

Abstract
In humans, prenatal alcohol exposure can result in significant impairments in several types of learning and memory, including declarative and spatial memory. Animal models have been useful for confirming that many of the observed effects are the result of alcohol exposure, and not secondary to poor maternal nutrition or adverse home environments. Wagner and Hunt (2006) reported that rats exposed to ethanol during the neonatal period (postnatal days [PD] 4–9) exhibited impaired trace fear conditioning when trained as adolescents, but were unaffected in delay fear conditioning. The present series of three experiments represent a more detailed analysis of ethanol-induced deficits in trace conditioning. In Experiment 1 the dose of ethanol given to neonates was varied (3.0, 4.0 or 5.0 g/kg/day). There was a dose-dependent reduction in trace conditioning, with the poorest performance observed in animals treated with the highest dose. In Experiment 2 it was found that the impairment in trace conditioning resulting from neonatal ethanol exposure was dependent on the duration of the trace interval used for training; less learning was evident in ethanol-exposed animals trained with longer trace interval durations. These results confirm other reports of delay-dependent memory deficits. Finally, Experiment 3 determined that ethanol exposure limited to the first half of the neonatal period (PD 4–6) was more detrimental to later trace conditioning than exposure during the second half (PD 7–9). These results support the hypothesis that trace conditioning impairments resulting from early ethanol exposure are due to the drug’s teratogenic effects on the developing hippocampus, as the findings parallel those observed in animals with discrete hippocampal lesions. Comparisons between delay and trace fear conditioning performance in animals exposed to ethanol during the brain growth spurt provide a model system to study both selective learning impairments and possible treatment approaches for humans with fetal alcohol spectrum disorders.
Keywords: ethanol, fetal alcohol spectrum disorders, trace conditioning, hippocampus, temporal vulnerability, rat
Introduction
Individuals with a history of prenatal exposure to alcohol exhibit deficiencies in a variety of behavioral and cognitive domains, and many of the observed impairments persist well beyond the period of alcohol exposure. Individuals identified with fetal alcohol spectrum disorders (FASD) are known to exhibit pervasive deficits in attentional performance, learning, memory and executive functioning (Kodituwakku, 2007; Streissguth, 2007). A variety of factors are known to contribute to alcohol’s teratogenicity and the likelihood of FASD. Among these are maternal age and parity, socioeconomic status, and patterns and timing of alcohol consumption (Abel and Hannigan, 1995). Binge drinking appears to be particularly detrimental to fetal development (for review see Maier and West, 2001) and published reports indicate continuing trends toward increased binge drinking by pregnant women (Ebrahim et al., 1999).
There is now recognition that the learning and memory impairments of alcohol-exposed individuals are not global in their character. For example, Mattson and Riley (1999) have shown that children with prenatal exposure to alcohol exhibit deficits on explicit, but not implicit, memory tasks. Relatively selective alcohol effects on measures of declarative, but not procedural, memory have also been reported (Carmichael Olson et al., 1998). Uecker and Nadel (1998) noted impairments in spatial, but not object, memory in children diagnosed with FASD (see also Hamilton et al., 2003). The scope of memory impairments observed in these studies suggests a significant impact of alcohol on the developing hippocampus. This speculation is supported by the literature on hippocampal-dependence of declarative, explicit, and spatial memory performance (Eichenbaum, 2001; Redish, 1999).
There are numerous reports of learning and memory impairments in animal subjects exposed to ethanol during the perinatal (pre- and/or postnatal) period. In many respects, the results parallel the findings from clinical literature on human FASD. Animals exposed to alcohol perinatally are often reported to be slower to learn a task (Reyes et al., 1989; Schneider et al., 2001), similar to results obtained from humans with FASD (Kaemingk et al., 2003; Mattson and Roebuck, 2002). Eyeblink conditioning deficits, indicative of cerebellar dysfunction, are also evident in both human FASD (Jacobson et al., 2008) and animal models (Brown et al., 2007), as are spatial learning impairments (Goodlett and Johnson, 1997; Uecker and Nadel, 1998).
Research designed to characterize learning and memory deficits in animals exposed to alcohol have primarily focused on performance using tasks that are dependent on the hippocampus. Moreover, studies of hippocampal-dependent memory impairments following early alcohol insult have been almost exclusively within the realm of spatial memory. For the most part, these studies have assessed spatial memory processes using tasks such as the Morris water maze, radial arm maze, and T-maze alternation. Altered spatial memory performance has been observed in both young and adult rats following neonatal ethanol exposure (e.g. Goodlett and Johnson, 1997; Nagahara and Handa, 1999; Reyes et al., 1989; Thomas et al., 2000). There are sparse reports of the consequences of alcohol exposure on hippocampal-dependent nonspatial memory performance (but see Green et al., 1992). While FASD individuals do exhibit spatial learning deficits (Uecker and Nadel, 1998), they are also impaired in their learning and memory abilities outside of the spatial domain. One such task that has been widely studied, appears to involve declarative memory processes (Clark et al., 2001), and provides an additional assay of hippocampal function is Pavlovian trace conditioning.
There are many variants of the Pavlovian conditioning paradigm; in the present experiments a fear conditioning procedure was employed. In what is called the delay procedure, the offset of a conditioned stimulus (CS), such as a tone or a light, is coincident with presentation of an unconditioned stimulus (US; e.g. electric shock). The amygdala is the brain structure most often associated with delay fear conditioning (LeDoux, 2000). However, in a trace procedure the offset of the CS and onset of the US are separated by a stimulus-free period known as the trace interval. Interference with normal hippocampal function has been shown to affect acquisition of Pavlovian trace conditioning, while sparing delay conditioning. Data supporting a role of the hippocampus in trace conditioning comes from several sources, including results from hippocampal-lesioned animals (Moyer et al., 1990; Quinn et al., 2002), administration of pharmacological agents that alter hippocampal cholinergic activity (Kaneko and Thompson, 1997; Moye and Rudy, 1987), and the ontogenetic emergence of trace conditioning (Barnet and Hunt, 2005; Ivkovich et al., 2000).
Wagner and Hunt (2006) examined ethanol-induced changes in memory processes in an animal model of FASD using these two fear conditioning paradigms. Neonates that were exposed to ethanol or sham intubations on postnatal days (PD) 4–9 were later trained as adolescents in either delay or trace fear conditioning. Subjects were tested 24 h later for CS-elicited freezing, a common measure of acquired fear. The differences observed in learning abilities in the delay versus trace paradigm afford a behavioral assay of hippocampal dysfunction resulting from neonatal alcohol exposure. The results were quite striking in showing that alcohol-exposed subjects were severely impaired in acquiring trace conditioning, but did not differ from controls in delay conditioning. The finding that ethanol-treated subjects did not differ from controls in delay conditioning implies that the subjects were not deficient in CS or US processing, basic associative learning abilities, or had difficulties with the expression of the freezing response.
The purpose of the present experiments was to more fully examine this ethanol-induced trace conditioning deficit by manipulating various aspects of our previous procedure. Specifically in Experiment 1 the dose of ethanol administered during the neonatal period was manipulated. In Experiment 2 changes in the duration of the trace interval used for training were evaluated. Finally, in Experiment 3 the timing of ethanol administration during the neonatal period was varied in order to determine the specific period of vulnerability to ethanol’s deleterious effects on trace fear conditioning. Results from these experiments serve to increase our understanding of the detrimental effects of neonatal alcohol exposure on short-term memory processing linked to hippocampal function.
Experiment 1
Wagner and Hunt (2006) reported that a high dose of ethanol (5.25 g/kg/day) given to rats on PD 4–9 resulted in poor trace fear conditioning performance in subjects trained as adolescents. This ethanol treatment had no observable effect on subjects’ acquisition of delay fear conditioning. The purpose of Experiment 1 was to conduct a dose-response analysis of neonatal ethanol exposure on trace fear conditioning. Several groups of subjects were treated on PD 4–9. Three ethanol-exposed groups were administered doses of 3.0, 4.0 or 5.0 g/kg/day ethanol during this time. Two control groups were also included: Sham-intubated and Unhandled. All animals were trained using a trace fear conditioning procedure similar to the one employed by Wagner and Hunt (2006).
Materials and Methods
Subjects
A total of 70 male and female Sprague-Dawley-derived rats representing 12 litters served as subjects in this experiment. Sixty animals (7 litters) were treated neonatally with 3.0, 4.0 or 5.0 g/kg/day ethanol or sham intubations, and 10 additional animals (5 litters) served as unhandled controls. Animals were born and reared in the Psychology Department vivarium at the College of William & Mary. Male and female breeders (Charles River Laboratories, Wilmington, MA) were housed in 50.8 × 40.6 × 21.6 cm clear polycarbonate cages with wire lids and pine chip bedding. Animals had free access to water and high-protein rodent chow (LabDiet Formula 5008). Cages were checked daily for the presence of pups, and the day of birth was designated as Postnatal Day 0 (PD 0). Litters were culled to 8–10 pups on PD 2. Pups remained with the dam until PD 21, at which time they were weaned and group-housed with siblings in identical polycarbonate cages for the remainder of the experiment. The vivarium was maintained on a 14:10 h light:dark cycle with light onset at 0600 h. All procedures occurred during the light portion of the cycle and were approved by the College of William and Mary’s Institutional Animal Care and Use Committee that follows guidelines established by the NIH.
Apparatus
Training occurred in two identical 38.0 × 26.0 × 22.0 cm modified Skinner boxes. The two shorter walls were made of aluminum and the two longer walls and top were made of clear Plexiglas. The floor was constructed of 5-mm stainless-steel bars spaced 1.5 cm apart (center-to-center). The 0.5 mA, 1-s shock unconditioned stimulus (US) was generated by a custom-built shock generator and delivered through the grid floor. The conditioned stimulus (CS) was produced by a 25-W white bulb, the center of which was located 12 cm above the floor and 8.5 cm from one of the longer walls of the chamber. The CS was 10 s in duration and flashed at a rate of two times per s. The chambers were housed in sound-attenuating shells measuring 67.0 × 71.5 × 71.0 cm. A 4-W red bulb was mounted inside the shell to provide constant low-level illumination. A PC computer was used to interface Coulbourn Instruments (Allentown, PA) software and hardware and controlled all stimulus presentations.
Testing for CS-elicited freezing occurred in a novel context located in a different room of the laboratory. Subjects were tested in two identical 29.0 × 21.5 × 46.5 cm clear Plexiglas chambers with open tops and bottoms. The lower 11 cm of the chambers was cut out and fitted with horizontally-mounted steel bars (0.5 mm diameter, 1.5 cm center-to-center). The chambers rested on a Plexiglas floor covered with brown paper. The chambers were individually housed in sound-attenuating shells (IAC; Industrial Acoustics, NY, NY) with a 7-W white bulb mounted on an inner wall to provide constant low-level illumination. Specifications of the CS used during the test were identical to those described for training. Test sessions were videotaped using Sony video cameras (Model CCD-TRV67).
Procedure
Ethanol administration
The ethanol administration procedure was based on that originally described by Pierce et al. (1993) and used in our previous studies. Beginning on PD 4, pups were removed from the home cage as a group and placed in a 35.3 × 21.9 × 13.0 cm opaque holding cage with pine chip bedding, maintained at 34°C by a heating pad placed beneath. Animals in the ethanol-exposed groups (EtOH) were given ethanol intragastrically in the form of an 11.9% v/v ethanol solution dissolved in Similac® (Abbott Laboratories, North Chicago, IL). The daily ethanol dose was divided into two administrations that were given each day, separated by a 2 h interval. The total daily dose of ethanol administered to the pups was 3.0, 4.0 or 5.0 g/kg. Ethanol administrations were achieved by using 15 cm lengths of polyethylene tubing (PE-10; Clay Adams, Parsippany, NJ) attached to 1 ml syringes, and the varying doses were achieved through different volumes of the solution. Two hours following the second ethanol administration, subjects were given a third vehicle-only feeding to at least partially overcome the weight loss typically seen in ethanol-treated pups. Sham intubated controls were subjected to the tube-insertion procedure three times per day, but were not given any fluid. Administration of large quantities of milk to young pups results in abnormal weight gain (Goodlett and Johnson, 1997). Intubations continued daily through PD 9. Unhandled controls received no treatment during the neonatal period, and were experimentally-naïve at the time of training on PD 30.
Trace fear conditioning
Conditioning occurred on PD 30 (+/− 1 day). Subjects were placed individually into one of the two conditioning chambers and allowed 5 min to adapt. Animals were then given 5 trials during which the offset of the 10 s light CS was separated from onset of the shock US by a 10 s stimulus-free trace interval. Inter-trial intervals ranged from 200–300 s, and conditioning sessions lasted 30 min. Animals were returned to the home cage immediately after training.
Testing
Testing for CS-elicited freezing occurred in a novel context approximately 24 h after conditioning. Subjects were placed into the test chamber and, after a 5 min period of adaptation, were presented with three 10 s light CSs that were separated by 90–120 s intervals. No shocks occurred during the test. Videotaped records of the test sessions were later scored by an observer blind to the experimental history of the subject. During the 10 s period prior to onset of each CS and during the 10 s presentation of the stimulus, animals were briefly observed every 2 s. At each observation period a judgment was made as to whether or not the subject was freezing (e.g. Fanselow, 1980). Freezing was defined as the absence of all observable movements except those required for respiration. The percentage of intervals scored as freezing during each epoch was calculated (0 – 100%) and data were converted into difference scores (%CS – %pre-CS) for analysis. Difference scores reflect relatively “pure” measures of fear to the CS, as levels of nonspecific freezing are subtracted out (Thompson and Rosen, 2000). Data were averaged across the three test trials for analysis.
Results and Discussion
Four animals (one in each of the neonatal treatment groups) were lost because of improper intubations and an additional three animals were lost because of technical complications during training or testing. Test data from the remaining 63 animals were analyzed. Ethanol and sham groups were composed of 13–14 subjects each and the unhandled group consisted of 9 subjects. There were approximately equal numbers of males and females in each group.
Body weights
Body weights recorded during the ethanol administration period were analyzed using a 4 (neonatal treatment) × 2 (sex) × 6 (day) mixed-design Analysis of Variance (ANOVA). Body weight data from unhandled subjects were not obtained during this time. This analysis revealed a significant main effect of day [F (5, 255) = 2139.00, p < .001] and a Neonatal Treatment × Day interaction [F (15, 255) = 26.28, p < .001]. Post hoc comparisons were conducted using Fisher tests (p < .05; Keppel, 1982) to determine the nature of the interaction. All subjects gained weight during the neonatal treatment period. In comparison with sham controls, the 3.0 g/kg group did not differ on any of the treatment days. Subjects treated with 4.0 g/kg weighed less than sham subjects on days 8 and 9, while subjects treated with 5.0 g/kg/day ethanol weighed less than sham controls on PD 5–9. In addition, the group treated with 5.0 g/kg/day weighed less than the 3.0 and 4.0 g/kg groups beginning on PD 5. Thus, ethanol treatment with the higher doses resulted in significantly less weight gain during the neonatal treatment phase. Finally, there were no sex differences in body weights.
Body weights recorded on the day of training (PD 30) were analyzed using a 5 (neonatal treatment) × 2 (sex) ANOVA that yielded a main effect of sex [F (1, 53) = 15.67, p < .001]. At PD 30 males weighed more than females (Mmales = 122.58 +/− 1.8 g; Mfemales = 110.40 +/− 2.5 g). There was no effect of neonatal treatment, indicating that body weights that had been reduced by ethanol treatment during the neonatal period had recovered by adolescence. Ethanol-treated and control subjects weighed the same on the day of training. Body weight data are shown in Table 1.
Table 1.
Mean (+/− SEM) body weight (g) recorded from subjects of Experiment 1 during the neonatal treatment phase (postnatal days [PD] 4–9) and on the day of training (PD 30). On PD 4–9 subjects were administered 3.0, 4.0 or 5.0 g/kg/day i.g. or were given sham intubations. The Unhandled group received no treatment until they were trained on PD 30.
| Postnatal Day | |||||||
|---|---|---|---|---|---|---|---|
| Neonatal Treatment | 4 | 5 | 6 | 7 | 8 | 9 | 30 |
| 3.0 g/kg | 14.6 (0.8) |
17.2 (0.9) |
19.9 (1.0) |
22.7 (1.1) |
25.6 (1.1) |
28.7 (1.2) |
118.1 (4.0) n = 13 |
| 4.0 g/kg | 14.8 (0.8) |
17.1 (0.9) |
19.1 (1.0) |
21.7 (1.1) |
23.7 (1.1) |
26.4 (1.2) |
118.4 (3.1) n = 14 |
| 5.0 g/kg | 14.9 (0.9) |
15.0 (1.0) |
16.9 (1.1) |
19.1 (1.2) |
20.9 (1.2) |
22.9 (1.3) |
117.1 (3.5 n = 13 |
| Sham | 13.8 (0.8) |
16.5 (0.9) |
19.2 (1.0) |
22.0 (1.1) |
24.8 (1.1) |
28.1 (1.2) |
115.5 (3.3) n = 14 |
| Unhandled | --- | --- | --- | --- | --- | --- | 120.4 (5.1) n = 9 |
Trace fear conditioning
The one way ANOVA conducted on the pre-CS freezing data revealed no group differences, F < 1. All groups showed equivalent levels of pre-CS freezing in the novel test context (M +/− SEM % pre-CS freezing: unhandled 33.3 +/− 13.7%, sham 14.3 +/− 11.0%, 3.0 g/kg 21.5 +/− 11.4%, 4.0 g/kg 28.5 +/− 10.9%, 5.0 g/kg 30.7 +/− 11.4%). The ANOVA conducted on the Difference scores (%CS - %pre-CS) revealed a main effect of neonatal treatment [F (4, 53) = 2.62, p < .05]. The data are shown in Figure 1. Post hoc comparisons (Fisher tests) indicated that subjects treated with 5.0 g/kg/day displayed lower levels of freezing to the light CS than unhandled, sham and 3.0 g/kg subjects. Subjects in the 4.0 g/kg/day group showed reduced levels of freezing as well, but this group’s performance only differed from the unhandled control group. Finally, freezing recorded from unhandled, sham-intubated, and the 3.0 g/kg/day groups did not differ.
Figure 1.
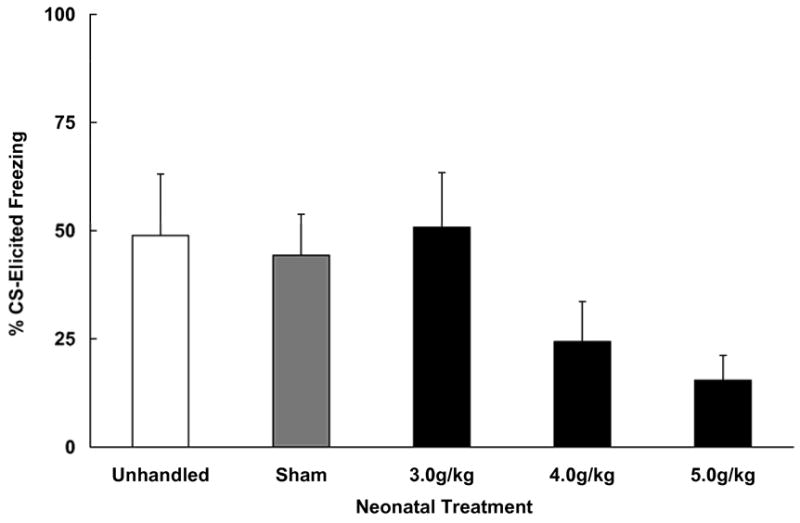
Mean (+/− SEM) freezing (%CS - %Pre-CS) in response to a trace conditioned CS in adolescent-age subjects. Animals were treated on postnatal days (PD) 4–9 with one of three doses of ethanol (3.0, 4.0 or 5.0 g/kg/day), received sham intubations (Sham) or were Unhandled. Higher doses of ethanol resulted in more profound deficits in later trace conditioning.
These results confirm those of Wagner and Hunt (2006) that rats exposed to ethanol on PD 4–9 exhibit deficits in trace fear conditioning as adolescents. In addition, the effect was found to be dose-dependent, with the greatest impairment observed in animals treated with the 5.0 g/kg/day dose of ethanol. The lowest dose of ethanol used in this experiment (3.0 g/kg/day) had no effect on subsequent trace conditioning, and animals treated with this dose did not differ in CS-elicited freezing from either the unhandled or sham control groups.
Experiment 2
A common finding in the literature on neonatal ethanol exposure effects is a decrement in memory performance with imposed delays, and this may also serve as an indication of hippocampal dysfunction (Clark et al., 2000). Nagahara and Handa (1999), for example, trained rats on a delayed alternation task in which there was a forced-run trial followed by a test trial. On the test run, the subject was rewarded for entering the arm opposite to that of the forced run. Ethanol-exposed animals had little difficulty with the task when the delay between the forced and choice run was minimal (0 or 20 s). However, memory performance of ethanol subjects declined dramatically with longer delays (60 and 180 s). Similarly, Girard et al. (2000) reported more severe memory impairments in ethanol-exposed rats in a delayed matching-to-place task with long delays. There are several other studies that report time-dependent decrements in memory and impairments are typically more severe in ethanol-exposed subjects (e.g. Clements et al., 2005; Green et al., 1992; but see Schneider et al., 2001).
The purpose of Experiment 2 was to evaluate the consequences of neonatal ethanol exposure on trace conditioning as a function of the duration of the trace interval used for training. Hippocampal lesions can oftentimes result in severely impaired responding to a trace conditioned CS, but this effect is dependent upon the memory demands of the task. Specifically, animals with hippocampal lesions can acquire trace conditioned responding provided that the trace interval is relatively short, but exhibit increasing impairment as the trace interval duration is lengthened (Chowdhury et al., 2005; Moyer et al., 1990). These findings suggest that the role of the hippocampus in trace conditioning lies along a continuum that is defined by trace interval length. Our working hypothesis of the effects of neonatal ethanol on trace conditioned performance is that this treatment compromises the functioning of the hippocampus. If this is true, and ethanol-exposed subjects resemble animals with more discrete lesions of the hippocampus, then neonatal treatment with ethanol would be expected to produce reductions in trace conditioning that would worsen with increases in trace interval duration (cf. Girard et al., 2000; Nagahara and Handa, 1999).
Materials and Methods
Subjects
The subjects in this experiment were 104 Sprague-Dawley derived rats, representing 13 litters. Animals were maintained as described in Experiment 1.
Procedures
On PD 4, pups were randomly assigned to one of two neonatal treatment groups. One group was given 5.0 g/kg/day ethanol on PD 4–9 and the other was given sham intubations, as described in Experiment 1. On PD 30 all animals were given 5 trace conditioning trials during a 30-min training session. The trace interval separating CS offset from US onset differed between groups of subjects. Animals were trained with trace interval durations of 0, 1, 3 or 10 s, and the CS-to-CS interval was equated for all groups. These trace interval durations were chosen largely on the basis of those used by Chowdhury et al. (2005). Twenty-four hours after training animals were tested for CS-elicited freezing as previously described.
Results and Discussion
Seven subjects were lost during the neonatal treatment phase of the experiment, and an additional two animals were lost due to experimenter error during testing. Test data from the remaining 95 animals were analyzed, and groups included 11–12 subjects each. There were approximately equal numbers of males and females in each treatment group.
Body weights
Body weights recorded on the day of training were analyzed using a 2 (neonatal treatment) × 2 (sex) ANOVA. The analysis yielded main effects of neonatal treatment [F (1, 91) = 12.22, p < .01] and sex [F (1, 91) = 41.72, p < .001]. On PD 30, males weighed more than females and, in contrast to results of Experiment 1, ethanol-treated subjects weighed slightly less than sham-treated controls (Methanol = 115.6 +/− 1.6 g; Msham = 123.5 +/− 1.6 g).
Trace fear conditioning
Trace conditioning performance of all groups is depicted in Figure 2. A 2 (neonatal treatment) × 4 (trace interval) × 2 (sex) ANOVA conducted on pre-CS freezing data indicated no differences in levels of freezing prior to CS onset during the test, and freezing was relatively low in all groups (range 10.7 – 25.3%). The ANOVA conducted on the Difference scores yielded significant main effects of neonatal treatment [F (1, 79) = 8.88, p < .01] and trace interval [F (3, 79) = 4.91, p < .01]. Separate one-way ANOVAs were conducted on the data obtained from each neonatal treatment group. There was an effect of trace interval duration for ethanol-treated subjects [F (3, 44) = 6.15, p < .01], but not for sham controls (F < 1). Post hoc comparisons (Fisher tests) conducted on the data obtained from ethanol-treated subjects indicated that responding in the group trained with the 10 s trace interval duration was lower than that obtained in the 0 s group. Those subjects trained with a 3 s trace interval exhibited an intermediate level of CS-elicited freezing, but freezing in this group did not differ from that observed in either the 0 s or the 10 s trace interval groups.
Figure 2.

Mean (+/− SEM) freezing (%CS - %Pre-CS) to a visual CS in adolescent-age subjects treated with 5.0 g/kg/day ethanol (EtOH) or Sham intubations on PD 4–9. For training, the duration of the trace interval (time from CS offset to US onset) was varied: 0, 1, 3 or 10 s. Ethanol-treated rats exhibited impaired trace conditioning with increasing trace interval durations. There was no effect of trace interval length on responding in Sham subjects.
The results of this experiment confirmed the prediction that ethanol-exposed animals would exhibit a trace interval-dependent decline in conditioned responding. These findings are similar to the results of Chowdhury et al. (2005) who manipulated the duration of the trace interval used for training adult rats with discrete excitotoxic lesions of the hippocampus. In that study, hippocampal lesions disrupted trace conditioned performance, but only when a relatively long trace interval was used. Lesioned animals trained with shorter trace interval durations showed much less of a deficit. The same pattern of results is seen in the present experiment with animals exposed to ethanol on PD 4–9; performance decreased with increases in length of the trace interval. These data are in line with the hypothesis that neonatal ethanol exposure disrupts trace fear conditioning by interfering with the functioning of the hippocampus.
Experiment 3
The timing of fetal alcohol exposure has been associated with the severity of cognitive and behavioral impairments observed (Goodlett and Johnson, 1999). For example, Brown et al. (2007) examined eyeblink conditioning in weanling rats that were treated with ethanol during the entire “third-trimester equivalent” period (PD 4–9) or during the second half only (PD 7–9). It was found that acquisition of eyeblink conditioning, which critically depends on cerebellar circuitry, was reduced by ethanol exposure on PD 4–9, but was not affected by ethanol exposure that was confined to the PD 7–9 period. These behavioral findings are corroborated by anatomical evidence showing greater cerebellar Purkinje cell loss following ethanol exposure on PD 4–6 relative to PD 7–9 (Goodlett and Lundahl, 1996).
Goodlett and Johnson (1997) investigated the timing of ethanol exposure on hippocampus-dependent spatial memory performance. Adolescent animals were trained in a spatial version of the Morris water maze task following exposure to ethanol on PD 4–9, 4–6 or 7–9. Performance in the water maze was more severely compromised by ethanol exposure during the second half of this neonatal period. That is, rats treated with ethanol on PD 7–9 exhibited more severe disruption in water maze performance than animals treated with ethanol on PD 4–6, and performance of subjects in the PD 7–9 group did not differ significantly from that observed in animals given ethanol during the entire period (PD 4–9).
In the present study, ethanol exposure during specific periods of the brain growth spurt was examined in order to define critical periods of vulnerability for trace fear conditioning. Animals were exposed to ethanol on PD 4–6, 7–9 or 4–9. It was predicted that ethanol exposure confined to PD 7–9 would be more detrimental to trace conditioning than exposure limited to PD 4–6, based upon results of Goodlett and Johnson (1997) using another hippocampus-dependent task.
Materials and Methods
Subjects
The subjects were 40 Sprague-Dawley rats derived from 8 different litters.
Procedures
On PD 4 animals were assigned to one of four treatment groups: E4-6, E4-9, E7-9 or S4-9 that differed in whether they were administered ethanol (E) or were given sham intubations (S), and the days on which these treatments were given. S4-9 and E4-9 were treated as described previously on PD 4–9, and ethanol was administered at a dose of 5.0 g/kg/day. Group E4-6 was treated with 5.0 g/kg/day ethanol on days 4–6 and received sham intubations on days 7–9 while the reverse was true for group E7-9 (sham intubations on PD 4–6 and 5.0 g/kg/day ethanol on PD 7–9). Trace conditioning occurred on PD 30 and all animals were trained using a 10 s trace interval. Subjects were tested for CS-elicited freezing 24 h later.
Results and Discussion
A total of 8 subjects were lost because of improper intubations or equipment malfunction, leaving 32 subjects for which behavioral data were collected (ns = 8/group). An equal number of male and female subjects was included in each group.
Body weights
The analysis of body weights recorded on the day of training revealed a main effect of sex [F (1, 24) = 36.75, p < .001], but no effect of neonatal treatment. Males weighed more than females (Mmales = 130.0 +/− 2.9 g; Mfemales = 111.0 +/− 1.9 g). The groups treated with ethanol did not differ from sham controls.
Trace fear conditioning
Analysis of pre-CS freezing was accomplished using a 4 (neonatal treatment) × 2 (sex) ANOVA, and revealed no significant effects. All animals exhibited relatively low levels of freezing in the test context (range 15.0 – 25.3%). The analysis of the Difference scores yielded a significant main effect of neonatal treatment [F (3, 24) = 4.41, p < .05]. These data are presented in Figure 3. Post hoc comparisons (Fisher tests) indicated that ethanol treatment on either PD 4–6 or 4–9 resulted in significantly less trace conditioned responding than that observed in subjects given sham intubations. Furthermore, ethanol treatment restricted to PD 7–9 had no effect on CS-elicited freezing, as group E7-9 did not differ from group S4-9. It could be argued that differences in blood-alcohol levels obtained in the E4-6 and E7-9 groups contributed to the differences in trace fear conditioning observed. While blood-alcohol levels were not measured in this experiment, other laboratories that have varied the timing of ethanol exposure have not reported substantially different alcohol levels in groups given ethanol on PD 4–6 (measured on PD 6) and PD 7–9 (measured on PD 9) (Goodlett and Johnson, 1997; Goodlett and Lundahl, 1996). Therefore it is unlikely that different levels of alcohol exposure contributed to the behavioral outcome. These data instead suggest that the critical period for ethanol’s detrimental effects on trace fear conditioning is the first half of the third trimester equivalent. This is a different pattern of results from those reported using the Morris water maze task (Goodlett and Johnson, 1997).
Figure 3.
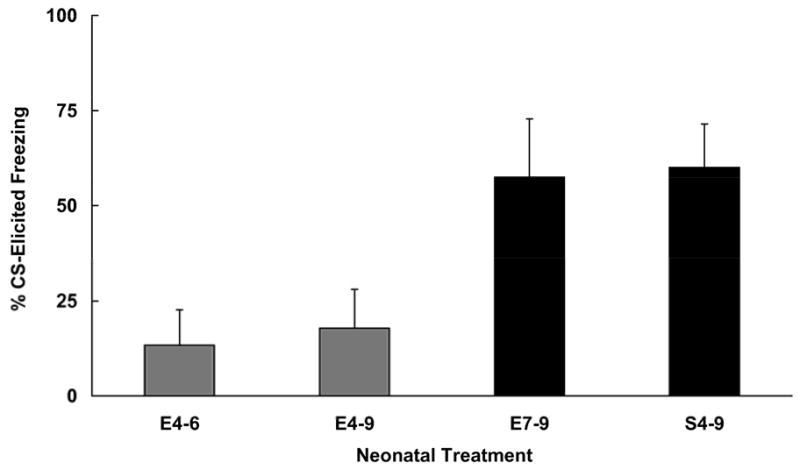
Mean (+/− SEM) freezing (%CS - %Pre-CS) to the trace CS by subjects exposed to ethanol (E) on postnatal days 4–6, 4–9 or 7–9. Groups were compared to a control group that received sham intubation on postnatal days 4–9 (S4-9). Ethanol given on postnatal days 4–6 produced an effect as severe as ethanol given during the entire third trimester equivalent (4–9). Ethanol exposure limited to PD 7–9 did not affect trace conditioning.
General Discussion
The results of these experiments confirm the negative consequences of neonatal ethanol exposure on acquisition of trace fear conditioning (Wagner and Hunt, 2006). The effects of early ethanol exposure were found to be (1) dose-dependent, (2) parallel to the effects of hippocampal lesions on trace conditioning with different trace interval durations and (3) more detrimental to performance when ethanol exposure occurs during the first half of the third trimester equivalent period.
Neonatal ethanol exposure has been reported to result in hyperactivity (Melcer et al., 1994; Thomas et al., 2004), visual disturbances (Medina et al., 2005; Phillips et al., 1991), and changes in stress responsivity (Slone and Redei, 2002; Weinberg et al., 2008), any of which could affect subject’s ability to learn or express trace fear conditioning to a visual CS. Increased responsiveness to a stressor could impact the animal’s perception of the shock US, although the prediction here would be that a perception of a more intense US would increase learning to the CS. Changes in visual acuity could alter the perception of the CS, perhaps making the CS less detectable. And finally, increases in general activity levels might interfere with the expression of the behavioral response, freezing, that could be misinterpreted as less learning. The results of Experiment 2 however can be used to discount these effects of ethanol exposure on the phenomenon observed here. In Experiment 2 the ethanol-exposed and sham control groups that were trained with a 0 s trace interval expressed equivalent levels of CS freezing (se also Wagner and Hunt, 2006). If neonatal ethanol exposure resulted in changes in the perception of the CS or the US, or a reduced ability to respond with immobility/freezing, then the ethanol-exposed subjects trained with the 0 s trace interval would show different levels of responding compared with sham controls. This was clearly not the case.
Given the correspondence between the effects of neonatal ethanol and results obtained from adult animals with discrete lesions to the hippocampus, the results suggest that trace conditioning impairments following neonatal ethanol exposure are the result of altered functioning of the hippocampus. Current hypotheses concerning the role of the hippocampus in trace conditioning suggest that it serves to preserve a memory representation of the CS during the trace interval, and may be important for the encoding of critical temporal information about the CS and US (e.g. Cole et al., 1995; Rodriguez and Levy, 2001). Research has shown that hippocampal neurons exhibit altered firing rates during the trace interval that may serve as a neural code/memory of the CS (e.g. Green and Arenos, 2007; McEchron et al., 2003). However, the effects of post-training manipulations suggest that the hippocampus also contributes to memory processing occurring after training, perhaps by promoting the consolidation of the trace memory (Quinn et al., 2002).
The hippocampus is significantly affected by perinatal ethanol exposure in terms of both morphology and function. The number of pyramidal neurons in area CA1 is reduced following neonatal exposure (Green et al., 1992; Livy et al., 2003; Tran and Kelly, 2003), as is the number of granule cells in the dentate gyrus (Livy et al., 2003; Miller, 1995). In addition to producing structural changes, early ethanol exposure has been found to alter several neurochemical systems, including central cholinergic systems, that are highly implicated in hippocampus-dependent memory tasks (Maier et al., 1996; Valles et al., 1995). For example, Kelly et al. (1989) reported a change in muscarinic cholinergic receptor number in the hippocampus following neonatal ethanol exposure, and Swanson et al. (1995) reported a decrease in choline acetyltransferase activity in the septo-hippocampal system. Others report alterations in dose-response functions for cholinergic drugs (Nagahara and Handa, 1999; but see Hannigan et al., 1993). Thus, changes in cell structure and number in hippocampal regions may not necessarily be the primary culprits in terms of memory deficits but rather, altered function in other brain regions that influence the activity of the hippocampus could be the underlying mechanism.
Ethanol has widespread effects within the central nervous system, and although hippocampal function is altered by ethanol, this structure is not necessarily the only site of ethanol’s deleterious consequences on trace conditioned responding. Another possible site for ethanol’s effects on trace fear conditioning is the frontal cortex. Recent studies have implicated the medial prefrontal cortex (mPFC) as a storage site for trace memories (McLaughlin et al., 2002). Neonatal ethanol exposure alters the structure and activity of the frontal lobes in animals. Inomata et al. (1987) observed a reduction in the number of synapses in the frontal cortex following prenatal ethanol exposure. Similarly, Mihalick et al. (2001) reported significant cell loss in the medial prefrontal cortex that was correlated with reversal learning impairments. It is thus possible that the trace conditioning impairments observed here are due to ethanol’s actions in the frontal cortex as opposed to, or in addition to, its known actions within the hippocampus (cf. Takahara et al., 2003).
The present experiments used a model of third-trimester ethanol exposure (Pierce et al., 1993). There are varied reports of consequences arising from drug exposure in relation to the three trimesters of human gestation. Some studies have identified effects following very early alcohol exposure, during the first trimester (e.g. Astley et al., 1999; Clarren et al., 1992). Others, however, report what appear to be more significant cognitive impairments following alcohol exposure that spans all three trimesters (Coles et al., 1991; Korkman et al., 2003). While total duration of exposure is obviously a confounding factor in this analysis, results of the latter studies suggested that exposure to alcohol during the latter part of gestation may be particularly devastating to neurobehavioral outcome. Regional differences in brain vulnerability to alcohol insult depend on the timing of ethanol exposure relative to critical periods of brain development (e.g. Brown et al., 2007; Goodlett and Johnson, 1999; Tran and Kelly, 2003). Development of several brain regions, in particular those known to be critical for many types of learning and memory, undergo substantial anatomical and functional development during the third trimester brain growth spurt (Altman and Bayer, 1975).
In the rat, a comparable period of brain growth occurs during the first two postnatal weeks (Dobbing and Sands, 1979). In order to model neurobehavioral changes resulting from drug exposure, ethanol can be given during targeted periods of neural development (Goodlett and Johnson, 1999). As described above, ethanol exposure limited to PD 4–6 has significant effects on anatomical and functional development of the cerebellum (Brown et al., 2007; Goodlett and Lundahl, 1996), while a somewhat later period (PD 7–9) produces effects within the hippocampus (Goodlett and Johnson, 1997). The data from the present Experiment 3 contrast somewhat with the results reported by Goodlett and Johnson (1997). Results of this experiment clearly demonstrate that the deleterious effects of neonatal ethanol exposure on trace fear conditioning are due to ethanol exposure earlier in development (PD 4–6, as opposed to PD 7–9). This result was surprising because performance in both tasks (trace conditioning and spatial learning) appear to rely on similar anatomical and neurochemical systems. The dorsal hippocampus, for example, has been found to be critical for both trace conditioning (Burman et al., 2006) and spatial navigation (Pothuizen et al., 2004). Moreover, neurotransmitter systems within the hippocampus that are critical for trace conditioning and spatial memory also overlap, and include the cholinergic (Herrera-Morales et al., 2007; Hunt and Richardson, 2007) and glutamatergic systems (Bast et al., 2005; Wanisch et al., 2005). Given the similarity in neuroanatomical and neurochemical substrates of trace and spatial learning, it is not clear why the pattern of temporal vulnerability to neonatal ethanol exposure differs so dramatically for these two tasks (Wagner and Hunt, 2006; Goodlett and Johnson, 1997). Clearly what is needed is a more detailed analyses of subregion-specific populations of neurons within the dorsal hippocampus, their specific roles in learning, and their developmental vulnerability to ethanol insult.
Animal models of FASD have been valuable for demonstrating the consequences of early alcohol exposure. In particular, anatomical and neurochemical alterations resulting from alcohol exposure can be clearly demonstrated and more causally linked to deficits in cognitive and behavioral performance. Furthermore, the potential benefits of a variety of intervention and treatment approaches can be thoroughly explored (e.g. Klintsova et al., 1999; Thomas et al., 2000). Trace fear conditioning deficits may provide another useful paradigm for further studies in these and other areas (Wagner and Hunt, 2006).
Acknowledgments
This research was supported by grant AA015343 from the National Institute on Alcohol Abuse and Alcohol, a grant from the Virginia Tobacco Settlement Foundation, and a Howard Hughes Medical Institute grant through the Undergraduate Biological Sciences Education Program to the College of William & Mary.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel EL, Hannigan JH. Maternal risk factors in fetal alcohol syndrome: Provocative and permissive influences. Neurotoxicol Teratol. 1995;17:445–462. doi: 10.1016/0892-0362(95)98055-6. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer S. Postnatal development of the hippocampal dentate gyrus under normal and experimental conditions. In: Isaacson RL, Pribram KH, editors. The Hippocampus: Structure and Development. Vol. 1. Plenum; New York: 1975. pp. 95–122. [Google Scholar]
- Astley SJ, Magnuson SI, Omnell LM, Clarren SK. Fetal alcohol syndrome: Changes in craniofacial form with age, cognition and timing of ethanol exposure in the macaque. Teratology. 1999;59:163–172. doi: 10.1002/(SICI)1096-9926(199903)59:3<163::AID-TERA8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Barnet RC, Hunt PS. Trace and long-delay fear conditioning in the developing rat. Learn Behav. 2005;33:437–443. doi: 10.3758/bf03193182. [DOI] [PubMed] [Google Scholar]
- Bast T, da Silva BM, Morris RGM. Distinct contributions of hippocampal NMDA and AMPA receptors to encoding and retrieval of one-trial place memory. J Neurosci. 2005;25:5845–5856. doi: 10.1523/JNEUROSCI.0698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KL, Calizo LH, Goodlett CR, Stanton ME. Neonatal alcohol exposure impairs acquisition of eyeblink conditioned responses during discrimination learning and reversal in weanling rats. Dev Psychobiol. 2007;49:243–257. doi: 10.1002/dev.20178. [DOI] [PubMed] [Google Scholar]
- Burman MA, Starr MJ, Gewirtz JC. Dissociable effects of hippocampus lesions on expression of fear and trace fear conditioning memories in rats. Hippocampus. 2006;16:103–113. doi: 10.1002/hipo.20137. [DOI] [PubMed] [Google Scholar]
- Carmichael Olson H, Feldman JJ, Streissguth AP, Sampson PD, Bookstein FL. Neuropsychological deficits in adolescents with fetal alcohol syndrome: Clinical findings. Alcohol Clin Exp Res. 1998;22:1998–2012. [PubMed] [Google Scholar]
- Chowdhury N, Quinn JJ, Fanselow MS. Dorsal hippocampus involvement in trace fear conditioning with long, but not short, trace intervals in mice. Behav Neurosci. 2005;119:1396–1402. doi: 10.1037/0735-7044.119.5.1396. [DOI] [PubMed] [Google Scholar]
- Clark RE, Manns JR, Squire LR. Trace and delay eyeblink conditioning: Contrasting phenomena of declarative and nondeclarative memory. Psychol Sci. 2001;12:304–308. doi: 10.1111/1467-9280.00356. [DOI] [PubMed] [Google Scholar]
- Clarren SK, Astley SJ, Gunderson VM, Spellman D. Cognitive and behavioral deficits in nonhuman primates associated with very early embryonic binge exposures to ethanol. J Pediatr. 1992;121:789–796. doi: 10.1016/s0022-3476(05)81917-1. [DOI] [PubMed] [Google Scholar]
- Clements KM, Girard TA, Ellard CG, Wainwright PE. Short-term memory impairment and reduced hippocampal c-Fos expression in an animal model of fetal alcohol syndrome. Alcohol Clin Exp Res. 2005;29:1049–1059. doi: 10.1097/01.alc.0000171040.82077.e. [DOI] [PubMed] [Google Scholar]
- Cole RP, Barnet RC, Miller RR. Temporal encoding in trace conditioning. Anim Learn Behav. 1995;23:144–153. [Google Scholar]
- Coles CD, Brown RT, Smith IE, Platzman KA, Erickson S, Falek A. Effects of prenatal alcohol exposure at school age: Physical and cognitive development. Neurotoxicol Teratol. 1991;13:357–367. doi: 10.1016/0892-0362(91)90084-a. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Ebrahim SH, Diekman ST, Floyd L, Decoufle P. Comparison of binge drinking among pregnant and non-pregnant women, United States, 1991–1995. Am J Obstet Gynecol. 1999;180:1–7. doi: 10.1016/s0002-9378(99)70139-0. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. The hippocampus and declarative memory: Cognitive mechanisms and neural codes. Behav Brain Res. 2001;127:199–207. doi: 10.1016/s0166-4328(01)00365-5. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Conditional and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Girard TA, Xing HC, Ward GR, Wainwright PE. Early postnatal alcohol exposure has long-term effects on the performance of male rats in a delayed matching-to-place task in the Morris water maze. Alcohol Clin Exp Res. 2000;24:300–306. [PubMed] [Google Scholar]
- Goodlett CR, Johnson TB. Neonatal binge ethanol exposure using intubation: Timing and dose effects on place learning. Neurotoxicol Teratol. 1997;19:435–446. doi: 10.1016/s0892-0362(97)00062-7. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Johnson TB. Temporal windows of vulnerability within the third trimester equivalent: Why “knowing when” matters. In: Hannigan JH, Spear LP, Spear NE, Goodlett CR, editors. Alcohol and alcoholism: Effects on brain and development. Mahwah NJ: Erlbaum; 1999. pp. 59–91. [Google Scholar]
- Goodlett CR, Lundahl KR. Temporal determinants of neonatal alcohol-induced cerebellar damage and motor performance deficits. Pharmacol Biochem Behav. 1996;55:531–540. doi: 10.1016/s0091-3057(96)00248-1. [DOI] [PubMed] [Google Scholar]
- Green JT, Arenos JD. Hippocampal and cerebellar single-unit activity during delay and trace eyeblink conditioning in the rat. Neurobiol Learn Mem. 2007;87:269–284. doi: 10.1016/j.nlm.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PL, Diaz-Granados JL, Amsel A. Blood ethanol concentrations from early postnatal exposure: Effects of memory-based learning and hippocampal neuroanatomy in infant and adult rats. Behav Neurosci. 1992;106:51–61. doi: 10.1037//0735-7044.106.1.51. [DOI] [PubMed] [Google Scholar]
- Hamilton DA, Kodituwakku P, Sutherland RJ, Savage DD. Children with fetal alcohol syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav Brain Res. 2003;143:85–94. doi: 10.1016/s0166-4328(03)00028-7. [DOI] [PubMed] [Google Scholar]
- Hannigan JH, Cortese BM, DiCerbo JA, Radford LD. Scopolamine does not differentially affect Morris maze performance in adult rats exposed prenatally to alcohol. Alcohol. 1993b;10:529–535. doi: 10.1016/0741-8329(93)90077-2. [DOI] [PubMed] [Google Scholar]
- Herrera-Morales W, Mar I, Serrano B, Bermudez-Rattoni F. Activation of hippocampal postsynaptic muscarinic receptors is involved in long-term spatial memory formation. Eur J Neurosci. 2007;25:1581–1588. doi: 10.1111/j.1460-9568.2007.05391.x. [DOI] [PubMed] [Google Scholar]
- Hunt PS, Richardson R. Pharmacological dissociation of trace and long-delay fear conditioning in young rats. Neurobiol Learn Mem. 2007;87:86–92. doi: 10.1016/j.nlm.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Inomata K, Nasu F, Tanaka H. Decreased density of synaptic formation in the frontal cortex of neonatal rats exposed to ethanol in utero. Int J Dev Neurosci. 1987;5:455–460. doi: 10.1016/0736-5748(87)90023-2. [DOI] [PubMed] [Google Scholar]
- Ivkovich D, Paczkowski CM, Stanton ME. Ontogeny of delay versus trace eyeblink conditioning in the rat. Dev Psychobiol. 2000;36:148–160. doi: 10.1002/(sici)1098-2302(200003)36:2<148::aid-dev6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Jacobson SW, Stanton ME, Molteno CD, Burden MJ, Fuller DS, Hoyme HE, Robinson LK, Khaole N, Jacobson JL. Impaired eyeblink conditioning in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 2008;32:365–372. doi: 10.1111/j.1530-0277.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- Kaemingk KL, Mulvaney S, Halverson PT. Learning following prenatal alcohol exposure: Performance on verbal and visual multitrial tasks. Arch Clin Neuropsychol. 2003;18:33–47. [PubMed] [Google Scholar]
- Kaneko T, Thompson RF. Disruption of trace conditioning of the nictitating membrane response in rabbits by central cholinergic blockade. Psychopharmacology. 1997;131:161–166. doi: 10.1007/s002130050279. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Black AC, West JR. Changes in the muscarinic cholinergic receptors in the hippocampus of rats exposed to ethyl alcohol during the brain growth spurt. J Pharmacol Exp Ther. 1989;249:798–804. [PubMed] [Google Scholar]
- Keppel G. Design and analysis: A researcher’s handbook. 2. Englewood Cliffs, NJ: Prentice Hall; 1982. [Google Scholar]
- Klintsova AY, Goodlett CR, Greenough WT. Therapeutic motor training ameliorates cerebellar effects of postnatal binge alcohol. Neurotoxicol Teratol. 1999;22:125–132. doi: 10.1016/s0892-0362(99)00052-5. [DOI] [PubMed] [Google Scholar]
- Kodituwakku PW. Defining the behavioral phenotype in children with fetal alcohol spectrum disorders: A review. Neurosci Biobehav Rev. 2007;31:192–201. doi: 10.1016/j.neubiorev.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Korkman M, Kettunen S, Autti-Ramo I. Neurocognitive impairment in early adolescence following prenatal alcohol exposure of varying duration. Child Neuropsychol. 2003;9:117–128. doi: 10.1076/chin.9.2.117.14503. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: Effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Maier SE, Chen WJ, West JR. Prenatal binge-like alcohol exposure alters neurochemical profiles in fetal rat brain. Pharmacol Biochem Behav. 1996;55:521–529. doi: 10.1016/s0091-3057(96)00282-1. [DOI] [PubMed] [Google Scholar]
- Maier SE, West JR. Drinking patterns and alcohol-related birth defects. Alcohol Res Health. 2001;25:168–174. [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, Riley EP. Implicit and explicit memory functioning in children with heavy prenatal alcohol exposure. J Int Neuropsychol Soc. 1999;5:462–471. doi: 10.1017/s1355617799555082. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Roebuck TM. Acquisition and retention of verbal and nonverbal information in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res. 2002;26:875–882. [PubMed] [Google Scholar]
- McEchron MD, Tseng W, Disterhoft JF. Single neurons in CA1 hippocampus encode trace interval duration during trace heart rate (fear) conditioning in rabbit. J Neurosci. 2003;23:1535–1547. doi: 10.1523/JNEUROSCI.23-04-01535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, Skaggs H, Churchwell J, Powell DA. Medial prefrontal cortex and Pavlovian conditioning: Trace versus delay conditioning. Behav Neurosci. 2002;116:37–47. [PubMed] [Google Scholar]
- Medina AE, Krahe TE, Ramoa AS. Early alcohol exposure induces persistent alteration of cortical columnar organization and reduced orientation selectivity in the visual cortex. J Neurophysiol. 2005;93:1317–1325. doi: 10.1152/jn.00714.2004. [DOI] [PubMed] [Google Scholar]
- Melcer T, Gonzalez D, Barron S, Riley EP. Hyperactivity in preweanling rats following postnatal alcohol exposure. Alcohol. 1994;11:41–45. doi: 10.1016/0741-8329(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Mihalick SM, Crandall JE, Langlois JC, Krienke JD, Dube WV. Prenatal ethanol exposure, generalized learning impairment, and medial prefrontal cortical deficits in rats. Neurotoxicol Teratol. 2001;23:453–462. doi: 10.1016/s0892-0362(01)00168-4. [DOI] [PubMed] [Google Scholar]
- Miller MW. Generation of neurons in the rat dentate gyrus and hippocampus: effects of prenatal and postnatal treatment with ethanol. Alcohol Clin Exp Res. 1995;19:1500–1509. doi: 10.1111/j.1530-0277.1995.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Moye TB, Rudy JW. Visually mediated trace conditioning in young rats: Evidence for cholinergic involvement in the development of associative memory. Psychobiol. 1987;15:128–136. doi: 10.1002/dev.420200405. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Deyo RA, Disterhoft JF. Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behav Neurosci. 1990;104:243–252. doi: 10.1037//0735-7044.104.2.243. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Handa RJ. Fetal alcohol-exposed rats exhibit differential response to cholinergic drugs on a delay-dependent memory task. Neurobiol Learn Mem. 1999;72:230–243. doi: 10.1006/nlme.1999.3909. [DOI] [PubMed] [Google Scholar]
- Phillips DE, Krueger SK, Rydquist JE. Short- and long-term effects of combined pre- and postnatal ethanol exposure (three trimester equivalency) on the development of myelin and axons in rat optic nerve. Int J Dev Neurosci. 1991;9:631–647. doi: 10.1016/0736-5748(91)90025-h. [DOI] [PubMed] [Google Scholar]
- Pierce DR, Serbus DC, Light KE. Intragastric administration of alcohol during postnatal development of rats resulted in selective cell loss in the cerebellum. Alcohol Clin Exp Res. 1993;17:1275–1280. doi: 10.1111/j.1530-0277.1993.tb05241.x. [DOI] [PubMed] [Google Scholar]
- Pothuizen HHJ, Zhang W, Jongen-Relo AL, Feldon J, Yee B. Dissociation of function between the dorsal and ventral hippocampus in spatial learning abilities of the rat: A within-subject, within-task comparison of reference and working spatial memory. Eur J Neurosci. 2004;19:704–712. doi: 10.1111/j.0953-816x.2004.03170.x. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Oommen SS, Morrison GE, Fanselow MS. Post-training excitotoxic lesions of the dorsal hippocampus attenuate forward trace, backward trace, and delay fear conditioning in a temporally specific manner. Hippocampus. 2002;12:495–504. doi: 10.1002/hipo.10029. [DOI] [PubMed] [Google Scholar]
- Redish AD. Beyond the cognitive map: From place cells to episodic memory. Cambridge, MA: MIT Press; 1999. [Google Scholar]
- Reyes E, Wolfe J, Savage DD. The effects of prenatal alcohol exposure on radial arm maze performance in adult rats. Physiol Behav. 1989;46:45–48. doi: 10.1016/0031-9384(89)90319-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Levy WB. A model of hippocampal activity in trace conditioning: Where’s the trace? Behav Neurosci. 2001;115:1224–1238. doi: 10.1037//0735-7044.115.6.1224. [DOI] [PubMed] [Google Scholar]
- Schneider ML, Moore CF, Kraemer GW. Moderate alcohol during pregnancy: Learning and behavior in adolescent rhesus monkeys. Alcohol Clin Exp Res. 2001;25:1383–1392. [PubMed] [Google Scholar]
- Slone JL, Redei EE. Maternal alcohol and adrenalectomy: Asynchrony of stress response and forced swim behavior. Neurotoxicol Teratol. 2002;24:173–178. doi: 10.1016/s0892-0362(01)00186-6. [DOI] [PubMed] [Google Scholar]
- Streissguth AP. Offspring effects of prenatal alcohol exposure from birth to 25 years: The Seattle Prospective Longitudinal Study. J Clin Psychol Med Settings. 2007;14:81–101. [Google Scholar]
- Swanson DJ, King MA, Walker DW, Heaton MB. Chronic prenatal ethanol exposure alters the normal ontogeny of choline acetyltransferase activity in the rat septohippocampal system. Alcohol Clin Exp Res. 1995;19:1252–1260. doi: 10.1111/j.1530-0277.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Kirino Y. Time-dependent reorganization of the brain components underlying memory retention in trace eyeblink conditioning. J Neurosci. 2003;23:9897–9905. doi: 10.1523/JNEUROSCI.23-30-09897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Garrison M, O’Neill TM. Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicol Teratol. 2004;26:35–45. doi: 10.1016/j.ntt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Thomas JD, La Fiette MH, Quinn VRE, Riley EP. Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicol Teratol. 2000;22:703–711. doi: 10.1016/s0892-0362(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Thompson BL, Rosen JB. Effects of TRH on acoustic startle, conditioned fear and active avoidance in rats. Neuropeptides. 2000;34:38–44. doi: 10.1054/npep.1999.0785. [DOI] [PubMed] [Google Scholar]
- Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicol Teratol. 2003;25:519–528. doi: 10.1016/s0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Uecker A, Nadel L. Spatial but not object memory impairments in children with fetal alcohol syndrome. Am J Ment Retard. 1998;103:12–18. doi: 10.1352/0895-8017(1998)103<0012:SBNOMI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Valles S, Felipo V, Montoliu C, Guerri C. Alcohol exposure during brain development reduces 3H-MK-801 binding and enhances metabotropic glutamate receptor-stimulated phosphoinositide hydrolysis in rat hippocampus. Life Sci. 1995;56:1373–1383. doi: 10.1016/0024-3205(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Wagner AF, Hunt PS. Impaired trace fear conditioning following neonatal ethanol: Reversal by choline. Behav Neurosci. 2006;120:482–487. doi: 10.1037/0735-7044.120.2.482. [DOI] [PubMed] [Google Scholar]
- Wanisch K, Tang J, Mederer A, Wotjak CT. Trace fear conditioning depends on NMDA receptor activation and protein synthesis within the dorsal hippocampus of mice. Behav Brain Res. 2005;157:63–69. doi: 10.1016/j.bbr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Weinberg J, Sliwowska JH, Lan N, Hellemans KGC. Prenatal alcohol exposure: Foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome. J Neuroendocrinol. 2008;20:470–488. doi: 10.1111/j.1365-2826.2008.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]