

Abstract
Ubiquitination regulates a host of cellular processes by labeling proteins for degradation, but also by functioning as a regulatory, nonproteolytic posttranslational modification. Proteome-wide strategies to monitor changes in ubiquitination profiles are important to obtain insight into the various cellular functions of ubiquitination. Here we describe generation of stable cell lines expressing a tandem hexahistidine-biotin tag (HB-tag) fused to ubiquitin for two-step purification of the ubiquitinated proteome under fully denaturing conditions. Using this approach we identified 669 ubiquitinated proteins from HeLa cells, including 44 precise ubiquitin attachment sites on substrates and all seven possible ubiquitin chain-linkage types. To probe the dynamics of ubiquitination in response to perturbation of the ubiquitin/proteasome pathway, we combined ubiquitin profiling with quantitative mass spectrometry using the stable isotope labeling with amino acids in cell culture (SILAC) strategy. We compared untreated cells and cells treated with the proteasome inhibitor MG132 to identify ubiquitinated proteins that are targeted to the proteasome for degradation. A number of proteasome substrates were identified. In addition, the quantitative approach allowed us to compare proteasome targeting by different ubiquitin chain topologies in vivo. The tools and strategies described here can be applied to detect changes in ubiquitination dynamics in response to various changes in growth conditions and cellular stress and will contribute to our understanding of the ubiquitin/proteasome system.
Keywords: HB-ubiquitin, ubiquitin profiling, SILAC, MG132, tandem affinity purification
Introduction
Ubiquitin is a 76-amino acid protein that is ubiquitously distributed and highly conserved throughout eukaryotic organisms. Regulation of proteins by post-translational modification with ubiquitin plays an important role in a variety of cellular processes including protein degradation, stress response, cell-cycle regulation, protein trafficking, endocytosis, signaling, and transcriptional regulation.1,2
The active form of ubiquitin is generated from a high molecular weight precursor by the action of ubiquitin C-terminal hydrolases (UCH), which release the mature 8 kDa protein. After cleavage, ubiquitin exposes glycine 76, which forms an isopeptide bond with the ∈-amino-group of a lysine residue of substrate proteins. Ubiquitin conjugates are formed by the sequential catalytic actions of E1-activating and E2-conjugating enzymes and E3-ligases. Whereas only two E1-activating enzymes are involved in the ubiquitination of all target proteins, distinct E2-conjugating enzymes appear to be dedicated to the ubiquitination of different substrates. E3-ligases stimulate E2 conjugation activity and provide protein target specificity by bridging the substrate/E2 interaction. Like other posttranslational modifications, ubiquitination is a reversible modification due to the function of ubiquitin hydrolases.
The ubiquitin molecule can be found free or conjugated to protein substrates. When conjugated to substrate proteins one distinguishes between mono, multi, and poly ubiquitinated substrates. While the former two describe the linkage of single ubiquitin molecules to one or more lysine residues in substrates, polyubiquitination involves the formation of ubiquitin chains, which is achieved by linking additional ubiquitin molecules to lysine residues in substrate-attached ubiquitin molecules. All seven internal lysine residues of ubiquitin (K6, K11, K27, K29, K33, K48, and K63) can in principle be used for chain formation, and the various resulting ubiquitin chain topologies have been detected in vivo in yeast and mammalian cells.3-5
Mono ubiquitination does generally not target proteins to the proteasome for degradation, but has regulatory functions that remain to be described at a mechanistic level. However, the importance of monoubiquitination of histone molecules in regulation of chromatin structure and the function of mono and multiubiquitination in receptor down regulation is evident from physiological studies.6
The most common ubiquitin chain linkage, through lysine-48, provides primarily a signal for proteolysis by the 26S proteasome. In contrast, lysine-63 linked ubiquitin chains are thought to have signaling function and are not recognized by the proteasome. The function of other ubiquitin chain topologies is not known.
Ubiquitination is involved in most if not all cellular processes. To obtain a global understanding of the role of ubiquitination, proteome-wide approaches are desirable. Most of the global strategies have been applied in the model system yeast,3,5,7-9 but some studies have demonstrated feasibility in mammalian systems.4,10-13 We describe here the development of a strategy that utilizes cell lines stably expressing tandem 6xHis-biotin-tagged ubiquitin for purification of the ubiquitinated proteome under fully denaturing conditions. Furthermore, we combined this approach with SILAC-based quantitative mass spectrometry for sensitive detection of changes in global ubiquitin profiles in response to inhibition of the proteasome.
Methods
Plasmids, Cloning and Expression of HB-Ubiquitin, Cell Culture, and Transfections
The retroviral vector pQCXIP (BD Biosciences) was used to express yeast HB-ubiquitin (pQCXIP-HB-ubi). Viral particles were generated in 293 GP2 packaging cells and used to transduce HeLa cells according to standard protocols in order to establish a stable cell line expressing HB-ubiquitin. Stable cell lines were periodically maintained with puromycine for selection.
HeLa cells were cultured in DMEM supplemented with 10% FCS, 1 μM biotin and 1% antibiotic-antimycotic agent (Invitrogen) in 5% CO2 at 37 °C. All cell lines were tested for mycoplasma contaminations and periodically treated with plasminogen (InvivoGen, San Diego).
To differentially label MG132 treated and untreated cells expressing HB-ubiquitin, a SILAC DMEM medium was used (Thermo Scientific, Rockford, IL), lacking the two essential amino acids arginine and lysine. Heavy media were supplemented with 0.028 mg/mL 13C6 15N4 arginine (isotopic purity > 98 atom %) and 0.073 mg/mL 13C6 15N2 lysine (isotopic purity > 98 atom %) (Cambridge Isotope Labeling, Andover, MA) and the same amount of 12C14N-arg/lys was added to the light medium.
To inhibit proteasome activity, cells were treated with 10 μM MG132 (American Peptide, Sunnyvale, CA) for 90 min at 37 °C. The control cells were treated with the solvent (DMSO) in parallel.
Tandem Affinity Purification of Ubiquitinated Proteins from Cell Lysates
Cells were grown in 150 mm dishes (15 plates in experiment 1–3; 20 plates in the SILAC experiments 4 and 5). Cells attached to plates were washed twice with ice cold 1× PBS, pH 7.4, and harvested on plates with buffer A (8 M urea, 300 mM NaCl, 50 mM NaH2PO4, 0.5% NP-40), pH 8.0, and 1 mM PMSF.
Lysates were centrifuged at 15 000g, 30 min, 20 °C, and the clarified supernatant was used for purification. 35 μL of Ni2+ sepharose beads (GE Healthcare) were used for each 1 mg of protein lysates and were incubated overnight at room temperature in buffer A with 10 mM imidazole on a rocking platform. Beads were pelleted by centrifugation at 100 × g for 1 min and washed sequentially with 20 bead volumes of buffer A (pH 8.0), buffer A (pH 6.3), and buffer A (pH 6.3) with 10 mM imidazole. After washing the beads, proteins were eluted twice with 5 bead volumes of buffer B (8 M Urea, 200 mM NaCl, 50 mM Na2HPO4, 2% SDS, 10 mM EDTA, 100 mM Tris, 250 mM imidazole) pH 4.3. The pH of the elute was adjusted to pH 8.0. Ubiquitinated proteins were bound to 7 μL streptavidin sepharose beads (Thermo Scientific, Rockford, IL) for each 1 mg of initial protein lysate by incubation on a rocking platform overnight at room temperature. Streptavidin beads were washed sequentially with 2 × 25 bead volumes of buffer C (8 M Urea, 200 mM NaCl, 2% SDS, 100 mM Tris, pH 8.0) and buffer D (8 M Urea, 1.2 M NaCl, 0.2% SDS, 100 mM Tris, 10% EtOH, 10% Isopropanol, pH 8.0). After washing the beads 3 times with 25 mM NH4HCO3, pH 8, the proteins were released by on-bead digestion with trypsin at 37 °C for 12–16 h on a rocking platform as described.5,14 Tryptic peptides were extracted 3 times using 25% (v/v) acetonitrile (ACN), 0.1% (v/v) formic acid (FA) and subsequently separated by strong cation exchange (SCX) chromatography, as previously described.15 Twelve fractions were manually collected, desalted, concentrated, and analyzed by LC–MS/MS as described.15
Liquid Chromatography, Tandem Mass Spectrometry, and Data Processing
LC–MS/MS was carried out by nanoflow reverse phase liquid chromatography (RPLC) (Eksigent, CA) coupled online to a Linear Ion Trap (LTQ)-Orbitrap XL mass spectrometer (Thermo-Electron Corp). Briefly, the LC separation was performed using a capillary column (100 μm ID × 150 mm long) packed with C18 resin (GL sciences) and the peptides were eluted using a linear gradient from 2 to 5% B over 5 min and 5 to 25% B over 90 min at a flow rate of 350 nL/min (solvent A: 100% H2O/0.1% formic acid; solvent B: 100% acetonitrile/0.1% formic acid). Nanoelectrospray was achieved using a pulled capillary tip with 10 μm ID (New Objectives, Woburn, MA) mounted on a packed tip stand manufactured by Thermoelectron Corp.; 1.7kV was applied on the tip. A cycle of one full FT scan mass spectrum (350–2000 m/z, resolution of 60 000 at m/z 400) was followed by 10 data-dependent MS/MS acquired in the linear ion trap with normalized collision energy (setting of 35%). Target ions already selected for MS/MS were dynamically excluded for 30 s.
Monoisotopic masses of parent ions and corresponding fragment ions, parent ion charge states and ion intensities from the tandem mass spectra (MS/MS) were obtained using an in-house software with Raw_Extract script from Xcalibur v2.4. Following automated data extraction, resultant peak lists for each LC–MS/MS experiment were submitted to the development version 5.0 of Protein Prospector (UCSF) for database searching similarly as described.16
A concatenated SwissProt (2007.11.07) database generated from the normal database and its reversed form (34,972 entries) was used for database searching. Trypsin was set as the enzyme with a maximum of two missed cleavage sites. The mass tolerance for parent ion was set as ± 20 ppm, whereas ±1 Da tolerance was chosen for the fragment ions. Following chemical modifications were selected as variable modifications during database search: protein N-terminal acetylation, methionine oxidation, N-terminal pyroglutamine, and deamidation of asparagine. Maximal modifications on a given peptide was set as 3. The Search Compare program in Protein Prospector was used for summarization, validation and comparison of results. To determine the expectation value cutoff that corresponds to a percent false positive (% FP) rate, the plot of the expectation values versus % FP rate for each search result was automatically obtained using the Search Compare Program. Based on these results, we chose an expectation value cutoff for all peptides corresponding to ≤1% FP. General protein identification is based on at least two peptides. For the SILAC samples, two additional variable modifications were included: 13C6 15N4-labeled arginine and 13C6 15N2-labeled lysine.
To quantify protein relative abundance changes, the Search Compare function was used to determine the light/heavy (L/H) ratios based on the intensities of the monoisotopic masses of the parent ion peptide pairs. Search Compare also corrects for the isotopic purity of the heavy SILAC amino acids, which was set to 98% purity with the signal/noise threshold set at 10. The peptide peak intensities were averaged across the elution profile (30 s) as described.16 For peptides matching to multiple members of a protein family, only the protein containing at least one unique peptide was reported. In the case that none of the proteins contain at least one unique peptide, all of the possibilities are reported with a “/” separating the protein names.
After protein identification, a second search was performed for each sample against the list of proteins identified with the given cutoff threshold to identified ubiquitination sites. During the second search, the following variable modifications were added: carbamylation of lysine, dioxidation of tryptophan, GlyGly modification of lysine, and phosphorylation of serine, threonine, or tyrosine.
All peptides with precise ubiquitination sites and ubiquitin chain linkage types were manually confirmed, considering the following 4 steps:
Each MS/MS spectrum for validation was individually submitted for a database search, using the Swissprot concantenated database without species restriction.
All of the b and y ion series were inspected manually to ensure the sensible interpretation based on peptide fragmentation rules (e.g., favorable cleavage at P site).17
A series of consecutive y and/or b ions should be observed and all of the major ions have to be interpreted.
MS/MS spectra of modified- and unmodified peptides have to be compared.
Cell Proliferation Assay
The growth rate of HeLa cells and HeLa cells expressing HB-tagged ubiquitin was determined by a colorimetric cytotoxicity assay, which measures the cellular protein content of cell cultures.18 In brief, cells were fixed with 0.4% trichloroacetic acid (wt/vol) and stained with sulforhodamine B dissolved in 1% acetic acid. The protein bound dye was extracted with 10 mM Tris base and the optical density was measured at 564 nm (Figure 1C).
Figure 1.
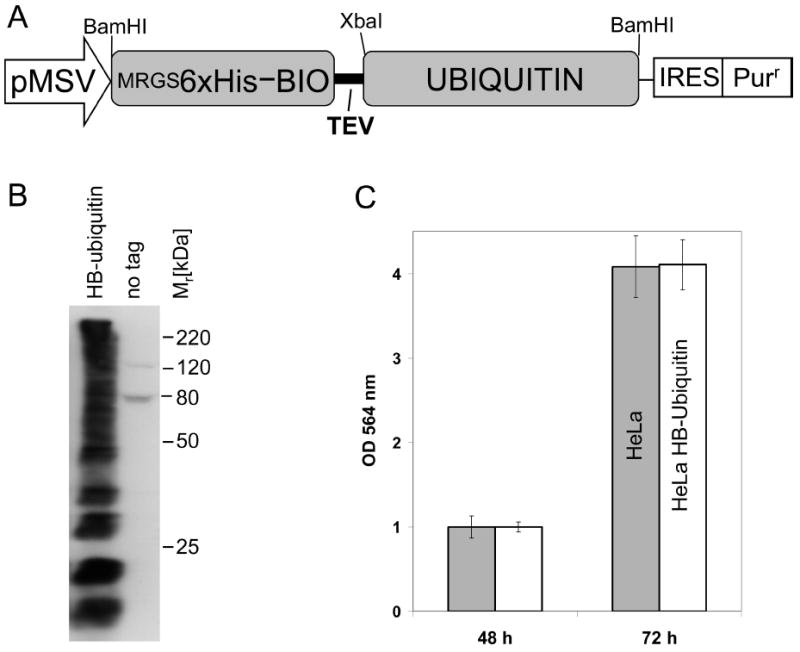
HeLa cells stably expressing HB-tagged ubiquitin. (A) Schematic depiction of the HB-ubiquitin expression construct. The RGS6xHis epitope combined with a bacterially derived in vivo biotinylation signaling peptide was fused to ubiquitin. Expression was driven by the CMV type I enhancer and a MSV promoter. The puromycine resistance marker (Purr) was coexpressed using an internal ribosomal entry site (IRES). (B) Total cell lysates from HeLa cells expressing HB-ubiquitin or no tag were analyzed by Western blotting using a HRP-streptavidin conjugate to detect the HB-tag. (C) Cell proliferation of HeLaHB-ubi cells and HeLa cells expressing no tag was compared using a sulforhodamine-B based assay. The median values with standard deviations obtained from 5 independent experiments are shown.
Immunoblot Analysis
Proteins were separated by SDS-PAGE and transferred to PVDF membranes using a semidry blotting apparatus. Membranes were blocked in TBS containing 0.2% Tween-20 and 5% milk for 60 min and incubated in primary antibodies overnight at 4 °C. The RGS4His antibody (Qiagen, Valencia, CA) for detection of the RGS6xHis tag was diluted 1:2000 in TBS-Tween-20, 5% milk. HRP-conjugated secondary antibodies were used at a dilution of 1:15 000 in TBS-Tween-20, 5% milk. To detect the biotinylated portion of the HB-tag the membrane was incubated for 1–2 h at room temperature with HRP-conjugated streptavidin (1:10 000 in TBS-Tween) (Fisher, Pittsburgh, PA). Immunodetection was performed with SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Results and Discussion
HeLa Cells Stably Expressing HB-Tagged Ubiquitin
To detect ubiquitinated proteins in human cells, we fused a tandem affinity tag to ubiquitin (HB-ubiquitin), and generated stable cell lines expressing HB-ubiquitin (Figure 1A, 1B). The HB-tag allows two-step purification under fully denaturing conditions such as 8 M urea.5,14,19 These stringent conditions preserve ubiquitination and avoid copurification of proteins that bind to ubiquitinated proteins but are not ubiquitinated themselves. Expression of HB-ubiquitin in the stable HeLa cell line was confirmed by immunoblot analysis with a streptavidin-HRP conjugate (Figure 1B). A smear pattern typical for ubiquitinated proteins was detected in the total lysates of HB-ubiquitin expressing HeLa cells (HeLaHB-ubi) but not in control cell lines without the tagged ubiquitin. Detection of high-molecular weight ubiquitin signals confirmed previous results that HB-ubiquitin is functional and is conjugated to proteins.5 The two distinct bands detected in the control cell lines are endogenous biotinylated proteins (Figure 1B). Expression of HB-ubiquitin had no effect on the growth rate of HeLa cells, indicating that HB-tagged ubiquitin does not significantly interfere with cellular processes regulated by ubiquitination (Figure 1C). Together these results demonstrate that HB-tagged ubiquitin is conjugated to other proteins in vivo and expression of HB-ubiquitin has no obvious adverse effects on cellular pathways.
Purification and Identification of Ubiquitinated Proteins
To identify proteins covalently modified with HB-ubiquitin in mammalian cells, we sequentially purified proteins by Ni2+-chelate choromatography and binding to streptavidin agarose as described previously.5 Ubiquitinated proteins were purified from between 41 and 60 mg of total HeLa cell lysates. Endogenous biotinylated proteins were eliminated in the first purification step. Binding to streptavidin beads is virtually irreversible allowing for stringent wash conditions, but prevented elution of the purified ubiquitinated proteins. Therefore, samples still bound to streptavidin beads were digested with trypsin for MS analyses. We followed the purification of ubiquitinated proteins by immunoblotting using antibodies against the RGS6His epitope that is part of the HB-tag (Figure 2). Importantly, similar to what has been observed in yeast,5 the HB-tag was quantitatively biotinylated in vivo in HeLa cells because a very high fraction of proteins carrying the RGS6His epitope bound to streptavidin agarose indicating that they had been biotinylated in vivo (Figure 2, compare lanes 4 and 5).
Figure 2.
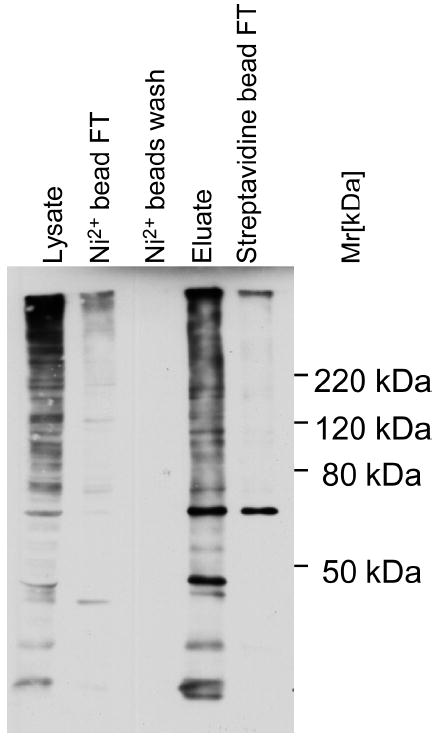
Tandem-affinity purification of ubiquitinated proteins from HeLa cells. Purification efficiency was monitored by immuno blotting using an anti-RGS4H antibody directed against the HB-tag. Protein samples were separated on a 10% SDS-polyacrylamide gel and processed for immunoblotting. FT: flow-through.
Purified samples were digested “on-bead” with trypsin, separated by ion exchange chromatography and about 12 –22 fractions were collected and analyzed by LC–MS/MS. Data were processed using the developmental version of Protein Prospector at UCSF. Three independent experiments were performed (experiments 1–3, Supplementary Table 1, Supporting Information). Using a false-positive rate setting of <1%, we identified 535 potential ubiquitination substrates by at least two unique peptides in at least two out of three experiments (Supplementary Table 1, Supporting Information). A total of 669 putative ubiquitinated proteins were identified when we included proteins identified by a single peptide in at least two out of the three independent experiments (experiments 1–3 Supplementary Table 1, Supporting Information). Purification from an equivalent amount of protein lysates prepared from cells expressing no HB-tagged ubiquitin showed that nonspecifically purified background was very low. Twenty-five proteins could be clearly identified as background because the number of identified unique peptides was similar or higher in the control (untagged ubiquitin) purification as compared to the HeLaHB-ubi experiment. Sixty-two additional proteins were identified in both the control experiment and the three HeLaHB-ubi experiments, indicating that these might be potential background proteins. However, these 62 proteins were generally high abundant proteins such as Actin, tubulin, ribosomal proteins, heatshock proteins, and histones and only very few peptides were detected in the control purification whereas substantially more unique peptides were detected in the HeLaHB-ubi purification. In addition, for a number of them we could detect ubiquitinated lysine residues, which strongly suggests that these proteins are genuinely ubiquitinated. We think that these proteins are likely ubiquitination substrates and thus included them in the list of ubiquitinated proteins, but marked them to indicate that they were also detected in the control purification (Supplementary Table 1, Supporting Information).
We identified known short-lived proteins that have been demonstrated to be substrates of the ubiquitin proteasome system, such as cyclins, cyclin-dependent kinase inhibitors, E3 ubiquitin ligases, the hypoxia-inducible factor 1α (HIF-1α), and several DNA replication licensing factors (MCM proteins).20,21 In addition, we also identified monoubiquitinated proteins such as histones, and the Fanconi anemia proteins FANC-D2 and FANC-I.22,23 Together these results demonstrate that stable cell lines expressing HB-tagged ubiquitin combined with the purification and analysis strategy presented here is an effective approach for system-level identification of a wide-spectrum of ubiquitination substrates.
Precise Ubiquitin Attachment Sites and Ubiquitin Chain Topology
In addition to the identification of 669 proteins as potential ubiquitination substrates we identified 44 precise ubiquitination sites, based on a 114 Da mass shift due to a double glycine that remains attached to the modified amino acid after trypsin cleavage3 (Table 1). Among them are lysines 119 and 121 in histone H2A and H2B, respectively (Figure 3A-B). These lysine residues in histones have been shown in directed studies to be monoubiquitinated.24 Furthermore, we identified lysine 538 in HIF-1α as a ubiquitin attachment site in vivo (Figure 3C). This lysine residue has previously been suggested as an ubiquitination site based on mutational analysis.25 These examples demonstrate that the global strategy reported here leads to biologically relevant information and also shows that our approach is efficient enough for identification of monoubiquitinated substrates. All identified ubiquitin attachment sites were lysine residues. We did not detect ubiquitination of cysteine, or the N-terminal amino group in this global approach, which has been reported for selected proteins and in the case of cysteine residues could have been detected as an activated thioester intermediate on E1, E2, or HECT domain E3s.26-29
Table 1.
Summery of All Peptides Containing Ubiquitination Sides Identified by LC–MS/MS
| Swiss-Prot ID |
peptide sequence | position of ubiquitinated lysine |
protein name | number uniquea |
|---|---|---|---|---|
| P51665 | DIK(GlyGly)DTTVGTLSQR | K180 | 26S proteasome non-ATPase regulatory subunit 7 | 4 |
| P60866 | DTGK(GlyGly)TPVEPEVAIHR | K8 | 40S ribosomal protein S20 | 3 |
| P23396 | FGFPEGSVELYAEK(GlyGly)VATR | K90 | 40S ribosomal protein S3 | 13 |
| P23396 | K(GlyGly)PLPDHVSIVEPK | K202 | 40S ribosomal protein S3 | 13 |
| P08195 | IK(GlyGly)VAEDEAEAAAAAK | K46 | 4F2 cell-surface antigen heavy chain | 17 |
| P27635 | FNADEFEDM(Oxidation)VAEK(GlyGly)R | K188 | 60S ribosomal protein L10 | 6 |
| P36404 | LNIWDVGGQK(GlyGly)SLR | K71 | ADP-ribosylation factor-like protein 2 | 3 |
| Q9Y3E7 | ILFEITAGALGK(GlyGly)APSK | K183 | Charged multivesicular body protein 3 | 2 |
| Q13619 | TNGLT(Phospho)KPAALAAAPAK(GlyGly)PGGAGGSK | K33 | Cullin-4A | 3 |
| P04844 | LSK(GlyGly)EETVLATVQALQTASHLSQQADLR | K154 | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase 63 kDa subunit precursor | 7 |
| O14602 | DYQDNK(GlyGly)ADVILK | K88 | Eukaryotic translation initiation factor 1A, Y-chromosomal | 2 |
| Q13347 | VK(GlyGly)GHFGPINSVAFHPDGK | K282 | Eukaryotic translation initiation factor 3 subunit 2 | 7 |
| P62826 | FNVWDTAGQEK(GlyGly)FGGLR | K71 | GTP-binding nuclear protein Ran | 8 |
| P16403 | M(Met-loss+Acetyl)SETAPAAPAAAPPAEK(GlyGly)APVK | K17 | Histone H1.2 | 8 |
| P04908 | VTIAQGGVLPNIQAVLLPK(GlyGly)K | K119 | Histone H2A type 1-B | 11 |
| P23527 | AVTK(GlyGly)YTSSK | K121 subtype 1-O | Histone H2B subtype | 16 |
| Q09028 | M(Met-loss+Acetyl)ADK(GlyGly)EAAFDDAVEER | K4 | Histone-binding protein RBBP4 | 4 |
| Q16576 | M(Met-loss+Acetyl)ASK(GlyGly)EMFEDTVEER | K4 | Histone-binding protein RBBP7 | 7 |
| Q15047 | KS(Phospho)SSQDLHK(GlyGly)GTLSQMSGELSK | K182 | Histone-lysine N-methyltransferase SETDB1 | 11 |
| Q16665 | LELVEK(GlyGly)LFAEDTEAK | K538 | Hypoxia-inducible factor 1 alpha | 9 |
| Q01650 | ALAAPAAEEK(GlyGly)EEAR | K19 | Large neutral amino acids transporter small subunit 1 | 7 |
| Q01650 | M(Oxidation)LAAK(GlyGly)SADGSAPAGEGEGVTLQR | K30 | Large neutral amino acids transporter small subunit 1 | 7 |
| O43684 | VAVEYLDPSPEVQK(GlyGly)K | K216 | Mitotic checkpoint protein BUB3 | 5 |
| P62937 | VSFELFADK(GlyGly)VPK | K28 | Peptidyl-prolyl cis–trans isomerase A | 6 |
| Q99755 | GAIQLGITHTVGSLSTK(GlyGly)PER | K103 | Phosphatidylinositol-4-phosphate 5-kinase type-1 alpha | 6 |
| P07737 | TFVNITPAEVGVLVGK(GlyGly)DR | K54 | Profilin-1 | 7 |
| P25786 | LVSLIGSK(GlyGly)TQIPTQR | K115 | Proteasome subunit alpha type-1 | 5 |
| P25786 | ETLPAEQDLTTK(GlyGly)NVSIGIVGK | K208 | Proteasome subunit alpha type-1 | 5 |
| Q16186 | M(Oxidation)SLK(GlyGly)GTTVTPDK | K34 | ADRM1 | 4 |
| Q01105 | VLSK(Label:13C6 15N2+GlyGly)EFHLNESGDPSSK(Label:13C6 15N2) | K154 | SET | 3 |
| Q8N0X7 | TRPSSDQLK(GlyGly)EASGTDVK | K362 | Spartin | 7 |
| P63279 | K(GlyGly)DHPFGFVAVPTK | K18 | SUMO-conjugating enzyme UBC9 | 2 |
| Q9H3M7 | IVVPK(Label:13C6 15N4)+GlyGly)AAIVAR(Label:13C6 15N4) | K212 | Thioredoxin-interacting protein | 4 |
| P51571 | VQNM(Oxidation)ALYADVGGK(GlyGly)QFPVTR | K73 | Translocon-associated protein subunit delta precursor | 4 |
| P68363 | VGINYQPPTVVPGGDLAK(GlyGly)VQR | K370 subtype 1B | Tubulin alpha-subtype | 17 |
| P68363 | GDVVPK(GlyGly)DVNAAIATIK | K326 subtype 1B | Tubulin alpha-subtype | 17 |
| P07437 | ISVYYNEATGGK(GlyGly)YVPR | K58 | Tubulin beta chain | 20 |
| O94888 | QEILVEPEPLFGAPK(GlyGly)R | K99 | UBX domain-containing protein 7 | 10 |
| P54725 | EDK(GlyGly)SPSEESAPTTSPESVSGSVPSSGSSGR | K122 | UV excision repair protein RAD23 homologue A | 6 |
| O94833 | IAT(Phospho)TAEPADKVK(GlyGly)ILK | K3058 | Bullous pemphigoid antigen 1, isoforms 6/9/10 | |
| P07437 | M(Oxidation)SM(Oxidation)K(GlyGly)EVDEQM(Oxidation)LNVQNK | K324 | Tubulin beta chain | |
| P15531 | VM(Oxidation)LGETNPADSK(Label:13C6 15N2)+GlyGly)PGTIR(Label:13C6 15N4) | K100 | Nucleoside diphosphate kinase A/kinase B | |
| P55061 | K(GlyGly)INFDALLK | K7 | Bax inhibitor 1 | |
| P63092 | QDLLAEK(GlyGly)VLAGK | K360 | Guanine nucleotide-binding protein G(s) subunit alpha isoforms short | |
| P67809 | GAEAANVTGPGGVPVQGSK(GlyGly)YAADR | K137 | Nuclease sensitive element-binding protein 1 |
Number of unique peptides identified for each protein. MS/MS spectra for each reported peptide can be found in Supplementary Figure 1 (Supporting Information).
Figure 3.
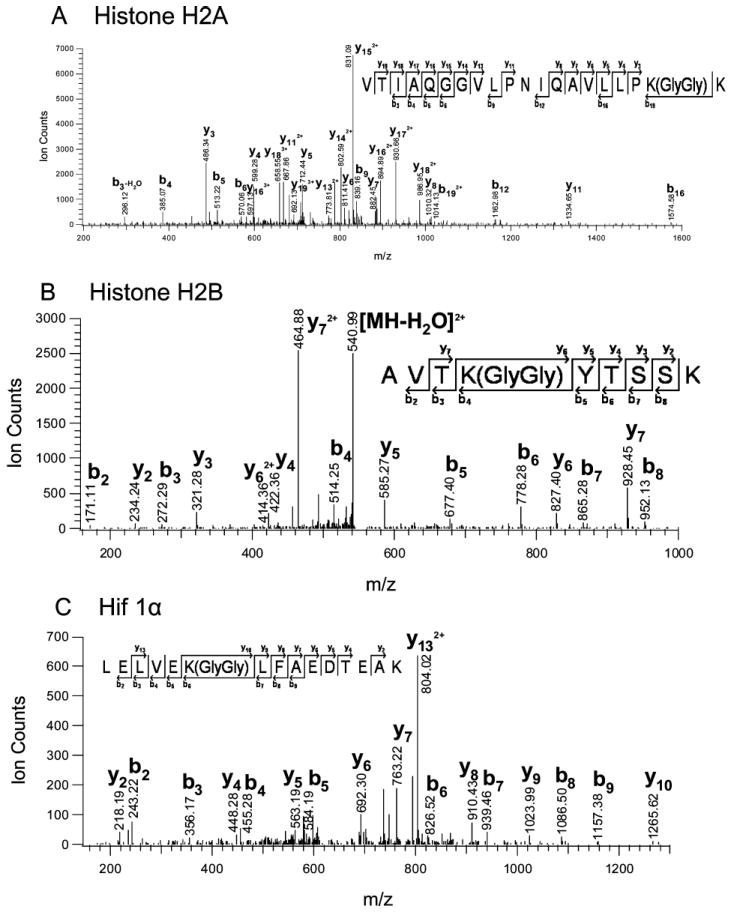
Ubiquitin acceptor lysines. MS/MS spectra of tryptic peptides containing Gly/Gly linked to lysine. (A) Histone H2A; MH33+ 725.11; VTIAQGGVLPNIQAVLLPK(GlyGly)K+3. (B) Histone H2B; MH22+ 549.79; AVTK(GlyGly)YTSSK+2. (C) Hypoxia- inducible factor 1 alpha; MH33+ 619.99; LELVEK(GlyGly)LFAEDTEAK+3.
Precise ubiquitin attachment sites are based on a double glycine remnant linked to lysine after trypsin digestion. Similar double glycine remnants are indicative for modification with the ubiquitin-like protein Nedd8.30 This mass spectrometric strategy can thus not differentiate between neddylation and ubiquitination sites. However, because we analyzed samples enriched for ubiquitinated proteins, the lysine residues presented in Table 1 are most likely ubiquitination sites.
Ubiquitin chains are formed by isopeptide links between the C-terminal carboxyl group of one ubiquitin molecule with any of seven lysine residues in another ubiquitin molecule. Depending on the ubiquitin chain architecture the chain can send different biological signals. Our global analyses found in vivo evidence for ubiquitin chain linkages through any of the seven possible lysine residues in ubiquitin (Table 2). Similar results have been reported in yeast and to some extend in mammalian cells.3-5,13
Table 2.
Representative Peptides of Identified Ubiquitin-Chain Linkages by LC–MS/MSa
| position of ubiquitinated lysine | peptide sequence | m/z (observed) | expectation value39 |
|---|---|---|---|
| K6 | MQIFVK(GlyGly)TLTGK | 690.3899 | 2.50E–02 |
| K11 | TLTGK(GlyGly)TITLEVEPSDTIENVK | 1201.6399 | 8.80E–06 |
| K27 | TITLEVEPSDTIENVK(GlyGly)AK | 701.0390 | 1.40E–03 |
| K29 | AK(GlyGly)IQDK(Carbamyl)EGIPPDQQR | 940.4865 | 2.00E–02 |
| K33 | IQDK(GlyGly)EGIPPDQQR | 819.4203 | 1.70E–04 |
| K48 | LIFAGK(GlyGly)QLEDGR | 730.9002 | 3.20E–05 |
| K63 | TLSDYNIQK(GlyGly)ESTLHLVLR | 1122.6052 | 7.40E–06 |
MS/MS spectra for each reported peptide can be found in the Supplementary Figure 2 (Supporting Information).
Finally, we asked whether ubiquitination targets are enriched in any biological processes. Using the data reported in Supplemental Table 1 (Supporting Information), we found that ubiquitinated proteins were significantly overrepresented in several categories including expected processes like proteolysis, cell cycle, mitosis, chromatin packaging, chromosome segregation and protein metabolism (Figure 4).
Figure 4.

Biological process analysis. Analysis was performed with the online software PANTHER (http://www.pantherdb.org/tools), using the data set reported in Supplementary Table 1 (Supporting Information). The p value was set to >0.05, the Bonferroni correction for multiple testing’s was used. Only categories with significant differences are shown.
Quantitative Analysis of Ubiquitinated Proteins in Response to Proteasome Inhibition
We aimed to apply the strategy outlined above to detect quantitative differences in global ubiquitination profiles in response to perturbation of normal cellular function. We chose to analyze changes in ubiquitination profiles in response to inhibition of 20S proteasome activity using the proteasome inhibitor MG132. We expected to observe significant shifts in ubiquitination profiles upon inhibition of proteasome activity because marking proteins for proteasomal degradation is one of the major roles for protein ubiquitination. In addition, we were interested to identify ubiquitinated proteins where ubiquitination does not target for degradation by the proteasome but presents a different signal. To identify quantitative changes in global ubiquitination patterns we used the SILAC strategy31-33 and labeled all proteins with heavy lysine and arginine. Cells grown in heavy medium were treated with 10 μM MG132 for 90 min prior to cell lysis and cells in light medium were mock treated with DMSO. To exclude effects of supplementation with heavy lysine and arginine and to evaluate reproducibility of this quantitative approach we performed a label-switch experiment where proteasome inhibitor was added to cells grown in light medium and cells grown in heavy medium where mock-treated.
Twenty-three milligrams and 39 mg of total cell lysates (experiment 4 and 5 in Supplementary Table 2), respectively, were analyzed. Light and heavy lysates for each experiment were mixed in a 1:1 ratio after cell lysis. Ubiquitinated proteins were tandem-affinity purified and analyzed as described above.
Using a false-positive rate setting of <1% we identified 381 proteins (191 proteins with at least two unique peptides) in experiment 4, and 362 proteins (187 proteins with at least two unique peptides) in experiment 5 (label switch experiment). The complete lists with all proteins can be found in Supplementary Table 2 (Supporting Information); 230 proteins were identified in both experiments and were used for quantitative comparison.
We next determined the relative abundance changes induced by proteasome inhibition. Light to heavy (L/H) ratios were determined for all proteins with at least one high-quality, quantifiable peptide detected in both experiments. The reciprocal L/H value was calculated for experiment 5. L/H ratios over 5-fold and under 0.2-fold were set to 5 and 0.2, because peak height measurements for the low abundant peptide partner in the pairs was no longer reliable. Following these criteria we detected 220 ubiquitinated proteins, 51 of them were up-regulated and 15 down-regulated in response to MG132 treatment considering a ratio between 0.75 and 1.33 as neither significantly up- or down-regulated. 97% of the proteins identified in experiment 4 and 5 were also identified as ubiquitination substrates in experiments 1 to 3, which were performed without proteasome inhibition. (Supplementary Tables 1 and 2, Supporting Information).
L/H ratios were visualized in a log–log plot to the base 2 (Figure 5). As expected, quantitation showed an increase in the abundance of many ubiquitinated proteins in response to proteasome inhibition. Surprisingly, a significant number of proteins showed a highly reproducible decrease in the ubiquitinated form in response to MG132 treatment. Among them were various histones. For example, histone H2A.Z had a L/H ratio of 2.7 in experiment 4 (MG132 added to the heavy sample) and a matching reciprocal L/H ratio of 0.38 in experiment 5 (MG132 in the light sample). This phenomenon can be explained by the fact that most of the cellular ubiquitin is trapped in ubiquitin chains upon proteasome inhibition, which has been proposed to cause ubiquitin stress and induction of release of ubiquitin from ubiquitinated histones.34,35 Our quantitative analyses of proteome-wide ubiquitination is in agreement with the model of redistribution of cellular ubiquitin pools.
Figure 5.
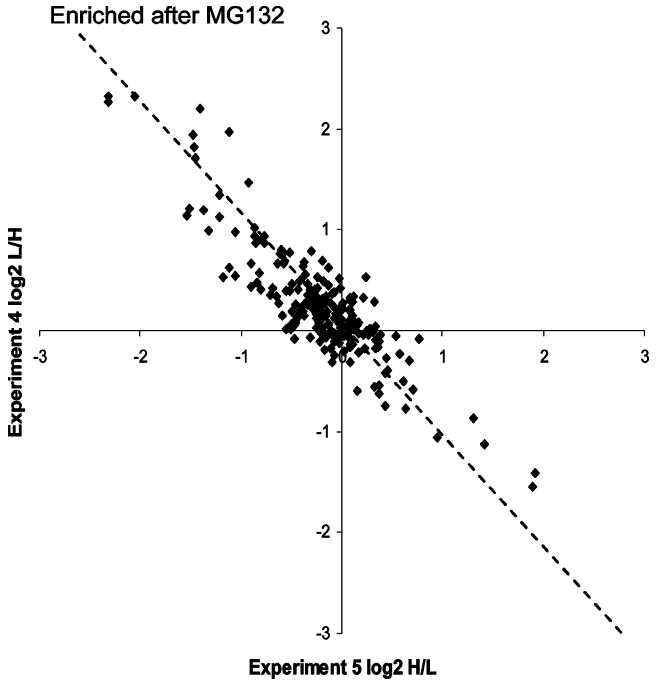
Abundance changes of ubiquitinated proteins in response to proteasome inhibition. SILAC ratios of identified ubiquitinated proteins from two independent experiments are compared on a log–log plot to the base 2. Each black square represents a single protein. The dotted line indicates the position of exact matching data points from both experiments.
The HB-ubiquitin expressed in HeLa cells was derived from the budding yeast UBI4 gene, which encodes for ubiquitin that differs at three positions from human ubiquitin. Two of the three amino acid changes are located on a single tryptic peptide and thus allowed us to distinguish between HB-tagged ubiquitin and the endogenous untagged human ubiquitin. The third amino acid change affects position 28, which is flanked by lysine residues and the corresponding tryptic peptide could not be detected. As expected, the HB-ubiquitin specific L/H peptide ratios were close to 1 (Table 3), demonstrating that neither purification efficiency nor HB-ubiquitin expression was affected by treatment with MG132. The human-specific tryptic ubiquitin peptide was used to quantify the increase of ubiquitin chains after inhibition of the proteasome because the untagged ubiquitin will only be purified when it is part of an ubiquitin chain that also contains HB-ubiquitin. Based on the L/H ratios for the human-specific ubiquitin peptide we can therefore conclude that the abundance of ubiquitin chains increased 3 fold in response to MG132 treatment (Table 3). The vast majority of tryptic ubiquitin peptides do not allow to differentiate between HB-ubiquitin and endogenous ubiquitin. The L/H rations for these peptides were 2.24, which is significantly lower than the 3-fold increase measured for total ubiquitin chains. This reduced ratio observed for peptides that cannot distinguish between the tagged and the endogenous ubiquitin is due to the fraction of tagged ubiquitin incorporated into chains because the L/H ratio of HB-ubiquitin remains at 1, even after proteasome inhibition (table 3). We can thus use the differences in L/H ratios obtained for human-specific and for general ubiquitin peptides to calculate that HB-ubiquitin was expressed at about 38% of the endogenous ubiquitin levels in these cell lines. Immunoblot analyses were consistent with these results suggesting a 2-fold excess of endogenous ubiquitin over HB-ubiquitin.
Table 3.
L/H Peptide Ratios for the HB-Tag, Ubiquitin, and Specific Ubiquitin-Chain Typesa
| experiment 4 |
experiment 5 |
|||
|---|---|---|---|---|
| peptides | L/H ratio | StDev. | L/H ratio | StDev. |
| HB-tag | 0.99 | 0.20 (n = 1368) | 0.99 | 0.13 (n = 1611) |
| yeast ubiquitin | 0.96 | 0.05 (n = 394) | 0.99 | 0.04 (n = 307) |
| human ubiquitin | 3.08 | 0.35 (n = 196) | 0.34 | 0.05 (n = 269) |
| other ubiquitin | 2.24 | 0.38 (n = 220) | 0.37 | 0.10 (n = 472) |
| K6 | 2.92 | n/a | 0.21 | n/a |
| K11 | 2.82 | n/a | 0.34 | n/a |
| K27 | 6.12 | n/a | 0.24 | n/a |
| K33 | 4.35 | n/a | 0.20 | n/a |
| K48 | 2.43 | n/a | 0.42 | n/a |
| K63 | 1.74 | n/a | 0.64 | n/a |
n/a = not available.
Quantitative Analysis of Ubiquitin Chain Topologies
We next asked how the different ubiquitin chain topologies contribute to the overall increase in ubiquitin chains after proteasome inhibition.
We determined L/H ratios of the signature peptides for K6, K11, K27, K33, K48, and K63 ubiquitin/ubiquitin linkages (Figure 6, table 3). The reciprocal values from the label-switch experiment were almost identical suggesting that the observed changes in these ubiquitin chain types in response to proteasome inhibition were specific and highly reproducible. We did not detect signature peptides for K29 chains that could be quantitated in both experiments to generate data for these linkage types. Surprisingly, all six types of ubiquitin chains that could be analyzed increased in response to proteasome inhibition (Figure 6, Table 3). The K63 linkage showed the smallest increase consistent with its proteasome-independent functions. Nevertheless, the 1.7-fold increase demonstrates that at least some proteins containing K63 chains are degraded. These experiments cannot determine the context of the detected linkage types and it is possible that proteins contain mixed chain topologies, which has been proposed from in vitro studies.36 In agreement with our global studies, K11, K48, and K63 ubiquitin chains have previously been reported to increase in response to proteasome inhibition in mammalian cells.11
Figure 6.
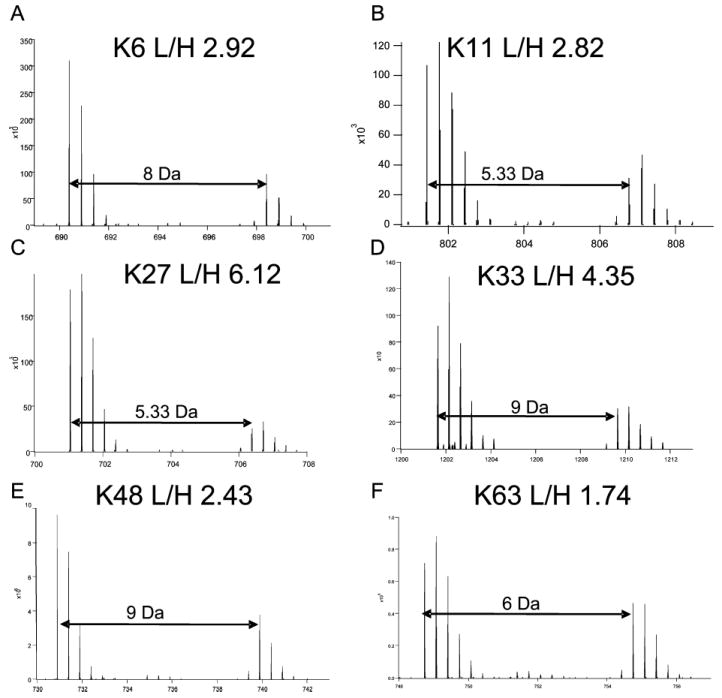
Quantitative comparison of ubiquitin chain topologies after proteasome inhibition. MS spectra of peptide pairs characteristic for ubiquitin chain topologies as indicated. Light/heavy ratios (L/H) were determined using the Search Compare function of Protein Prospector. The peptide peak intensities were averaged across the elution profile (30 s). Ubiquitin linkage types: (A) K6; MH22+ 698.41; MQIFVK (Label: 13C6 15N2+GlyGly)TLTGK (Label: 13C6 15N2)+2. (B) K11; MH33+ 806.77; TLTGK (Label: 13C6 15N2+GlyGly) TITLEVEPSDTIENVK (Label: 13C6 15N2)+3. (C) K27; MH33+ 706.39; TITLEVEPSDTIENVK(Label: 13C6 15N2+GlyGly)AK (Label: 13C6 15N2)+3. (D) K33; MH22+ 828.43; IQDK (Label: 13C6 15N2+GlyGly) EGIPPDQQR (Label: 13C6 15N4)+2. (E) K48; MH22+ 739.91; LIFAGK (Label: 13C6 15N2+GlyGly) QLEDGR (Label: 13C6 15N4)+2. (F) K63; MH33+ 754.75; TLSDYNIQK (Label: 13C6 15N2+GlyGly) ESTLHLVLR (Label: 13C6 15N4)+3.
K48 chains represent the most abundant chain topology3 and consistent with it being the predominant chain linkage, the K48 linkage increased at approximately the same level as total ubiquitin chains did in response to MG132 treatment (Figure 6, Table 3).
Unexpectedly, K6, K11, K27, and K33 were significantly more up-regulated after MG132 treatment than other chains (Figure 6, Table 3), suggesting that these chains are either part of or represent very active proteasome targeting signals. Consistent with this result, K11 linked chains have recently been implicated in APC-controlled degradation of cell cycle regulators.37
Quantitation of chain topologies and their changes in response to proteasome inhibition suggested that all chain types are either themselves degradation signals or that they are present in mixed chains that signal degradation. An alternative, although in our opinion unlikely interpretation is that a substrate is modified by two distinct types of chains and only one of them signals degradation. One also needs to consider that ubiquitin chain steady state levels are determined by both protein degradation and deubiquitination. Because proteasome inhibition significantly increases the total chains in the cell, an overall reduction of deubiquitination by substrate competition could occur under these conditions. Nevertheless, K48 chains are the canonical degradation signals and their observed 2.7 fold accumulation in response to MG132 treatment suggests that L/H ratios above this value are strong indicators for proteasome targeting functions. Our results therefore suggest that K6, K11, K27, K33, and K48 are part of proteasome targeting signals in mammalian cells. We could not reliably determine K29 chains in these experiments. This is probably due to technical issues, because tryptic digestion releases only a very short peptide for the K29 linkage type. Results from yeast demonstrated that only K48 and K29 chains accumulated upon proteasome inhibition,38 suggesting differences in proteasome signals between yeast and mammals.
Together, the quantitative analysis demonstrated that our approach can reproducibly describe changes in ubiquitination dynamics in response to cellular processes and should be a useful tool to probe various aspects of ubiquitin biology.
Conclusions
We have developed stable cell lines expressing HB-tagged ubiquitin and a purification strategy for system-level approaches to ubiquitination in mammalian cells. We applied this approach to identify proteins covalently modified with ubiquitin and demonstrated sensitivity by identification of 669 potential ubiquitination substrates as well as several precise ubiquitin attachment sites. Combination of proteome-wide ubiquitin profiling with SILAC-based quantitative mass spectrometry provided insight into ubiquitin-profile changes in response to proteasome inhibition and the role of individual ubiquitin chain topologies in proteasome targeting. The tools and approaches presented here can generally be applied to study the dynamics of the ubiquitin system in response to perturbation of cellular pathways or in response to extracellular signals.
Supplementary Material
Acknowledgments
This work was supported by the California Breast Cancer Research Program (9IB-0124) and NIH (GM66164) to P.K. L.H. acknowledges support from NIH (GM074830 and IS10RR023552). D.M. is an Erwin Schrödinger fellow supported by the FWF Austria. We thank P. L. Chen, E. Lee, and Y. Li for helpful advice and reagents and K. Lin for help with generating pathway categories.
Footnotes
Supporting Information Available: Supplementary Table 1 : sheet 1 (protein list experiment 1, 2, and 3) and sheet 2 (background proteins).
Supplementary Table 2 : sheet 1 (protein list experiment 4), sheet 2 (protein list experiment 5), and sheet 3 matching proteins with L/H ratios.
Supplementary Figure 1 : MS/MS spectra for ubiquitination sites listed in Table 1.
Supplementary Figure 2: MS/MS spectra for ubiquitination sites listed in Table 2.
This material is available free of charge via the Internet at http://pubs.acs.org and https://webfiles.uci.edu/xythoswfs/webui/_xy-5651887_1-t_uYaSIHQL.
References
- 1.Pickart CM. Back to the future with ubiquitin. Cell. 2004;116(2):181–90. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67(425):425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21(8):921–6. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto M, Hatakeyama S, Oyamada K, Oda Y, Nishimura T, Nakayama KI. Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 2005;5(16):4145–51. doi: 10.1002/pmic.200401280. [DOI] [PubMed] [Google Scholar]
- 5.Tagwerker C, Flick K, Cui M, Guerrero C, Dou Y, Auer B, Baldi P, Huang L, Kaiser P. A tandem affinity tag for two-step purification under fully denaturing conditions: application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol Cell Proteomics. 2006;5(4):737–48. doi: 10.1074/mcp.M500368-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2(3):195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 7.Mayor T, Graumann J, Bryan J, MacCoss MJ, Deshaies RJ. Quantitative profiling of ubiquitylated proteins reveals proteasome substrates and the substrate repertoire influenced by the Rpn10 receptor pathway. Mol Cell Proteomics. 2007;6(11):1885–95. doi: 10.1074/mcp.M700264-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Mayor T, Lipford JR, Graumann J, Smith GT, Deshaies RJ. Analysis of polyubiquitin conjugates reveals that the Rpn10 substrate receptor contributes to the turnover of multiple proteasome targets. Mol Cell Proteomics. 2005;4(6):741–51. doi: 10.1074/mcp.M400220-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Hitchcock AL, Auld K, Gygi SP, Silver PA. A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc Natl Acad Sci U S A. 2003;100(22):12735–40. doi: 10.1073/pnas.2135500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falsone SF, Gesslbauer B, Rek A, Kungl AJ. A proteomic approach towards the Hsp90-dependent ubiquitinylated proteome. Proteomics. 2007;7(14):2375–83. doi: 10.1002/pmic.200600996. [DOI] [PubMed] [Google Scholar]
- 11.Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448(7154):704–8. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 12.Vasilescu J, Smith JC, Ethier M, Figeys D. Proteomic analysis of ubiquitinated proteins from human MCF-7 breast cancer cells by immunoaffinity purification and mass spectrometry. J Proteome Res. 2005;4(6):2192–200. doi: 10.1021/pr050265i. [DOI] [PubMed] [Google Scholar]
- 13.Kirkpatrick DS, Weldon SF, Tsaprailis G, Liebler DC, Gandolfi AJ. Proteomic identification of ubiquitinated proteins from human cells expressing His-tagged ubiquitin. Proteomics. 2005;5(8):2104–11. doi: 10.1002/pmic.200401089. [DOI] [PubMed] [Google Scholar]
- 14.Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics. 2006;5(2):366–78. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, Huang L. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry. 2007;46(11):3553–65. doi: 10.1021/bi061994u. [DOI] [PubMed] [Google Scholar]
- 16.Baker PR, Chalkley RJ, Wang X, Jen N, Huang L. SILAC and iTRAQ Quantitation on an Orbitrap Using Protein Prospector. The proceedings of 56th ASMS (American Society of Mass Spectrometry); American Society of Mass Spectrometry; Denver, CO. 2008. [Google Scholar]
- 17.Medzihradszky KF. Peptide sequence analysis. Methods Enzymol. 2005;402:209–44. doi: 10.1016/S0076-6879(05)02007-0. [DOI] [PubMed] [Google Scholar]
- 18.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 19.Tagwerker C, Zhang H, Wang X, Larsen LS, Lathrop RH, Hatfield GW, Auer B, Huang L, Kaiser P. HB tag modules for PCR-based gene tagging and tandem affinity purification in Saccharomyces cerevisiae. Yeast. 2006;23(8):623–32. doi: 10.1002/yea.1380. [DOI] [PubMed] [Google Scholar]
- 20.Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4(11):855–64. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 21.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249–62. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 23.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, Elledge SJ. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29(6):653–63. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 25.Paltoglou S, Roberts BJ. HIF-1alpha and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene. 2007;26(4):604–9. doi: 10.1038/sj.onc.1209818. [DOI] [PubMed] [Google Scholar]
- 26.Scheffner M, Nuber U, Huibregste JM. Protein ubiquitination involving E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- 27.Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309(5731):127–30. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- 28.Ravid T, Hochstrasser M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat Cell Biol. 2007;9(4):422–7. doi: 10.1038/ncb1558. [DOI] [PubMed] [Google Scholar]
- 29.Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115(1):71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- 30.Jones J, Wu K, Yang Y, Guerrero C, Nillegoda N, Pan ZQ, Huang L. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J Proteome Res. 2008;7(3):1274–87. doi: 10.1021/pr700749v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29(2):124–30. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhu H, Pan S, Gu S, Bradbury EM, Chen X. Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun Mass Spectrom. 2002;16(22):2115–23. doi: 10.1002/rcm.831. [DOI] [PubMed] [Google Scholar]
- 33.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 34.Groothuis TA, Dantuma NP, Neefjes J, Salomons FA. Ubiquitin crosstalk connecting cellular processes. Cell Div. 2006;1:21. doi: 10.1186/1747-1028-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dantuma NP, Groothuis TA, Salomons FA, Neefjes J. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol. 2006;173(1):19–26. doi: 10.1083/jcb.200510071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8(7):700–10. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 37.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133(4):653–65. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu P, Cheng D, Duong DM, Rush J, Roelofs J, Finley D, Peng J. A Proteomic Strategy for Quantifying Polyubiquitinn Chain Topologies. Isr J Chem. 2006;46(2):171–182. [Google Scholar]
- 39.Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame AL. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M8000021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.