

Abstract
Cytokines such as interleukin-6 induce tyrosine and serine phosphorylation of Stat3 that results in activation of Stat3-responsive genes. We provide evidence that Stat3 is present in the mitochondria of cultured cells and primary tissues, including the liver and heart. In Stat3−/− cells, the activities of complexes I and II of the electron transport chain (ETC) were significantly decreased. We identified Stat3 mutants that selectively restored the protein's function as a transcription factor or its functions within the ETC. In mice that do not express Stat3 in the heart, there were also selective defects in the activities of complexes I and II of the ETC. These data indicate that Stat3 is required for optimal function of the ETC, which may allow it to orchestrate responses to cellular homeostasis.
Binding of cytokines to their cell surface receptors activates the Jak protein tyrosine kinases that phosphorylate conserved tyrosine residues on Stat proteins (1). Tyrosine-phosphorylated Stats translocate to the nucleus and bind to the promoters of early response genes. Several of the Stats are also phosphorylated on a conserved serine residue.
GRIM-19 was identified as a protein involved in interferon-β (IFNβ)– and retinoic acid–stimulated apoptosis of breast cancer cells (2). GRIM-19 inhibits Stat3 transcriptional activity, and these proteins directly interact (3, 4). GRIM-19 was also purified as a component of complex I of the electron transport chain (ETC) (5). GRIM-19−/− mice are embryonically lethal, and in GRIM19−/− embryonic stem cells, assembly of complex I (of which there are 46 known components) is defective. The absence of GRIM-19 also influences the assembly and function of other complexes of the ETC (6). These observations indicate that Stat3 and GRIM-19 might colocalize in the mitochondria.
To address this possibility, we used SDS–polyacrylamide gel electrophoresis (SDS-PAGE) to separate isolated mitochondria and cytoplasm from the livers and hearts of mice, and we used Western blotting to detect Stat3 (Fig. 1A) (7). Stat3 was present in mitochondria and cytoplasmic fractions. The amount of Stat3 in the mitochondria (mStat3) was about one-tenth that in the cytosol. The blots were also probed for GRIM-19, tubulin as a cytoplasmic marker, calreticulin as an endoplasmic reticulum marker, and porin as a mitochondria marker. In most experiments, we detected little or no tubulin or calreticulin in the mitochondrial fraction. Although GRIM-19 is reported to be located in the nucleus and cytosol, we only observed the protein in mitochondria (3, 4). Similar results were obtained from mitochondria isolated from the mouse brain, kidney, and primary splenocytes, as well as several other mouse and human cell lines. We have yet to find a tissue or cell line that does not contain Stat3 in the mitochondrial fraction.
Fig. 1.
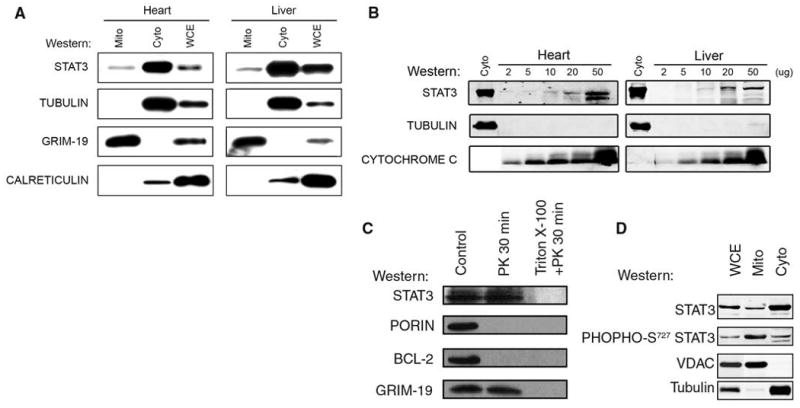
Stat3 in the mitochondria from mouse heart and liver. (A) Whole cell (WCE), cytoplasmic (Cyto), and mitochondrial (Mito) extracts were separated by SDS-PAGE. The blots were probed with antisera against Stat3, α-tubulin, calreticulin, and GRIM-19. (B) Increasing amounts of heart and liver mitochondria probed for Stat3, tubulin, and cytochrome c. (C) mStat3 is proteinase K–resistant. Mitochondria were incubated with (lanes 2 and 3) or without (lane 1) proteinase K (PK). To disrupt mitochondrial integrity, triton X-100 was added in the digestion buffer (lane 3). Samples were probed for Stat3, porin, Bcl-2, and GRIM-19. (D) mStat3 is serine phosphorylated. Purified mitochondria, as well as cytosolic and whole cell extracts were prepared from WT mice hearts. The immunoblots were probed for either total Stat3 or serine phosphorylated Stat3, as well as voltage-dependent anion channel (VDAC) and α-tubulin as controls for mitochondrial purity. A fluorescent conjugated secondary antibody was used to develop the blots allowing the relative amount of total and serine phosphorylated Stat3 to be measured. The ratio of total Stat3 to serine phosphorylated Stat3 in whole cell extracts was 2.5, in cytosolic extracts was 2.3, and in mitochondria extracts was 0.3.
To confirm that mStat3 did not represent contamination from the cytoplasm, we immunobloted proteins from increasing amounts of purified mitochondria for Stat3, tubulin, and cytochrome c (Fig. 1B). The ratio of Stat3 to tubulin in the cytoplasm was 0.9 in the heart and 1.0 in the liver. The ratio of Stat3 to tubulin in the heart mitochondria ranged from 13 to 19 and in the liver mitochondria ranged from 4 to 9. If the Stat3 detected in the mitochondria was due to cytosolic contamination, then the ratio of Stat3 to tubulin should have been ∼1.
To determine whether mStat3 was on the outer membrane, we incubated liver mitochondria with proteinase K in the presence or absence of triton X-100. Stat3 and GRIM-19 (an inner membrane protein) were resistant to proteinase K, whereas Bcl-2 and porin, both of which are localized on the outer membrane, were degraded (Fig. 1C). When proteinase K was added to mitochondria in the presence of triton X-100, all of the proteins were degraded. These results indicate that Stat3 does not adhere to the outer membrane of the mitochondria in a potentially nonspecific manner and is probably located in either the intermembrane space, the inner mitochondrial membrane, or the matrix.
We detected Stat3 in immunoprecipitates of mitochondrial extracts prepared from the liver with the use of a monoclonal antibody that captures all components of complex I of the ETC (Fig. 2) (8). We focused on complex I because GRIM-19 is a component of complex I, and Stat3 has been shown to interact with GRIM-19 (3, 4). As a control, identical samples were incubated with isotype-matched antibody under the same conditions. To ensure antibody specificity, the blots were also probed for known components of complex I. Immunoprecipitates of complex I contained Stat3, whereas no immunoreactive Stat3 or any other proteins were present in the immunoprecipitates using the immunoglobulin G (IgG)–matched control. As expected, NDUFA9 [NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 9, where NADH is the reduced form of nicotinamide adenine dinucleotide] and GRIM-19 (both components of complex I) were immunoprecipitated with the antibody. The complex I antibody did not immunoprecipitate complex III core protein 2, complex IV subunit I, or the 19-kD adenosine triphosphate (ATP) synthase, a component of complex V (5th, 6th, and 7th panels in Fig. 2). The complex II Fp subunit did immunoprecipitate with the complex I antibody (4th panel in Fig. 2). These results indicate the presence of Stat3 in complex I and possibly II of the ETC. Alternatively, there could be an interaction of complexes I and II of the ETC.
Fig. 2.
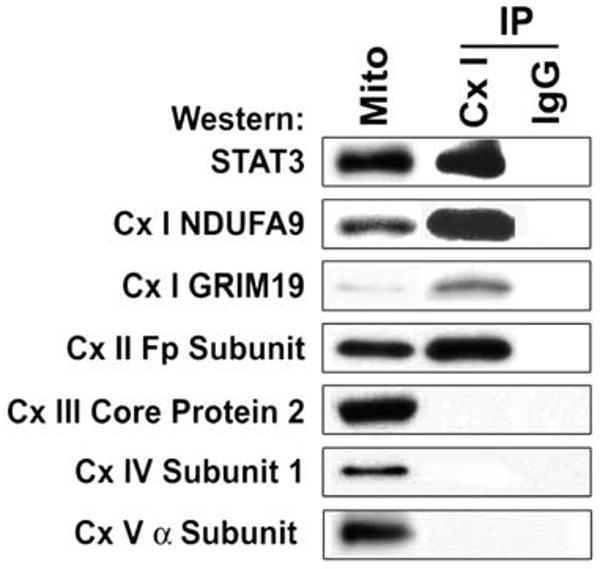
Stat3 in complex I immunoprecipitates (IP). Antibody to complex I (CxI) or a nonspecific isotype-matched IgG were incubated with liver mitochondrial extracts. Immunoprecipitates were resolved on SDS-PAGE and probed for Stat3, GRIM-19, NDUFA9, Cx II Fp subunit, Cx III core protein 2, Cx IV subunit I, and Cx V α subunit.
To examine whether mitochondrial Stat3 expression might modulate oxidative phosphorylation, we performed polarographic studies with the use of intact mitochondria isolated from Stat3+/+ and Stat3−/− pro-B cells. This assay evaluates the integrated function of the ETC coupled to ATP synthesis, membrane transport, dehydrogenase activities, and the structural integrity of the mitochondria. Glutamate was used to drive complex I–dependent electron transport. Succinate is the specific substrate that we used as the electron donor for complex II. We were not able to accumulate a sufficient amount of mitochondria to measure complex III activity. We measured complex IV activity using N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) with l-ascorbate.
Maximal state 3 oxidation rates with adenosine diphosphate (ADP) (2 mM) were reduced by 70% in Stat3−/− cells with glutamate as a substrate for complex I and by 50% in cells with succinate as a substrates for complex II (Table 1). Oxidation rates for TMPD/ascorbate (complex IV) were not substantially different, localizing defects to complexes I, II, and/or III of the ETC.
Table 1.
Maximal rates of state 3 respiration in mitochondria isolated from WT and Stat3−/− pro-B cells with glutamate, succinate, or TMPD/l-ascorbate as substrates. Results are expressed as nAtom O/min/mg of mitochondrial protein and are presented as mean ± SD. Significant differences between Stat3−/− and WT pro-B cells are shown in the table as the corresponding P value. N denotes the number of independent experiments.
| Electron transport chain complexes required | WT 2 mM ADP | Stat3−/− 2 mM ADP |
|---|---|---|
| Complexes I, III, IV | 52 ± 28 | 15 ± 9 |
| 20 mM glutamate | (N = 4) | (N = 4) |
| P = 0.049 | ||
| Complexes II, III, IV | 129 ± 21 | 64 ± 15 |
| 20 mM succinate | (N = 4) | (N = 4) |
| P = 0.005 | ||
| Complex IV | 521 ± 92 | 468 ± 40 |
| 1mM TMPD + 20 mM l-ascorbate | (N = 2) | (N = 2) |
Oxidase activities were measured in mitochondria permeabilized by freezing and thawing (9). The oxidase studies bypass potential defects in substrate carriers to exclude defects of substrate transport as causes of the decreased oxidation rates. NADH oxidase activity requires complexes I, III, and IV, and duroquinol oxidase (DHQ) requires complexes III and IV. Consistent with the respiration rates obtained with intact mitochondria, NADH oxidase activity was reduced by 65% in mitochondrial preparations from Stat3−/− pro-B cells as compared with that in wild-type (WT) pro-B cells (Fig. 3A). In contrast to that of NADH oxidase, the activities of DHQ (Fig. 3A) were similar in Stat3−/− and WT pro-B cells.
Fig. 3.

Depressed mitochondrial respiration in Stat3−/− pro-B cells. (A) Mitochondrial oxidase activity in WT and Stat3−/− cells: NADH oxidase (left) and DHQ oxidase (right). Oxygen consumption is expressed as nanogram atom of oxygen per minute per milligram of mitochondrial protein (nAtom O/min/mg) and is presented as mean ± SD. (B) ETC activities of complexes I, II, III, and IV. Activities are expressed as milliunits (nanomoles per minute) per milligram of mitochondrial protein and were normalized to citrate synthase activity. Results are presented as mean ± SD. Error bars indicate SD; asterisks indicate P < 0.05.
These results reveal a defect at complexes I and II of the ETC in mitochondria from Stat3−/− pro-B cells. To further localize the ETC defects, we measured enzyme activities of the electron transport chain complexes (ETC assays) in solubilized mitochondria. Consistent with the polarographic studies, the defects of complexes I were decreased by 40% in Stat3−/− pro-B cells as compared with WT pro-B cells (Fig. 3B). Complex II activity in Stat3−/− cells was also decreased by ∼85%. Complex III and IV activities for both WT and Stat3−/− pro-B cells were not affected (Fig. 3B).
To determine whether the deficiency in oxygen consumption in Stat3−/− cells was due to a decrease in mitochondrial content, we measured the activity of the mitochondrial enzyme citrate synthase in WT and Stat3−/− pro-B cells. This enzyme is an exclusive marker of the mitochondrial matrix, and its activity was similar in Stat3+/+ and Stat3−/− cells (fig. S1A). We also measured amounts of porin, cytochrome c, and several components of ETC complexes including NDUFA9, NDUFS3 [NADH dehydrogenase (ubiquinone) Fe-S protein 3], complex II subunit Fp (70 kD), complex II subunit I p (30 kD), and COX-I (cytochrome c oxidase-I) in WT and Stat3−/− cells by immunoblotting. All of these proteins were present in similar amounts in both cell types (fig. S1B).
The amount of mitochondrial DNA–encoded genes was similar between WT and Stat3−/− pro-B cells (fig. S1C), as were the concentrations of mitochondrial-encoded RNAs (fig. S1D). These results suggest that mitochondrial content is not affected in pro-B cells that lack Stat3.
To determine whether alterations in ETC were directly due to a loss of Stat3 expression, we reconstituted Stat3a into Stat3−/− B cells with a retrovirus expressing Stat3a and a downstream green fluorescent protein cDNA to allow identification of infected cells (10). Expression of Stat3a restored complex I and II activities (see Table 2, legend). Immunoblots of isolated mitochondria and cytosol from the reconstituted cells confirmed that Stat3a was expressed in both cellular fractions (fig. S2A).
Table 2.
Activities of complexes I and II of the respiratory chain in mouse Stat3−/− pro-B cells reconstituted with Stat3 mutants. Complex I and II activity in Stat3−/− pro-B cells and different mutants of Stat 3 expressed in terms of percentage values, considering the activity of complexes I and II being 100% in Stat3−/− pro-B cells reconstituted with WT Stat3a. (Stat3−/− cells reconstituted with Stat3a showed complex I activity of 111 ± 9 and complex II activity of 74 ± 29, as compared with that seen in WT pro-B cells.) Results are presented as mean ± SD. Significant differences with P values of <0.05 were observed between Stat3−/− +Stat3a and Stat3−/− cells reconstituted with Stat3 mutants.
| Cell type | Complex I | Complex II | |
|---|---|---|---|
| 1 | Stat3−/− + Stat3a | 100 (N = 7) | 100 (N = 3) |
| 2 | Stat3−/− + MSCV | 44 ± 26 (N = 7)* | 46 ± 26 (N = 3)* |
| 3 | Stat3−/− + MLS Stat3a | 82 ± 20 (N = 6) | 114 ± 42 (N = 3) |
| 4 | Stat3−/− + MLS Stat3a Y705F | 83 ± 34 (N = 4) | 124 ± 75 (N = 3) |
| 5 | Stat3−/− + MLS Stat3a Y705F/S727A | 28 ± 19 (N = 4)* | 39 ± 30 (N = 3)* |
| 6 | Stat3−/− + MLS Stat3a Y705F/S727D | 98 ± 20 (N = 3) | 115 ± 80 (N = 3) |
| 7 | Stat3−/− + MLS Stat3a E434A/E435A | 90 ± 6 (N = 3) | 126 ± 49 (N = 3) |
| 8 | Stat3−/− + Stat3CA | 61 ± 4 (N = 3)* | 16 ± 5 (N = 3)* |
P < 0.05
To determine whether Stat3, expressed only in mitochondria, restored respiration in Stat3−/− cells, we constructed a Stat3 that contains the mitochondrial targeting sequence of cytochrome c oxidase subunit VIII (MLS) fused to the N terminus of the protein (MLS Stat3). This sequence places the protein of interest in the inner mitochondrial membrane. Fractionation of MLS Stat3–expressing cells demonstrated that Stat3 was present only the mitochondria (fig. S2B). Cells expressing MLS Stat3 had about the same amount of Stat3 in the mitochondria as did WT cells, but they had less total Stat3 than WT cells. MLS Stat3 cells (Table 2, row 3) showed similar activities of complexes I and II of the ETC as Stat3+/+ or Stat3−/− cells that expressed Stat3a (Table 2, row 1).
To determine whether tyrosine and serine phosphorylation and DNA binding activity of Stat3 are important for regulation of the ETC, MLS Stat3 that contained mutations in its DNA binding domain (11), Tyr705 (Table 2, rows 4 and 7), or Ser727 and Tyr705 (Table 2, row 5) was expressed in Stat3−/− cells. Mutation in either Tyr705 or the DNA binding domain (Table 2, rows 4 and 7) restored activity of complexes I and II (Table 2). Because MLS Stat3 E434A/E435A (12) cannot form a dimer, it is possible that Stat3 exerts its actions in the mitochondria as a monomer (11). Consistent with this observation, Stat3−/− cells expressing a constitutively active Stat3 (Stat3a CA) that forms a dimer without being tyrosine phosphorylated (13) did not restore mitochondrial respiration (Table 2, row 8).
Expression of MLS Stat3 with serine 727 mutated to an alanine and tyrosine 705 mutated to a phenylalanine (row 5) also did not restore activity of complex I or II. To further characterize the role of Ser727, we mutated Tyr705 to a phenylalanine and Ser727 to either an alanine (Y705F/S727A) (row 5) or an aspartic acid (Y705F/S727D) (row 6). The former functions as a dominant negative and the latter as a mimetic of a constitutively serine-phosphorylated Stat3. MLS Stat3Y705F/S727D reconstituted complex I and II activities (Table 2, row 6), whereas MLS Y705F/S727A (Table 2, row 5) was ineffective, another indication that the actions of this protein in the mitochondria are distinct from its activity as a transcription factor. Interestingly, the relative concentration of serine-phosphorylated Stat3 was highly enriched in mouse mitochondria as compared with that present in the cytoplasm (Fig. 1D).
To confirm that MLS Stat3 and variants did not have transcriptional activity, we compared the expression of SOCS3, a Stat3-dependent gene, in cells incubated with or without IFNβ (14). Real-time polymerase chain reaction demonstrated that incubation of WT pro-B cells with IFNβ induced an eightfold increase in SOCS3 mRNA (fig. S3). However, Stat3−/− cells or those cells expressing MLS Stat3 or MLS Stat3 with a mutation in its DNA binding domain showed no increase in SOCS3 expression (fig. S3). Furthermore, cells that expressed constitutively active Stat3 in which respiration was not restored displayed robust IFNβ-stimulated expression of SOCS3 RNA. We obtained similar results when we examined IFNβ–induced expression of adenosine deaminase 1 RNA in pro-B cells (fig. S3). These findings are another independent support of the concept that MLS Stat3 controls the activity of the electron transport chain, and this effect of Stat3 is unrelated to its actions as a transcription factor.
To examine the role of Stat3 in mitochondrial respiration in vivo, we examined respiration in Stat3−/− mouse hearts. Mice that do not express Stat3 in cardiomyocytes develop cardiac inflammation with fibrosis, have dilated cardiomyopathy, and die prematurely of congestive heart failure (15). Female mice that do not express Stat3 in cardiomyocytes also develop postpartum cardiomyopathy, which is also seen in humans with reduced Stat3 expression in the myocardium (16).
Mitochondria from the hearts of Stat3flox/flox/cre (Stat3−/−) and Stat3flox/flox (wild type) mice were assayed for complex I and II activity. The mice used for these studies were 8 weeks old, at which time the hearts are normal as analyzed by histology and various physiological parameters (15). We used pyruvate and malate as complex I substrates (upper panels) and succinate as a complex II substrate (lower panels) to measure O2 consumption in WT and Stat3−/− heart mitochondria (Fig. 4). Rates of O2 consumption were decreased in Stat3−/− mitochondria with both substrates. Maximal rates of state 3 respiration (Vmax) stimulated by the addition of 2 mM ADP were blunted in Stat3−/− mitochondria. Pyruvate and malate Vmax in state 3 was decreased by ∼30% and succinate state 3 Vmax by ∼60% (table S1). ETC assays confirmed that Stat3−/− heart mitochondria had defects in complexes I and II but had normal activity of complex III (fig. S4).
Fig. 4.
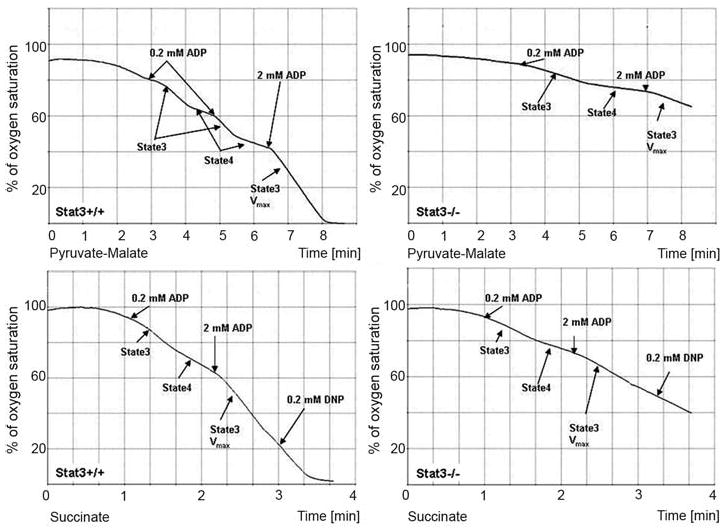
Decreased rates of O2 consumption in Stat3−/− heart mitochondria. Mitochondria isolated from littermates of WT (left) and Stat3−/− (right) hearts were incubated in an oxygen sensor chamber, and O2 consumption (y axis) as a function of incubation time (x axis) was recorded. In the upper panels, the mitochondria were incubated with pyruvate/malate (complex I substrates), and they were incubated with succinate (complex II substrate) in the lower panels. Different concentrations of ADP were added to the mitochondria to measure state 3 respiration (0.2 mM ADP) or state 3 Vmax rates of respiration (2 mM ADP). State 4 rates of respiration were calculated when 0.2mM ADP was depleted from the reaction.
The results presented here reveal a role of Stat3 as a modulator of mitochondrial respiration. The most likely mechanism by which Stat3 exerts its actions is not as a transcription factor that regulates nuclear gene expression, but rather through its localization in the mitochondria. The effects of Stat3 potentially represent a general mechanism by which this protein can influence cell metabolism. These studies are consistent with the concept of multidirectional communication between the mitochondria and the nucleus and changes in homeostasis detected at the plasma membrane. The precise mechanism by which Stat3 regulates complexes I and II remains to be determined.
Supplementary Material
Footnotes
References and Notes
- 1.Baker SJ, Rane SG, Reddy EP. Oncogene. 2007;26:6724. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- 2.Angell JE, Lindner DJ, Shapiro PS, Hofmann ER, Kalvakolanu DV. J Biol Chem. 2000;275:33416. doi: 10.1074/jbc.M003929200. [DOI] [PubMed] [Google Scholar]
- 3.Lufei C, et al. EMBO J. 2003;22:1325. doi: 10.1093/emboj/cdg135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, et al. Proc Natl Acad Sci USA. 2003;100:9342. [Google Scholar]
- 5.Fearnley IM, et al. J Biol Chem. 2001;276:38345. doi: 10.1074/jbc.C100444200. [DOI] [PubMed] [Google Scholar]
- 6.Huang G, et al. Mol Cell Biol. 2004;24:8447. doi: 10.1128/MCB.24.19.8447-8456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Materials and methods are available as supporting material on Science Online.
- 8.Murray J, et al. J Biol Chem. 2003;278:13619. doi: 10.1074/jbc.C300064200. [DOI] [PubMed] [Google Scholar]
- 9.Krahenbuhl S, Chang M, Brass EP, Hoppel CL. J Biol Chem. 1991;266:20998. [PubMed] [Google Scholar]
- 10.Gamero AM, et al. J Biol Chem. 2006;281:16238. doi: 10.1074/jbc.M509516200. [DOI] [PubMed] [Google Scholar]
- 11.Horvath CM, Wen Z, Darnell JE., Jr Genes Dev. 1995;9:984. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 12.Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr.
- 13.Bromberg JF, et al. Cell. 1999;98:295. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, et al. Mol Cell Biochem. 2006;288:179. doi: 10.1007/s11010-006-9137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacoby JJ, et al. Proc Natl Acad Sci USA. 2003;100:12929. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilfiker-Kleiner D, et al. Cell. 2007;128:589. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 17.This work was supported by NIH grants CA098924 (A.C.L.) and PO1AG15885 and the Office of Research and Development, Medical Research Service Department of Veterans Affairs (E.J.L.). We thank I. Scheffler for his many suggestions. DHQ was a kind gift from C. Hoppel. This manuscript is dedicated to Joseph Larner in honor of 60 years of scientific achievement and enthusiasm.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.