

Abstract
Formins are potent actin assembly factors. Diaphanous formins, including mDia1, mDia2, and mDia3 in mammals, are implicated in mitosis and cytokinesis but no chemical interactors have been reported. We developed an in vitro screen for inhibitors of actin assembly by mDia1, and identified an inhibitor of mDia1 and mDia2 that does not inhibit mDia3 at the concentrations tested. These results establish the druggability of mDia formins and introduce a first generation inhibitor.
The actin cytoskeleton is a vital component of at least 15 processes and cellular structures in mammalian cells. Individual actin-based structures are thought to be initiated by specific assembly factors, that accelerate actin filament nucleation and/or elongation (1). At least 18 mammalian actin assembly factors have been identified, including 15 isoforms of the formin protein family. A variety of evidence links formin proteins to several actin-based structures, including cytokinetic rings, filopodia, stress fibers, and phagocytic cups (1). However, a major challenge to the field has been to establish clearly the formin isoform or isoforms responsible for many of the above-mentioned structures. The availability of chemical reagents for inhibition of sub-sets of formins would allow for more detailed mechanistic studies in cells. Here we demonstrate that a class of actin assembly factors, the mammalian homologues of the Drosophila Diaphanous protein (mDia), can be inhibited by small molecules.
Mammals possess three mDia isoforms: mDia1, mDia2, and mDia3. The mDia proteins are strongly implicated in cell division, with mDia1 and/or mDia2 being necessary for cytokinesis (2, 3), and mDia3 enabling correct chromosome segregation during mitosis (4). Both mDia1 and mDia2 have been implicated in cell adhesion and cell migration as well (1).
Formins accelerate actin assembly in three ways: 1) they accelerate nucleation of new filaments; 2) they enhance elongation rate of these filaments, and 3) they inhibit capping of elongating filament “plus” ends by capping proteins (1). The Formin Homology 2 (FH2) domain mediates these effects. The FH2 domain is approximately 400 residues, and exists as a homodimer that binds to and moves with the elongating filament plus end, after having stabilized nucleation complexes. Structural and kinetic modeling studies suggest that FH2 domain dimers toggle between two or more conformations to regulate actin filament assembly (5, 6). This conformational switching relies on a flexible “linker” region in the FH2 domain (6). An attractive possibility is that small molecule inhibitors might trap transient intermediates between conformations, allowing for specific inhibition of formin isoforms.
To identify small-molecule inhibitors of actin assembly by the FH2 domain of mDia1, we developed a high-throughput screening assay utilizing actin labeled with the fluorophore pyrene, and recombinant mouse mDia1 FH2 domain (amino acids 748–1175). In this assay, actin polymerization is monitored by the 20-fold increase in the fluorescence of pyrene-actin monomers when incorporated into growing actin filaments. mDia1-FH2 (final concentration 6.7 nM) was aliquotted into wells of 384 well plates and individual compounds were transferred from dimethyl sulfoxide (DMSO)-solubilized stock plates by pin transfer to a final concentration of ~5 μM. Actin assembly was initiated by addition of monomeric actin (1 μM) and polymerization was monitored over ~ 10 minutes by pyrene fluorescence. Under these conditions, spontaneous actin polymerization occurs slowly, whereas mDia1-FH2 stimulates the kinetics of actin assembly by > 40-fold (see Figure 1A). The pyrene moiety has no effect on mDia1-mediated actin nucleation (7). 20,480 diverse, drug-like (largely Rule of 5-compliant) compounds were screened in duplicate and only compounds that met the hit criteria (see supplementary data) in both replicates were considered further.
Figure 1.

Pyrene-actin polymerization curves of 4 μM actin (5% pyrene labeled) with 2.5 nM mDia1-FH2 and the indicated μM concentrations of 1 (A) or 2 (B). (C) Dose-response curves for 1 and 2 on mDia1-FH2 activity, calculated from pyrene-actin curves. (D) Effects of compounds (50 μM) on polymerization of 4 μM actin (5% pyrene labeled). Compound 2 curve was normalized by a factor of 1.75, after subtracting fluorescence background. (E) Pyrene-actin assay with 2.5 nM mDia3-FH2 with or without 2 (50 μM). (F) Dose-response curves for 2.3 on pyrene-actin polymerization in the presence of 2.5 nM mDia2-FH2, 20 nM INF2-(FH1-FH2), 200 nM FRL1-(FH1-FH2), or 10 nM mDia3-FH2.
The resulting 134 active compounds (0.7% hit rate) were retested individually in single-cuvette pyrene-actin polymerization assays. 46 of these showed significant inhibitory activity at 10 μM and subsequent doseresponse experiments showed robust and dose-dependent inhibition at concentrations below 5 μM for 2 compounds (Table 1). Inhibition by 1 (2-(8-hydroxy-3, 6-disulfo-1- naphthylazo)-1,8-dihydroxy-naphthalene-3,6-disulfonic acid) is highly potent with an IC50 of 0.49 μM and a Hill coefficient of 2.4 (Figures 1A & 1C). In contrast, 2 (3-(2-amino-5-bromophenyl)-1H-quinoxalin-2-one) is somewhat less potent (2.1 μM IC50) but exhibits a strikingly sigmoidal inhibition curve (Hill coefficient of 7.6) (Figures 1B & 1C).
Table 1.
Potency (in μM) of 1, 2 and related compounds against mDia1-FH2 actin assembly.
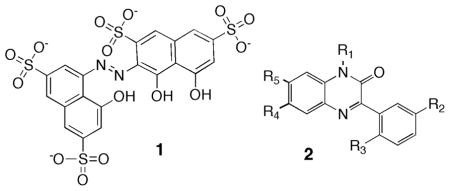 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | IC50 | |
| 1 | 0.5 | |||||
| 2 | H | Br | NH2 | H | H | 2.1 |
| 2.3 | H | NO2 | NH2 | H | H | 2.6 |
| 2.4 | H | NO2 | NH2 | CH3 | CH3 | 1.2 |
| 2.5 | H | Br | NHAc | H | H | > 50 |
| 2.6 | H | Br | NHMe | H | H | > 50 |
| 2.7 | CH3 | Br | NH2 | H | H | > 50 |
| 2.8 | H | -- | NH2 | H | H | > 50 |
| 2.9 | H | F | NH2 | H | H | > 50 |
| 2.10 | H | Cl | NH2 | H | H | > 50 |
To eliminate the possibility that the inhibitors targeted actin rather than mDia1-FH2, we assessed their ability to inhibit the slow, spontaneous polymerization of actin on its own. Neither 1 nor 2 shows any inhibitory effect toward polymerization of actin alone at concentrations up to 50 μM (Figure 1D). However, we observed that 2, but not 1, exhibited a dose-dependent quenching of pyrene-actin fluorescence. 2 has a broad absorption spectrum (max. at 380 nm) which overlaps pyrene’s emission spectrum (monitored at 410 nm here). Furthermore, addition of 2 to pre-polymerized pyrene-actin filaments causes a rapid drop in fluorescence (Supporting Information- Figure S1). This suggests fluorescence quenching as opposed to a shift in the monomer/filament equilibrium, which would be a slower process. The quenching effect is only detectable at concentrations of 2 above 12.5 μM (not shown), and is accounted for by correction factors in Figure 1D and subsequent experiments. Dose-response curves of 1 and 2 for a larger mDia1 construct, comprising the FH2 domain and the complete C terminus (FH2-C; amino acids 748–1255), show a similar inhibitory potency to those for mDia1 FH2 (not shown), indicating that the FH2 domain contains the entire binding site for each compound. We therefore conclude that 1 and 2 inhibit mDia-1-mediated actin assembly by targeting mDia1-FH2 and not actin.
1 is also known as Beryllon II and is widely used as a probe for a number of charged molecules (8). Therefore, due to the potential for non-selective effects of this compound, our subsequent studies focused on 2. 2 was repurchased from the supplier, and its inhibitory activity, chemical purity (> 95%), and chemical identity were confirmed by HPLC and 1H NMR (Supporting Information- Figure S2).
To test the reversibility of mDia-FH2 inhibition by 2, mDia1-FH2 was incubated with 50 μM 2 for 30 min and then the mixture was diluted 1000-fold (40 fold below the IC50 of 2) into the pyrene-actin assay. The diluted reactions showed full mDia1-mediated actin assembly activity (Supporting Information- Figure S3), indicating that compound 2 inhibits mDia1 reversibly.
To investigate the structure-activity relationship of 2, a variety of analogues of 2 were obtained commercially and their potency against mDia1-FH2-mediated actin polymerization was assessed (Table 1). Acetylation (2.5) or methylation (2.6) of the primary amine abolishes inhibition of mDia1, as does alkylation of the amide nitrogen (2.7). Removal of the Br group (2.8) or substitution with another halogen (2.9, 2.10) also eliminates inhibitory activity. However, substitution of the Br for a nitro group was well tolerated (2.3). In addition, alkylation of the quinoxalinone benzyl ring at two positions improved activity by ~ 2 fold, suggesting that substitutions at these positions may offer opportunities for potency enhancement in the future. Dose-response curves of 2.3 and 2.4 on actin assembly by mDia1-FH2 show that 2.3 inhibits with an IC50 of 2.6 μM and a Hill coefficient of 8.3, while 2.4 has an IC50 of 1.2 μM and a Hill coeffficient of 1.5.
The FH2 domain of mDia1 is closely related to two other paralogs, mDia2, which shares 56% sequence identity with the FH2 domain of mDia1, and mDia3 (60% identity). We therefore tested whether compound 2 and its analogues would inhibit the actin nucleation activity of the FH2 domains of these family members. Filament assembly by mDia2-FH2 is inhibited by 2 and the related active analogues 2.3, and 2.4 with potency and Hill coefficients comparable to their inhibition of mDia1-FH2 (Table 2). Remarkably no inhibitory activity of 2 (Figure 1E) or the related active analogues 2.3, and 2.4 (not shown) was observed toward actin assembly by mDia3-FH2 at any concentration tested. The lack of mDia3 inhibition supports a highly specific mechanism of action and demonstrates the feasibility of obtaining isoform selective inhibitors targeting this protein family.
Table 2.
Inhibitor selectivity (IC50/Hill coefficients in μM).
To test for direct binding of 2 to mDia1, we used equilibrium dialysis (Supporting Information, Figure S4). mDia1-FH2 effectively retained 2 in a dose-dependent manner, whereas mDia3-FH2 displayed no binding.
Finally, we tested whether 2 might exhibit inhibitory activity against more distantly related formin family members INF2 (9) and FRL1 (10), and the unrelated nucleation factor, Arp2/3 complex (11). Both 2 (not shown) and 2.3 inhibit actin assembly by two other formins we tested, INF2 and FRL1, but with lower potency than against mDia1 (Figure 1F). Interestingly, for INF2, even at saturating concentrations of 2 or 2.3, inhibition is partial, with inhibition never reaching greater than 75% of INF2 activity. For FRL1, inhibition is incomplete even at 50 μM 2 or 2.3. None of the compounds displayed any ability to inhibit actin nucleation by Arp2/3 complex (Supplementary Information, Figure S5).
The results presented here establish that members of the mDia family can be inhibited by small-molecules. The sigmoidal nature of inhibition by 2 and 2.3 is intriguing, and may speak to the mechanism of FH2-mediated actin nucleation. Since FH2 domains function as anti-parallel dimers (12), the current structural and kinetic models predict that two sets of equivalent actin binding sites exist in the FH2 dimer, and stabilize the association of two actin monomers in a productive state for filament elongation (5, 6). After nucleation, the FH2 dimer stays attached to the elongating actin filament end, processively walking with the filament end because one of the two binding sites to actin remains bound at all times (6). In current models, these binding sites fluctuate independently between high affinity and low affinity for actin, allowing the binding and release necessary for processive movement. We postulate that compound 2 binds to and stabilizes the low affinity state for one binding site, and raises the affinity of the other binding site for a second molecule of 2. More detailed experiments will probe the reason for the sharply sigmoidal inhibition curves for 2 and 2.3.
2 and its analogues may also be used for mDia inhibition in the cellular context. The abilities of these compounds to inhibit both mDia1 and mDia2 with equal potency provide the opportunity to inhibit these formins simultaneously in cells, thereby avoiding redundancy problems. At present, a low solubility in cell culture medium has made cellular studies problematic for these compounds. Future work will focus on improving the aqueous solubility of this lead inhibitor class.
Supplementary Material
Acknowledgments
Support from NIH grants NS059359, GM083025 (JRP) and GM069818 (HNH) is gratefully acknowledged.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Experimental details and supplementary figures are available free of charge at http://pubs.acs.org.
References
- 1.Chhabra ES, Higgs HN. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- 2.Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS. Mol Cell. 2000;5:13–25. doi: 10.1016/s1097-2765(00)80399-8. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe S, Ando Y, Yasuda S, Hosoya H, Watanabe N, Ishizaki T, Narumiya S. Mol Biol Cell. 2008;19:2328–38. doi: 10.1091/mbc.E07-10-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yasuda S, Oceguera-Yanez F, Kato T, Okamoto M, Yonemura S, Terada Y, Ishizaki T, Narumiya S. Nature. 2004;428:767–771. doi: 10.1038/nature02452. [DOI] [PubMed] [Google Scholar]
- 5.Otomo T, Otomo C, Tomchick DR, Machius M, Rosen MK. Nature. 2005;13:511–522. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 6.Paul AS, Pollard TD. Cell Motil Cytoskeleton. 2009;66:606–617. doi: 10.1002/cm.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Higgs HN. Curr Biol. 2003;13:1335–1340. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- 8.Dong L, Jia R, Li Q, Chen X, Hu Z. Analyst. 2001;126:707–711. doi: 10.1039/b100420o. [DOI] [PubMed] [Google Scholar]
- 9.Chhabra ES, Higgs HN. J Biol Chem. 2006;281:26754–26767. doi: 10.1074/jbc.M604666200. [DOI] [PubMed] [Google Scholar]
- 10.Harris ES, Rouiller I, Hanein D, Higgs HN. J Biol Chem. 2006;281:14383–14392. doi: 10.1074/jbc.M510923200. [DOI] [PubMed] [Google Scholar]
- 11.Higgs HN, Pollard TD. Annu Rev Biochem. 2001;70:649–676. doi: 10.1146/annurev.biochem.70.1.649. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Moseley J, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ. Cell. 2004;116:711–723. doi: 10.1016/s0092-8674(04)00210-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.