV). However, for the prediction of food effects, large prediction errors (105–172%) were observed and AUC and Cmax were overpredicted for the 450-mg dose given under fed conditions (Table IV). Simulations suggested that the true in vivo human solubility under fed conditions was likely lower (∼0.035 mg/mL) than the measured in vitro value (0.070 mg/mL). This case illustrates that biorelevant solubility data are crucial for accurate human predictive PK/PD modeling. Measuring relevant solubility remains a challenge (45), and more work is needed to refine the models, including precipitation kinetics (28).
Conclusion
A GastroPlus™ PBPK model was used to describe human IV and PO data for a BCS class II compound. In general, PBPK strategies can entail (a) prediction (in silico) of rat IV profiles, (b) prediction (in silico) of rat oral profiles, and (c) prediction (in silico) of human IV and oral profiles. CL estimates can be derived by including microsomal data or by allometric scaling of in vivo data. Fasted human PK parameters could be predicted with acceptable predictive errors of less than 33%. Projections for fed humans overestimated exposure and predictive errors were high (>63%), likely due to nonpredictive solubility data.
Case Study 4: Human PK/PD Projections Using Only Single Species Rat PK/PD Data
Purpose
In some cases, human efficacious dose projections need to be performed early in discovery with very limited data. Human PK/PD projections were requested at optimization stage for a cardiovascular backup candidate, backup C, and only rat PK/PD data were available. For frontrunner B, human oral and IV clinical data were available, in addition to rat PK/PD data. The following tasks had to be accomplished: (a) to predict the human PK and bioavailability of backup C using only rat data and (b) to establish PK/PD relationships for both compounds for differentiation.
Background
Frontrunner B is a weak base, with a low Caco-2 permeability (∼2 × 10−6 cm/s) and a solubility >1 mg/mL at physiological pH and thus is a BCS class III drug. Similarly, backup C is also a weakly basic BCS class III drug. Frontrunner B has a low rat bioavailability (∼25%), which is similar to human bioavailability (∼24%), likely due to incomplete absorption and high clearance. In contrast, the backup C has a rat bioavailability near 60–75%. Rat PK/PD data were available for both compounds. Since there was a time delay in the PD response, indirect response modeling with inhibition of input (46) was done with the GastroPlus PDPlus™ module. Since rat CL could predict human CL of frontrunner B, human CL for backup C was estimated using rat single species scaling (17,47). As frontrunner B’s Vss could be predicted within less than 1.1-fold of human clinical data via ADMET Predictor™ using in silico parameters as inputs, the same approach was used for backup C.
Methods
The following steps were taken for the PK/PD projection:
Human PK model validation with clinical frontrunner B: Projections were done retrospectively to validate the PK projection method with rat data as the rat is thought to be predictive of human absorption for many drugs (48). IV profile projections were done by a method proposed by Wajima et al. (26), which is a universal and easy-to-use approach (23). Human IV plasma concentration–time profile could be predicted based on rat plasma concentration–time profiles which were superimposable when they are mathematically transformed (not shown). Frontrunner B’s oral profile could be predicted with r2 = 0.74, even though the observed data were highly variable (Fig. 7a, solid circles).
Backup C human IV and oral profile projection were carried out analogous to steps performed for frontrunner B.
Fig. 7.

a Prediction of human profile for frontrunner B (reference) using only rat data. Observed oral plasma concentration profile after an 80 mg QD dose (left axis and solid circles) vs. predicted oral profiles (dotted line). The r 2 value was 0.74. The long-dashed line (right axis) represents the projected PD response, assuming that the response is conserved. b Predicted oral plasma concentration–time profile (left axis, dotted line) after 50 mg QD oral dosing for backup C. The long-dashed (right axis) represents the PD response. The targeted PD effect is above 0.55, or 55%
Results and Discussion
The results of projected PK/PD relationships for frontrunner B and backup C are shown in Fig. 7a, b. Predicted oral plasma concentration–time profile after a 80 mg quaque die (QD) dosing of frontrunner B (left axis, dotted line) are in good agreement with the observed clinical data (solid circles). As rat PD effect parameters were predictive for human PD (data not shown), rat PD parameters were used. A targeted PD response above 0.50, near 0.55, or 55% was achieved with an 80 mg dose and maintained for 24 h (Fig. 7a). While backup C has a lower potency (Table V), it has a higher projected bioavailability in human, based on rat PK and bioavailability data. A 50 mg QD dose of backup C was projected to have a similar PD effect when compared to 80 mg frontrunner B (Fig. 7b; Table V). Thus, backup C is anticipated to require a similar or slightly lower dose than frontrunner B. As both compounds are likely BCS class III drugs, formulation parameters were not included in this analysis.
Table V.
Case 4: Rat and Human PK and PD Parameters
| Parameter | Frontrunner B | Backup C |
|---|---|---|
| Rat IC50 (ng/mL) | 1.6 | 100 |
| Rat bioavailability (%) | 25 | 60–75 |
| Human bioavailability (%) | 24 (observed), 29 (predicted) | 70–91 (predicted) |
| Maximum PD response at steady state (%) | 55 | 60 |
| Projected QD dose to yield a ∼55% PD effect | 80 | 50 |
The human IC50 was defined as the rat IC50, as the rat is a predictive model for human. While backup C has lower potency, it has a higher projected bioavailability in human, based on rat PK data
IC50 50% inhibitory concentration, PD pharmacodynamic, QD quaque die
For IV profile projections, the Wajima et al. method has the advantage that it is easy to use with existing preclinical IV data and any value for human predicted CL or Vss can be used as input parameters (26), which makes this method ideal for a reverse pharmacology approach, unlike other methods (49). While IV data from at least two to three preclinical species are recommended (26) which can allow identification of species dependent PK, here, single species rat data were adequate since frontrunner data are available. Indeed, single species scaling using the rat has been recommended by others as a first-line approach for predicting human PK (17), and thus, this model allows to perform AHD at the lead optimization stage in drug development.
The reverse pharmacology PK/PD approach has several limitations. It assumes that rat CL is predictive for human CL (29) for backup C, as was the case for frontrunner B. It is further assumed that the rat pharmacology model and its generated PD parameters, e.g., IC50, are conserved in the human. Potential transporter involvements in clearance mechanism were not included in the model, as these data were not available at optimization development stages. Thus, the AHD approach should be verified when additional PK or PD data become available from higher species later in development.
Conclusion
Existing clinical data from frontrunner compounds can be used to aid in the prediction of PK/PD performance for backup candidates using rat data only. The Wajima et al. method is suitable to predict human PK profiles. Incorporation of rat PK/PD data allowed the early projection for human PK/PD for backup candidates in drug development.
SUMMARY DISCUSSIONS, MODELS LIMITATIONS, AND OUTLOOK
Human PK and PD projections should be carried out at early development stages to aid in compound differentiation, formulation risk identification, and estimation of drug supply needed for toxicokinetic and FIH studies (1,3). The case studies use practical and straightforward methods for CL, Vss, and F projections. Human CL and Vss can be obtained using various methods. Allometric methods including the rule of exponents (11–14,17,25) can be used to estimate human CL when in vivo data are available. In vitro methods based on the well-stirred model are commonly employed for hepatically cleared compounds (15,17). For Vss, the Oie–Tozer method can be used, among others (17). Single species allometry (rat) data or multispecies data can be used for predictions, and in some cases, single species data can be more predictive than multispecies allometry (17).
Successful PBPK strategies have now become mainstream with the availability of integrated softwares such as Simcyp™ and GastroPlus™ (20,50). Required human CL inputs are often derived from hepatocytes or microsomal systems (15,51), while tissue distribution for Vss can be predicted using mechanistic tissue composition equations (52–54) embedded in, e.g., GastroPlus™ PBPK modules. PBPK models can be used to predict concentration-time profiles. Plasma profiles can also be predicted using the Wajima et al. method (23,26). Formulation-dependent human PK/PD profiles can be readily generated using GastroPlus™ with its PBPK, and PK/PDPlus™ software modules (31), or other softwares, such as Simcyp™ (33). The integration of formulation parameters and biopharmaceutical properties into PK/PD projections using risk scenarios allows the identification of in vivo PK performance issues for all BCS class drugs. While integrated softwares have now become mainstream, unmet needs include (a) further refinement of mechanistic PBPK models for highly lipophilic compounds, (b) improvements in predicting food effects, and (c) integration of population PK/PD.
Human PK and PK/PD projections are a multidisciplinary approach that requires knowledge in all ADME fields including pharmacokinetics, pharmacology, formulation sciences, and physical pharmacy. In summary, (a) allometry-based methods and mechanistic PBPK approaches are “practical” and should both be employed, (b) metabolism-based IVIVE, species agreement, and concordance between methods can enhance confidence in PK/PD projections, (c) reverse pharmacology approaches utilizing the available preclinical and clinical data to predict accurate profile for the backup compounds should be used whenever possible, and (d) simulation approaches should include model scenarios as a risk assessment tool for PK/PD predictions.
CONCLUSIONS
The anticipation of human dose and PK/PD projections can be readily implemented at all stages in drug discovery and development. Commercial softwares now allow the practical integration of biopharmaceutical properties, PK, and PD to carry out formulation-dependent PK/PD modeling for preclinical and clinical settings. Conventional tools such as allometry, the Wajima et al. method, or PBPK models all can aid in the projection of CL, or Vss, which are required PK parameters to project human PK and PK/PD profiles. Prospective simulations using preclinical PK/PD data can be used to identify development risks. While plasma profiles from multiple species may be advantageous to identify species agreement or species-dependent metabolism/disposition (“disconnects”), human PK/PD projections can often be carried out using only rat data. In situations where clinical PK/PD data are available from frontrunners, a reverse pharmacology strategy can be used to establish links with preclinical PK/PD models to aid in selection of backup candidates. Integrated simulation tools in combination with established PK projection methods can greatly aid in the projection of formulation-dependent preclinical and human PK/PD profiles and can offer advantages of reducing animal studies, while optimizing PK/PD success potential in the clinic.
References
- 1.Lowe PJ, Hijazi Y, Luttringer O, Yin H, Sarangapani R, Howard D. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica. 2007;37:1331–1354. doi: 10.1080/00498250701648008. [DOI] [PubMed] [Google Scholar]
- 2.Thomas VH, Bhattachar S, Hitchingham L, Zocharski P, Naath M, Surendran N, et al. The road map to oral bioavailability: an industrial perspective. Expert Opin Drug Metab Toxicol. 2006;2:591–608. doi: 10.1517/17425255.2.4.591. [DOI] [PubMed] [Google Scholar]
- 3.Miller R, Ewy W, Corrigan Brian W, Ouellet D, Hermann D, Kowalski Kenneth G, et al. How modeling and simulation have enhanced decision making in new drug development. J Pharmacokinet Pharmacodyn. 2005;32:185–197. doi: 10.1007/s10928-005-0074-7. [DOI] [PubMed] [Google Scholar]
- 4.Huang C, Zheng M, Yang Z, Rodrigues AD, Marathe P. Projection of exposure and efficacious dose prior to first-in-human studies: how successful have we been? Pharm Res. 2008;25:713–726. doi: 10.1007/s11095-007-9411-4. [DOI] [PubMed] [Google Scholar]
- 5.Amidon GL, Lennernaes H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 6.Custodio JM, Wu C-Y, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, He H, Parthiban LJ, Yin H, Serajuddin ATM. IV–IVC considerations in the development of immediate-release oral dosage form. J Pharm Sci. 2005;94:1396–1417. doi: 10.1002/jps.20378. [DOI] [PubMed] [Google Scholar]
- 8.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2001;44:235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 9.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernaes H, Hussain AS, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 10.Gabrielsson J, Dolgos H, Gillberg PG, Bredberg U, Benthem B, Duker G. Early integration of pharmacokinetic and dynamic reasoning is essential for optimal development of lead compounds: strategic considerations. Drug Discov Today. 2009;14:358–372. doi: 10.1016/j.drudis.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Mahmood I. Prediction of human drug clearance from animal data: application of the rule of exponents and ‘fu corrected intercept method’ (FCIM) J Pharm Sci. 2006;95:1810–1821. doi: 10.1002/jps.20650. [DOI] [PubMed] [Google Scholar]
- 12.Mahmood I, Yuan R. A comparative study of allometric scaling with plasma concentrations predicted by species-invariant time methods. Biopharm Drug Dispos. 1999;20:137–144. doi: 10.1002/(SICI)1099-081X(199904)20:3<137::AID-BDD165>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 13.Tang H, Mayersohn M. A novel model for prediction of human drug clearance by allometric scaling. Drug Metab Dispos. 2005;33:1297–1303. doi: 10.1124/dmd.105.004143. [DOI] [PubMed] [Google Scholar]
- 14.Mahmood I. Interspecies pharmacokinetic scaling: allometric principles and applications. Rockville: Pine House; 2005. [Google Scholar]
- 15.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, Macintyre F, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 16.Obach RS, Lombardo F, Waters NJ. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab Dispos. 2008;36:1385–1405. doi: 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- 17.Hosea N, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, et al. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharm. 2009;49:513. doi: 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]
- 18.Oie S, Tozer TN. Effect of altered plasma protein binding on apparent volume of distribution. J Pharm Sci. 1979;68:1203–1205. doi: 10.1002/jps.2600680948. [DOI] [PubMed] [Google Scholar]
- 19.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RAHJ. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35:1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 20.Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45:511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 21.Luttringer O, Theil F-P, Poulin P, Schmitt-Hoffmann AH, Guentert TW, Lave T. Physiologically based pharmacokinetic (PBPK) modeling of disposition of epiroprim in humans. J Pharm Sci. 2003;92:1990–2007. doi: 10.1002/jps.10461. [DOI] [PubMed] [Google Scholar]
- 22.Parrott N, Jones H, Paquereau N, Lave T. Application of full physiological models for pharmaceutical drug candidate selection and extrapolation of pharmacokinetics to man. Basic Clin Pharmacol Toxicol. 2005;96:193–199. doi: 10.1111/j.1742-7843.2005.pto960308.x. [DOI] [PubMed] [Google Scholar]
- 23.Fura A, Vyas V, Humphreys W, Chimalokonda A, Rodrigues D. Prediction of human oral pharmacokinetics using nonclinical data: examples involving four proprietary compounds. Biopharm Drug Dispos. 2008;29:455–468. doi: 10.1002/bdd.632. [DOI] [PubMed] [Google Scholar]
- 24.Stoner CL, Cleton A, Johnson K, Oh D-M, Hallak H, Brodfuehrer J, et al. Integrated oral bioavailability projection using in vitro screening data as a selection tool in drug discovery. Int J Pharm. 2004;269:241–249. doi: 10.1016/j.ijpharm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Sinha VK, De Buck SS, Fenu LA, Smit JW, Nijsen M, Gilissen AHJ, et al. Predicting oral clearance in humans: how close can we get with allometry? Clin Pharmacokinet. 2008;47:35–45. doi: 10.2165/00003088-200847010-00004. [DOI] [PubMed] [Google Scholar]
- 26.Wajima T, Yano Y, Fukumura K, Oguma T. Prediction of human pharmacokinetic profile in animal scale up based on normalizing time course profiles. J Pharm Sci. 2004;93:1890–1900. doi: 10.1002/jps.20099. [DOI] [PubMed] [Google Scholar]
- 27.Dannenfelser R-M, He H, Joshi Y, Bateman S, Serajuddin ATM. Development of clinical dosage forms for a poorly water soluble drug I: application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–1175. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 28.Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10:516–525. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang H, Hussain A, Leal M, Mayersohn M, Fluhler E. Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metab Dispos. 2007;35:1886–1893. doi: 10.1124/dmd.107.016188. [DOI] [PubMed] [Google Scholar]
- 30.Tang H, Mayersohn M. A mathematical description of the functionality of correction factors used in allometry for predicting human drug clearance. Drug Metab Dispos. 2005;33:1294–1296. doi: 10.1124/dmd.105.004135. [DOI] [PubMed] [Google Scholar]
- 31.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Delivery Rev. 2001;50:S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 32.FDA. Estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers (draft guidance), 2002.
- 33.Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11:225–237. doi: 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19:359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 35.Gabrielsson J, Weiner D. Pharmacokinetic and pharmacodynamic data analysis: concepts and applications. 4. Baco Raton: CRC; 2000. [Google Scholar]
- 36.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 37.Lukacova V, Parrott NJ, Lavè T, Fraczkiewicz G, Bolger MB, Woltosz WS. Role of fraction unbound in plasma in calculations of tissue: plasma partition coefficients. AAPS National Meeting, Atlanta, Georgia, 2008.
- 38.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling: predicting the tissue distribution of moderate-to-strong bases. [Erratum to document cited in CA143:221762] J Pharm Sci. 2007;96:3151–3152. doi: 10.1002/jps.20856. [DOI] [PubMed] [Google Scholar]
- 39.Rodgers T, Rowland M. Physiologically-based pharmacokinetic modeling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2007;96:3153–3154. doi: 10.1002/jps.20857. [DOI] [PubMed] [Google Scholar]
- 40.Meibohm B, Derendorf H. Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modelling. Int J Clin Pharmacol Ther. 1997;35:401–413. [PubMed] [Google Scholar]
- 41.Kwan KC. Oral bioavailability and first-pass effects. Drug Metab Dispos. 1997;25:1329–1336. [PubMed] [Google Scholar]
- 42.Gao P, Morozowich W. Case studies: rational development of self-emulsifying formulations for improving the oral bioavailability of poorly soluble, lipophilic drugs. Drugs Pharm Sci. 2007;170:273–302. [Google Scholar]
- 43.Lave T, Coassolo P, Reigner B. Prediction of hepatic metabolic clearance based on interspecies allometric scaling techniques and in vitro–in vivo correlations. Clin Pharmacokinet. 1999;36:211–231. doi: 10.2165/00003088-199936030-00003. [DOI] [PubMed] [Google Scholar]
- 44.Hellriegel ET, Bjornsson TD, Hauck WW. Interpatient variability in bioavailability is related to the extent of absorption: implications for bioavailability and bioequivalence studies. Clin Pharmacol Ther. 1996;60:601–607. doi: 10.1016/S0009-9236(96)90208-8. [DOI] [PubMed] [Google Scholar]
- 45.Dressman J, Reppas C. Drug solubility: how to measure it, how to improve it. Adv Drug Deliv Rev. 2007;59:531–532. doi: 10.1016/j.addr.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Jusko WJ, Ko HC. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther. 1994;56:406–419. doi: 10.1038/clpt.1994.155. [DOI] [PubMed] [Google Scholar]
- 47.Adolph EF. Quantitative relations in the physiological constitution of mammals. Science. 1949;109:579–585. doi: 10.1126/science.109.2841.579. [DOI] [PubMed] [Google Scholar]
- 48.Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm Res. 1998;15:1792–1795. doi: 10.1023/A:1011981317451. [DOI] [PubMed] [Google Scholar]
- 49.Dedrick RL, Bischoff KB, Zaharko DS. Interspecies correlation of plasma concentration history of methotrexate (NSC-740) Cancer Chemother Rep. 1970;54:95–101. [PubMed] [Google Scholar]
- 50.Gibson CR, Bergman A, Lu P, Kesisoglou F, Denney WS, Mulrooney E. Prediction of phase I single-dose pharmacokinetics using recombinant cytochromes P450 and physiologically based modelling. Xenobiotica. 2009;1–12. http://www.informahealthcare.com/action/doSearch. [DOI] [PubMed]
- 51.Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22:103–112. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 52.Poulin P, Schoenlein K, Theil F-P. Prediction of adipose tissue:plasma partition coefficients for structurally unrelated drugs. J Pharm Sci. 2001;90:436–447. doi: 10.1002/1520-6017(200104)90:4<436::AID-JPS1002>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 53.Poulin P, Theil F-P. A priori prediction of tissue:plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci. 2000;89:16–35. doi: 10.1002/(SICI)1520-6017(200001)89:1<16::AID-JPS3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 54.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94:1259–1276. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 55.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45:1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]