

Abstract
Drug-induced hepatotoxicity is one of the major problems encountered in drug discovery and development. Selection of a candidate compound for pre-clinical studies in the drug discovery process is a critical step that can determine the speed and expenditure of clinical development. Because inhibition of human adenosine triphosphate-binding cassette transporter ABCB11 (SPGP/bile salt export pump) has severe consequences, which include intrahepatic cholestasis and hepatotoxicity, resulting from exposure to toxic xenobiotics or drug interactions, in vitro screening methods are necessary for quantifying and characterizing the inhibition of ABCB11. In line with such initiatives, we developed methods for in vitro high-speed screening and quantitative structure-activity relationship (QSAR) analysis to investigate the interaction of ABCB11 with a variety of compounds. We identified one set of chemical fragmentation codes closely linked with inhibition of ABCB11. Furthermore, the high-speed screening method enables us to analyze the kinetics of ABCB11-inhibition by test compounds and to distinguish competitive and non-competitive inhibitors. Troglitazone and novobiocin were found to be competitive inhibitors to taurocholate, whereas porphyrins were non-competitive inhibitors. Kinetics-based classification of inhibitors is considered important to improve the accuracy of our QSAR analysis. The present mini-review addresses technical pitfalls and improvements for high-speed screening and QSAR analysis in the ABCB11 inhibition study.
Key words: ABCB11, bile salt, inhibition, intrahepatic cholestasis, QSAR analysis
INTRODUCTION
Bile is a vital secretion essential for intestinal digestion and absorption of lipids. Moreover, bile is an important route of elimination for environmental toxins, carcinogens, xenobiotics including drugs, and their metabolites. Bile secretion is also a major route of excretion for endogenous compounds and metabolic products (endobiotics) such as cholesterol, bilirubin, and steroid hormones (1–3). Bile is primarily secreted by hepatocytes into minute channels arranged as a network of tubules or canaliculi located between adjacent hepatocytes. Canalicular bile accounts for approximately 75% of the daily bile production in humans as it passes along the bile ductules and ducts. In humans, bile is further concentrated up to five- to ten-fold in the gallbladder (4). The human bile salt pool amounts to 50 to 60 μmol/kg of body weight and averages 3 to 4 g (3).
Bile secretion is an osmotic process driven predominantly by the active excretion of organic solutes into bile canaliculi, followed by the passive inflow of water, electrolytes, and nonelectrolytes (e.g., glucose) from hepatocytes and across semipermeable tight junctions. The main organic bile solutes of bile are bile salts, phospholipids, and cholesterol, which form mixed micelles in bile. Extrusion of bile salts from the hepatocytes is also critical for liver cell viability, as the intracellular accumulation of bile salts may lead to cell death by apoptosis and/or necrosis (5).
In humans, the classical bile acid synthetic pathway predominates, with only about 10% of bile acids being produced via the alternative pathway. In humans, over 70% of the bile acid pool consists of cholic acid (approximately 50%) and its bacterial metabolite deoxycholic acid ( approximately 20%) resulting from the classical pathway, whereas chenodeoxycholic acid formed from the alternative pathway constitutes 30% of the total bile acid pool (2). After being synthesized in hepatocytes, bile acids are conjugated with glycine or taurine. Under normal condition, conjugated bile salts are excreted into bile via the bile salt export pump (BSEP), an adenosine triphosphate (ATP)-binding cassette (ABC) transporter localized in the canalicular or apical domain of the hepatocyte plasma membrane. BSEP is also called ABCB11 according to the international nomenclature of human ABC transporter genes.
MOLECULAR PROPERTIES OF ABCB11
In 1995, the BSEP was originally discovered as a sister gene of P-glycoprotein (Spgp) in the pig (6), although its function was not clear at that time. Successive studies revealed that Spgp transports various bile salts (7) and that disruption of the Spgp gene in mice caused persistent intrahepatic cholestasis (8). In humans, the BSEP is encoded by the ABCB11 gene, which belongs to the human ABC transporter gene family (9). Human ABCB11 is a 170-kDa glycoprotein that consists of 1,321 amino acid residues and plays a major role in the hepatobiliary excretion of conjugated bile salts (10,11). The amino acid sequences of ABCB11 and ABCB1 (P-glycoprotein/MDR1) share about 50% identity and about 70% similarity. While ABCB11 is closely related to ABCB1, it is more selective in tissue distribution and substrate recognition. Under normal conditions, conjugated bile salts are excreted into bile via ABCB11 with the rank order of taurochenodeoxycholate ≥ taurocholate > tauroursodeoxycholate > glycocholate (12). Several mutations in the ABCB11 gene located on chromosome 2q24 have been reported to be associated with progressive familial intrahepatic cholestasis type 2 (10,13,14). These patients have extremely low biliary concentrations of conjugated bile salts.
Inhibition of ABCB11 is a potential mechanism for the development of several acquired cholestatic liver diseases, including drug-induced intrahepatic cholestasis. Taurocholate transport by rat ABCB11 was competitively inhibited by the immunosuppressive drug cyclosporin A, the sulfonylurea antidiabetic drug glibenclamide (15,16), and the endothelin receptor antagonist bosentan (17). Use of the insulin sensitizer troglitazone for the treatment of type-2 diabetes has been shown to inhibit taurocholate transport in vivo as well as in canalicular membrane vesicles prepared from the rat liver (15).
QUALITY CONTROL OF PLASMA MEMBRANE VESICLES PREPARED FROM Sf9 CELLS
The isolated membrane vesicle system provides a practical tool for low cost and high-throughput analysis of ABC multidrug transporters. Baculovirus-infected insect cells have successfully been employed to give relatively high protein expression yields; for example, Spodoptera frugiperda (Sf9) cells are widely used to obtain membranes overexpressing various ABC transporters. We usually infect Sf9 cells (1 × 106 cells/ml) with human ABCB11-recombinant baculovirus and culture them at 26°C with gentle shaking. The baculovirus has a 130-kb double-stranded DNA genome, packaged in a cigar-shaped (25 × 260 nm) enveloped nucleocapsid. Baculovirus enters insect cells via receptor-mediated endocytosis (18). The viral fusion protein gp64 is responsible for acid-induced endosomal escape (19). In the cytoplasm, the nucleocapsid probably induces the formation of actin filaments, which provide a possible mode of transport toward the nucleus (20,21). Figure 1a (upper panel) shows electron microscopic images, demonstrating that baculovirus particles were transported into the nucleus of Sf9 cells 3 days after infection.
Fig. 1.

Electron microscopic observation of Sf9 cells infected with ABCG2-recombinant baculovirus (a), plasma membrane vesicles prepared from Sf-9 cells (b), and effect of leupeptine on ABCB11-mediated taurocholate (TC) transport (c). The cultured Sf9 cells were fixed with Karnovsky’s fixative (5% (v/v) glutaraldehyde, 80 mM cacodylate buffer, pH 7.4, 4% (w/v) performaldehyde), post-fixed with 2% osmium tetroxide at 4°C for 2 h and 0.05% ruthenium tetroxide at room temperature for 5 min in the dark. After being dried by t-butyl freeze drying, the specimens were coated with magnetron sputtering (platinum, 2 nm) and viewed with a high resolution scanning electron microscopy (SEM), S-4300 (Hitachi, Japan), at an acceleration voltage of 2 kV. Otherwise, the specimens were coated with 2-nm-thick platinum-carbon in a high vacuum freeze-etch unit BAF 301 by using an electron gun EK522 (Balzers, Liechtenstein) before observation and viewed with an ultra-high resolution (UHR)-low voltage (LV)-SEM S-900LV commercially available as Model S-900H (Hitachi, Japan) at an acceleration voltage of 2 kV. The plasma membrane vesicles were fixed with 2% (v/v) glutaraldehyde, 0.1 M cacodylate buffer, pH 7.4, containing 0.4 M sorbitol for 1 h at 4°C. The specimens were viewed with an UHR-SEM 5200 (Hitachi, Japan) at an acceleration voltage of 2 kV as described previously (37,38). To observe the ultrastructure of vesicles, the specimens prepared with negative staining were observed by TEM JEM 1200EX (JEOL, Japan) at an acceleration voltage of 80 kV. Negative staining was done with a 3% aqueous solution of uranyl acetate
The use of low ionic strength buffers during the membrane preparation steps promotes the formation of open membrane sheets and inside-out membrane vesicles. It is important to maintain high integrity of the plasma membrane vesicles used in the transport assay. In other words, the membrane vesicles have to be completely sealed. To examine the quality of plasma membrane vesicles prepared from Sf9 cells, we use transmission electron microscopy (TEM) and scanning electron microscopy (SEM) technologies and identify the optimal conditions required to prepare the membrane vesicles. The timing of harvesting Sf9 cells after baculovirus infection appears to be very critical. As demonstrated in Fig. 1a (lower panels), the membrane morphology of infected Sf9 cells changed greatly; in particular, numerous pores were observed on day 5. Membrane vesicles prepared from those cells (day 5) are useless for our purpose. Figure 1b demonstrates negatively stained TEM and SEM images of the well-sealed membrane vesicles whose average size was estimated to be about 200 nm. It is also important to prepare membrane vesicles in the presence of serine/cysteine protease inhibitors. Leupeptin (10 μg/ml) inhibited the degradation of ABCB11 protein in membrane vesicles prepared from baculovirus-infected Sf9 cells during repetitive freeze-thaw cycles (Fig. 1c).
Membrane vesicles (suspended in 250 mM sucrose and 10 mM Tris/4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4) can be stored at −80°C or in liquid nitrogen until used. For long-term (over 1 year) storage of membrane vesicles, however, we recommend substituting tolehalose for sucrose in the membrane vesicle preparations. Trehalose (α-D-glucopyranosyl α-D-glucopyranoside) is a non-reducing disaccharide composed of two glucose molecules joined by an α,α-1,1 linkage. Trehalose is a stress protectant in biological systems as it interacts with and directly protects lipid membranes and proteins from desiccation and during freezing (22,23). In the field of medicine, evidence is accumulating that trehalose is superior to other disaccharides for the preservation of organs (24). In fact, the transport activity of ABCB11 was maintained at high levels in membrane vesicles that had been preserved for 1 year with 250 mM trehalose (data not shown).
HIGH-SPEED SCREENING OF HUMAN ABCB11 INHIBITION
To discern the contribution of ABCB11 to drug interactions, it is critical to explore and demonstrate methods for characterizing and quantifying the inhibition of ABCB11-mediated transport. Recently, we developed a high-speed screening system by introducing 96-well MultiScreen™ plates and an automated multi-dispenser system (25–27). Figure 2a depicts the scheme for the high-speed screening of ATP-dependent [14C]taurocholate transport mediated by ABCB11. ABCB11-mediated [14C]taurocholate transport exhibits saturation kinetics with an apparent Km value of about 10 μM for taurocholate. We have used this system to investigate the interaction of ABCB11 with a variety of test compounds. We selected structurally diverse test compounds to examine our hypothesis that common chemical components may be involved in the inhibition of ABCB11-mediated taurocholate transport. The selected test compounds were classified into seven groups, i.e., (a) neurotransmitters, (b) vasodilators, (c) potassium channel modulators, (d) steroids, (e) non-steroidal anti-inflammatory drugs, (f) anti-cancer drugs, and (g) miscellaneous. Figure 2b summarizes the effects of those test compounds on ABCB11-mediated taurocholate transport. It is important to note that the inhibition profile for ABCB11 is very similar to the ATPase profile for ABCB1. Vasodilators, e.g., fendiline (B-5), prenylamine (B-6), and nicardipine (B-7) strongly inhibited ABCB11-mediated taurocholate transport. Those drugs also stimulated the ATPase activity of ABCB1 expressed in the plasma membrane fraction from Sf9 insect cells (28–30), suggesting a close relationship between ABCB11 and ABCB1 in terms of drug interactions. Conjugated bile salts are synthesized in the liver by the enzymatic modification of cholesterol and comprise a steroid nucleus and an aliphatic side chain. Steroids, such as dexamethasone (C-1), betamethasone (C-2), prednisolone (C-3), and cortisone (C-4), however, did not significantly inhibit ABCB11-mediated taurocholate transport. These findings suggest that ABCB11 preferentially recognizes conjugated bile salts as substrates.
Fig. 2.

Procedure to measure adenosine triphosphate (ATP)-dependent taurocholate transport mediated by ABCB11 (a) and inhibition of ABCB11-mediated taurocholate transport by test compounds and their profiling (b). The standard incubation medium contains the plasma membrane vesicles (50 μg of protein), 20 μM [14C]taurocholate, 0.25 M sucrose, 10 mM Tris/4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, 10 mM MgCl2, 1 mM ATP, 10 mM creatine phosphate, and 100 μg/ml creatine kinase in a final volume of 100 μl. The incubation should be carried out at 37°C. After a specified time (15 min for the standard condition), the reaction medium is mixed with 1 ml of ice-cold stop solution (0.25 M sucrose, 10 mM Tris/HEPES, pH 7.4, and 2 mM ethylenediaminetetraacetic acid (EDTA)) to terminate the transport reaction. The chelating agent EDTA traps Mg2+ prerequisite for the ATP-dependent transport function of ABCB11. Subsequently, aliquots (270 μl per well) of the resulting mixture are transferred to MultiScreen™ plates (Millipore Corp., Billerica, MA, USA). Under aspiration, each well of the plate is rinsed four times with the 0.25-M sucrose solution containing 10 mM Tris/HCl, pH 7.4, (4 × 200 μl for each well) in an EDR384S system (BioTec, Tokyo, Japan). The [14C]taurocholate thus incorporated into the vesicles can be measured by counting the radioactivity remaining on the filter of the MultiScreen™ plates. ATP-dependent [14C]taurocholate transport was measured in the presence of a test compound (100 μM) in the standard incubation medium (0.25 M sucrose and 10 mM Tris/HEPES, pH 7.4, 10 mM creatine phosphate, 100 μg/ml creatine kinase, 10 mM MgCl2). Inhibition (percent) is expressed as relative values compared with the transport activity measured without test compounds (0% inhibition). The test compounds used are classified into seven groups, i.e., (a), neurotransmitters, (b) vasodilators, (c) steroids, (d) potassium channel modulators, (e) non-steroidal anti-inflammatory drugs, (f) anti-cancer drugs, (g) miscellaneous: glycine (a-1), glutamic acid (a-2), dopamine (a-3), norepinephrine (a-4), epinephrine (a-5), GABA (a-6), histamine (a-7), serotonin (a-8), melatonin (a-9), verapamil (b-1), nifedipine (b-2), diltiazem (b-3), bepridil (b-4), fendiline (b-5), prenylamine (b-6), nicardipine (b-7), dexamethasone (c-1), betamethasone (c-2), prednisolone (c-3), cortisone (c-4), nicorandil (d-1), pinacidil (d-2), acetylsalicylic acid (e-1), indomethacin (e-2), acemetacin (e-3), ibuprofen (e-4), naproxen (e-5), mepirizole (e-6), vinblastine (f-1), etoposide (f-2), actinomycin D (f-3), daunorubicin (f-4), paclitaxel (f-5), methotrexate (f-6), doxorubicin (f-7), 5-fluorouracil (f-8), mitoxantrone (f-9), quinidine (g-1), p-aminohippuric acid (g-2), penicillin G (g-3), novobiocin (g-4), prazosin (g-5). These figures have been reproduced from our previous publication (26) with permission from the American Chemical Society
QSAR ANALYSIS USING CHEMICAL FRAGMENTATION CODES
To gain more insight into the relationship between the chemical structures of the test compounds and the inhibition of ABCB11-mediated taurocholate transport activity, we have recently developed a new quantitative structure-activity relationship (QSAR) analysis method by introducing the use of “chemical fragmentation codes” to describe the chemical structures of a variety of drugs and natural compounds (19).
The chemical fragmentation codes were originally created to answer the need for accessing information from the increasing numbers of chemical patents. These chemical fragmentation codes are a set of alphanumeric symbols, each representing a fragment of a chemical structure that was developed by Derwent Information, Ltd. (current name Thomas Reuters), as a structure-indexing language, which is suitable for describing chemical structures. The Markush TOPFRAG program is a tool for searching the chemical structures and structural information in Derwent’s online databases (31,32).
We have applied these chemical fragmentation codes to the QSAR analysis of ABCB11-drug interactions. Our approach is unique in that the extent of ABCB11-mediated taurocholate transport inhibition is described as a linear combination of chemical fragmentation codes, and that the coefficient “C(i)” for each chemical fragmentation code reflects the extent of the contribution of a specific chemical moiety to interactions with the ABCB11 protein. Namely, the chemical fragmentation coefficient is defined as the contribution to the activity (here the ability to inhibit taurocholate transport) that is attributable to the presence of a particular chemical moiety in the test compound. We formulate the extent of inhibition of ABCB11-mediated taurocholate transport as a linear combination of chemical fragmentation codes, each of which is weighted by the corresponding coefficient as follows:
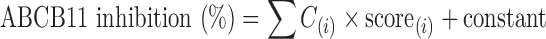 |
where the symbol (i) designates a specific chemical fragmentation (i). The “score” means the presence [≥1] or absence [0] of the corresponding chemical fragmentation code (i) in the chemical structure of a test compound (see Table I). Based on the chemical fragmentation codes thus obtained and in comparison with the observed inhibition of transport activity for each test compound, we have calculated chemical fragmentation coefficients, C(i), by multiple linear regression analysis that delineate a relationship between the structural components and the extent of ABCB11 inhibition. In this way, we could identify one set of chemical fragmentation codes that are closely related to the inhibition of ABCB11 transport activity. Explanations for these chemical fragmentation codes are also given in Table I. We use the descriptors of M132, ESTR, R-CC, H181, MN-HC, and OH-ALP to represent multiple chemical fragmentation codes. Based on the results of the multiple linear regression analysis, we calculated the values of predicted inhibition and compared them with the observed values. As demonstrated in Table I and Fig. 3a, the prediction of transport inhibition correlated well with the observed values of inhibition. The R2 value was estimated to be 0.952.
Table I.
Definition of Descriptors and Chemical Fragmentation Codes (CFC) Contributing to Inhibition of ABCB11-mediated Taurocholate Transport
| Descriptor | Coefficient (95% reliability) | CFC | Definition | Score |
|---|---|---|---|---|
| M132 | 35.07 (±8.99) | M132 | Ring-linking group containing one C atom (except for 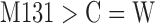 , W: hetero atom) , W: hetero atom) |
1 |
| ESTR | 31.47 (±4.97) | J211 | One ester (thioester) group bonded to heterocyclic 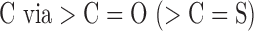
|
1 |
| J212 | Two or more ester (thioester) groups bonded to heterocyclic 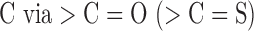
|
2 | ||
| R-CC | 14.41 (±2.91) | M530 | No carbocyclic system with at least one aromatic ring | 0 |
| M531 | One carbocyclic system with at least one aromatic ring | 1 | ||
| M532 | Two carbocyclic systems with at least one aromatic ring | 2 | ||
| M533 | Three or more carbocyclic systems with at least one aromatic ring | 3 | ||
| H181 | 10.95 (±4.86) | H181 | One amine bonded to aliphatic C | 1 |
| MN-HC | 9.66 (±.22) | M520 | No mononuclear heterocycle | 0 |
| M521 | One mononuclear heterocycle | 1 | ||
| M522 | Two mononuclear heterocycles | 2 | ||
| M523 | Three or more mononuclear heterocycles | 3 | ||
| OH-ALP | −15.23 (±5.06) | H481 | One −OH group bonded to aliphatic C | 1 |
| H482 | Two −OH groups bonded to aliphatic C | 2 | ||
| H483 | Three −OH groups bonded to aliphatic C | 3 | ||
| H484 | Four or more −OH groups bonded to aliphatic C | 4 | ||
| Constant | −9.50 | 1 |
R = 0.976; R 2 = 0.952; F(30,6) = 99.1; s = 6.67
Descriptors, CFC, coefficients, and constants were deduced from the inhibition of ABCB11-mediated taurocholate transport by test compounds
Data are from (26) with permission
Fig. 3.

Relationship between observed and predicted values in the inhibition of taurocholate transport by the test compounds (a) and chemical structure of troglitazone and its descriptors and chemical fragmentation codes associated with the inhibition of ABCB11-mediated taurocholate transport (b). The predicted values for the inhibition of ABCB11 by test compounds were deduced from the multiple linear regression analysis as described in the text. The inhibition by troglitazone is indicated by an open circle. These figures have been reproduced from our previous publication (26) with permission from the American Chemical Society
The structural components represented by the chemical fragmentation codes of M132 and H181 as well as by the descriptors of ESTR, R-CC, and MN-HC positively contribute to the inhibition, whereas the descriptor of OH-ALP (−OH groups bonded to aliphatic carbon) had a negative contribution. As summarized in Table I, the descriptor ESTR including chemical fragmentation codes of J211 and J212 had a relatively large positive coefficient, C(ESTR) = 31.47, suggesting that an ester (thioester) group bonded to a carbon of heterocyclic ring(s) is an important component for the interaction with the ABCB11 protein. In addition, the data for R-CC (Table I) suggest that carbocyclic systems with at least one aromatic ring are also important chemical moieties for the interaction with ABCB11. These QSAR profiles for ABCB11 are distinct from those for ABCG2 (27).
APPLICATION OF QSAR TO PREDICT INHIBITION OF ABCB11 BY TROGLITAZONE
In the case of troglitazone, the chemical fragmentation codes of M132, J211, M531, and M521 are involved in its chemical structure. Structural components corresponding to those chemical fragmentation codes are indicated in Fig. 3b. Based on the QSAR calculation 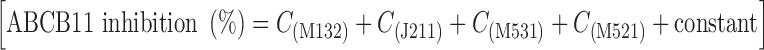 , the inhibition of taurocholate transport by troglitazone is estimated to be 81.11%, which is in accordance with the observed value of 90.06% (Fig. 3a).
, the inhibition of taurocholate transport by troglitazone is estimated to be 81.11%, which is in accordance with the observed value of 90.06% (Fig. 3a).
It has been reported that troglitazone sulfate was efficiently formed and accumulated in liver tissue and that it strongly inhibited rat ABCB11 (IC50 value of 0.4–0.6 μM; 15,16). Therefore, it is suggested that troglitazone sulfate is responsible for the interaction of conjugated bile salts with hepatobiliary export in rats. Based on those animal model and in vitro studies, inhibition of ABCB11 by troglitazone sulfate has been implicated as a potential cause for troglitazone-induced intrahepatic cholestasis in humans (15,16). Hitherto, extensive studies have been carried out to elucidate mechanisms underlying the troglitazone hepatotoxicity. Recent studies suggest that troglitazone is metabolized to a reactive metabolite that covalently binds to cellular macromolecules (33–35). Nevertheless, the mechanism for hepatotoxicity caused by troglitazone is still controversial.
ANALYSIS OF INHIBITION KINETICS
In order to obtain more insight into molecular mechanisms underlying the inhibition of ABCB11-mediated taurocholate transport, we have performed kinetic analyses. Figure 4a depicts Lineweaver–Burk plots, demonstrating that troglitazone inhibits ABCB11-mediated taurocholate transport in a competitive manner (Ki = 12 μM). These results suggest that taurocholate and troglitazone commonly share a substrate-binding site(s) in the ABCB11 protein. Conversely, pheophorbide a inhibited ABCB11-mediated taurocholate transport in a non-competitive manner (Ki = 4 μM; Fig. 4b). The non-competitive inhibition kinetics imply that pheophorbide a is bound to both ABCB11 and the ABCB11-taurocholate complex. The potential pheophorbide a-binding site may be distinct from the putative substrate-binding site. Recently the molecular structure of humen ABCB1 (P-glycoprotein/MDR1) has been analyzed by X-ray crystallography (36), demonstrating a poly-specific drug binding to the ABCB1 protein. Likewise, it is conceivable that human ABCB11 may also have multiple drug binding sites.
Fig. 4.

Lineweaver–Burk plots for the inhibition of ABCB11-mediated [14C]taurocholate transport by troglitazone (a) and pheophorbide a (b), as well as chemical structures of troglitazone, novobiocin, pheophorbide a, and hematoporphirin (c). ABCB11-mediated [14C]taurocholate transport was measured by a 15-min incubation at 37°C in the presence of either troglitazone (a) or pheophorbide a (b) at the different concentrations indicated in the figure. The concentrations of [14C]taurocholate were 5, 10, 20, or 40 μM in the incubation medium. In principle, the other experimental conditions and procedures were same as those described in Fig. 2a. The corresponding inhibition mechanisms are schematically illustrated under the Lineweaver–Burk plots. TC taurocholate, Trog troglitazone, Pheo pheophorbide a. Chemical structures of troglitazone, novobiocin, pheophorbide a, and hematoporphyrin (c). Experimentally observed and quantitative structure-activity relationship-based predicted values for the inhibition of taurocholate transport are compared. The observed values are expressed as mean values ± SD. (n = 3)
Since Lineweaver–Burk plots exhibit characteristic patterns depending on the type of inhibition kinetics, we are able to distinguish competitive and non-competitive inhibitors. Troglitazone and novobiocin were found to be competitive inhibitors to taurocholate, whereas porphyrins, e.g., pheophorbide a and hematoporphyrin, were non-competitive inhibitors (Fig. 4c). For pheophorbide a and hematoporphyrin, there are discrepancies between the experimental data and the predicted data based on the QSAR analysis (Fig. 4c), suggesting that it is important to perform QSAR analysis by considering the inhibition mechanism represented by kinetics patterns. In other words, inhibition kinetics should be analyzed by functional screening, and tested compounds (inhibitors) should be classified before QSAR analysis. Kinetics-based classification of inhibitors would improve the accuracy of QSAR analysis-based prediction and facilitate our understanding of molecular mechanisms involved in the inhibition of ABCB11-mediated bile salt transport by drugs.
CONCLUDING REMARKS
The high-speed screening method and QSAR strategy are practical and useful for the identification of inhibitors for ABCB11. To improve the accuracy of our QSAR analysis, however, we need to critically control the quality of the plasma membrane vesicles that are used for high-speed screening. In this review article, we have provided several hints and methods for preparing and maintaining the high-quality plasma membrane vesicles. In addition, kinetics-based classification of inhibitors is considered important to improve the accuracy of our QSAR analysis. By considering the kinetics of ABCB11 inhibition in QSAR analysis, we would be able to more accurately evaluate the interactions with ABCB11-mediated taurocholate transport.
There are multiple criteria in the decision of “go” or “no go” to candidate nomination for pre-clinical research or to new drug development. One of the criteria is the inhibition of ABCB11-mediated bile salt transport by a new chemical entity of interest. If a new chemical compound strongly inhibits ABCB11-mediated bile salt transport (IC50 < 1 μM), we would not recommend further development of the compound because of a risk of drug-induced intrahepatic cholestasis.
Acknowledgements
The study carried out in the authors’ laboratory was supported, in part, by the NEDO International Joint Research Grant program “International standardization of functional analysis technology for genetic polymorphisms of drug transporters” and research grants (No. 18201041 and No. 19659136) from the Japanese Society for Promotion of Science (JSPS). Hikaru Saito is a JSPS fellow. Under the license agreement (license number 2235670436990) with the American Chemical Society, Figs. 2 and 3 as well as Table I in the present review article have been reproduced from our previous study (26) published in Molecular Pharmaceutics.
References
- 1.Nathanson MH, Boyer JL. Mechanisms and regulation of bile secretion. Hepatology. 1991;14:551–66. doi: 10.1002/hep.1840140324. [DOI] [PubMed] [Google Scholar]
- 2.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–71. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 3.Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126:322–42. doi: 10.1053/j.gastro.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Carey MC, Small DM. The physical chemistry of cholesterol solubility in bile. Relationship to gallstone formation and dissolution in man. J Clin Invest. 1978;61:998–1026. doi: 10.1172/JCI109025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higuchi H, Gores GJ. Bile acid regulation of hepatic physiology: IV. Bile acids and death receptors. Am J Physiol Gastrointest Liver Physiol. 2003;284:G734–8. doi: 10.1152/ajpgi.00491.2002. [DOI] [PubMed] [Google Scholar]
- 6.Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995;55:2029–34. [PubMed] [Google Scholar]
- 7.Gerloff T, Stieger B, Hagenbuch B, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–50. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 8.Wang R, Salem M, Yousef IM, et al. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A. 2001;98:2011–6. doi: 10.1073/pnas.031465498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 10.Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–8. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 11.Arrese M, Ananthanarayanan M. The bile salt export pump: molecular properties, function and regulation. Pflugers Arch. 2004;449:123–31. doi: 10.1007/s00424-004-1311-4. [DOI] [PubMed] [Google Scholar]
- 12.Noe J, Stieger B, Meier PJ. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology. 2002;123:1659–66. doi: 10.1053/gast.2002.36587. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Soroka CJ, Boyer JL. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest. 2002;110:965–72. doi: 10.1172/JCI15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pauli-Magnus C, Lang T, Meier Y, et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14:91–102. doi: 10.1097/00008571-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Funk C, Ponelle C, Scheuermann G, Pantze M. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol. 2001;59:627–35. [PubMed] [Google Scholar]
- 16.Funk C, Pantze M, Jehle L, et al. Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology. 2001;167:83–98. doi: 10.1016/S0300-483X(01)00460-7. [DOI] [PubMed] [Google Scholar]
- 17.Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–31. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- 18.Wang P, Hammer DA, Granados RR. Binding and fusion of Autographa californica nucleopolyhedrovirus to cultured insect cells. J Gen Virol. 1997;78:3081–9. doi: 10.1099/0022-1317-78-12-3081. [DOI] [PubMed] [Google Scholar]
- 19.Blissard GW, Wenz JR. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J Virol. 1992;66:6829–35. doi: 10.1128/jvi.66.11.6829-6835.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanir LM, Volkman LE. Actin binding and nucleation by Autographa californica M nucleopolyhedrovirus. Virology. 1998;243:167–77. doi: 10.1006/viro.1998.9065. [DOI] [PubMed] [Google Scholar]
- 21.Whittaker GR, Helenius A. Nuclear import and export of viruses and virus genomes. Virology. 1998;246:1–23. doi: 10.1006/viro.1998.9165. [DOI] [PubMed] [Google Scholar]
- 22.Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13:17R–27. doi: 10.1093/glycob/cwg047. [DOI] [PubMed] [Google Scholar]
- 23.Furuki T, Oku K, Sakurai M. Thermodynamic, hydration and structural characterization of alpha, alpha-trehalose. Front Biosci. 2009;14:3523–35. doi: 10.2741/3468. [DOI] [PubMed] [Google Scholar]
- 24.Guo N, Puhlev I, Brown DR, et al. Trehalose expression confers desiccation tolerance on human cells. Nat Biotechnol. 2008;18:168–71. doi: 10.1038/72616. [DOI] [PubMed] [Google Scholar]
- 25.Ishikawa T, Sakurai A, Kanamori Y, et al. High-speed screening of human ATP-binding cassette transporter function and genetic polymorphisms: new strategies in pharmacogenomics. Methods Enzymol. 2005;400:485–510. doi: 10.1016/S0076-6879(05)00027-3. [DOI] [PubMed] [Google Scholar]
- 26.Hirano H, Kurata A, Onishi Y, et al. High-speed screening and QSAR analysis of human ATP-binding cassette transporter ABCB11 (bile salt export pump) to predict drug-induced intrahepatic cholestasis. Mol Pharmacol. 2006;3:252–65. doi: 10.1021/mp060004w. [DOI] [PubMed] [Google Scholar]
- 27.Saito H, Hirano H, Nakagawa H, et al. A new strategy of high-speed screening and quantitative structure-activity relationship analysis to evaluate human ATP-binding cassette transporter ABCG2-drug interactions. J Pharmacol Exp Ther. 2006;317:1114–24. doi: 10.1124/jpet.105.099036. [DOI] [PubMed] [Google Scholar]
- 28.Sakurai A, Onishi Y, Hirano H, et al. Quantitative SAR analysis and molecular dynamic simulation to functionally validate nonsynonymous polymorphisms of human ABC transporter ABCB1. Biochemistry. 2007;46:7678–93. doi: 10.1021/bi700330b. [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa T, Onishi Y, Hirano H, et al. Pharmacogenomics of drug transporters: a new approach to functional analysis of the genetic polymorphisms of ABCB1 (P-glycoprotein/MDR1) Biol Pharm Bull. 2004;27:939–48. doi: 10.1248/bpb.27.939. [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa T, Hirano H, Onoshi Y, Sakurai A, Tarui S. Functional evaluation of ABCB1 (P-glycoprotein) polymorphisms: high-speed screening and structure-activity relationship analyses. Drug Metab Pharmacokinet. 2004;19:1–14. doi: 10.2133/dmpk.19.1. [DOI] [PubMed] [Google Scholar]
- 31.Derwent T. World Patents Index, CPI Chemical Indexing Guidelines Indexing of Chemical and Pharmaceutical Patents. Available at: http://www.thomsonscientific.com/media/scpdf/chemical_index_guidelines.pdf
- 32.Thomson Reuters. Glossary of Thomson Scientific terminology. Available at: http://scientific.thomson.com/support/patents/patinf/terms/
- 33.Masubuchi Y. Metabolic and non-metabolic factors determining troglitazone hepatotoxicity: a review. Drug Metab Pharmacokinet. 2006;21:347–56. doi: 10.2133/dmpk.21.347. [DOI] [PubMed] [Google Scholar]
- 34.Smith MT. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol. 2003;16:679–87. doi: 10.1021/tx034033e. [DOI] [PubMed] [Google Scholar]
- 35.Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced hepatotoxicity. Annu Rev Pharmacol Toxicol. 2005;45:177–202. doi: 10.1146/annurev.pharmtox.45.120403.100058. [DOI] [PubMed] [Google Scholar]
- 36.Aller SG, Yu J, Ward A, Weng Y, Chittabonia ZR, Harrell PM, Trinh YT, Zheng Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osumi M, Yamada N, Kobori H, Taki A, Naito N, Baba M, Nagatani T. Cell wall formation in regenerating protoplasts of Schizosaccharomyces pombe: study by high resolution, low voltage scanning electron microscopy. J Electron Microsc (Tokyo) 1989;38:457–68. [PubMed] [Google Scholar]
- 38.Osumi M, Yamada N, Yaguchi H, Kobori H, Nagatani T, Sato M. Ultrahigh-resolution low-voltage SEM reveals ultrastructure of the glucan network formation from fission yeast protoplast. J Electron Microsc (Tokyo) 1995;44:198–206. [PubMed] [Google Scholar]