

Abstract
In the search for xenoestrogens within food additives, we have analyzed the Joint FAO-WHO expert committee database, containing 1500 compounds, using an integrated in silico and in vitro approach. This analysis identified 31 potential estrogen receptor α ligands that were reduced to 13 upon applying a stringent filter based on ligand volume and binding mode. Among the 13 potential xenoestrogens, four were already known to exhibit an estrogenic activity, and the other nine were assayed in vitro, determining the binding affinity to the receptor and biological effects. Propyl gallate was found to act as an antagonist, and 4-hexylresorcinol was found to act as a potent transactivator; both ligands were active at nanomolar concentrations, as predicted by the in silico analysis. Some caution should be issued for the use of propyl gallate and 4-hexylresorcinol as food additives.
Introduction
Estrogen receptors (ERs)1 are ligand-activated transcription factors belonging to the super family of nuclear receptors. ERs mediate a broad spectrum of physiological effects in different organs and tissues and are involved in a range of diseases, such as breast and endometrial cancer, osteoporosis, and prostate hypertrophy (1). Recently, estrogens and their receptors have also been implicated in cardiovascular and central nervous system disorders (2). Two receptor subtypes have been identified to date, ERα and ERβ. In some organs and tissues, they are expressed at similar levels, while in others, one subtype is predominant (3). Ligand binding promotes the dimerization of the receptor and stabilized ER in the cell nucleus where ERs interact with specific estrogen response cis DNA elements (ERE), triggering the transcription of specific genes. For its activity on transcription, ERs requires the interaction with coactivators or corepressors, allowing a fine modulation of the final response in different tissues (4, 5). In addition to binding to ERE, ERs can affect gene expression by protein—protein interaction with other transcription factors or with molecules involved in the signaling of membrane receptors. A variety of synthetic and natural chemicals are known to bind to ER, exerting a certain degree of estrogenic activity (6-8). These environmental estrogens, also called xenoestrogens, belong to the category of the endocrine disruptors, defined by the European Commission as “an exogenous substance or mixture that alters functions of the endocrine system and consequently causes adverse health effects in an intact organism or its progeny” (http://ec.europa.eu/environment). Because of the increasing concern regarding the possible adverse effects of these compounds, in 1996, the U.S. Congress passed the Food Quality Protection Act (6), while in 1999, the European Commission adopted the Community Strategy for Endocrine Disrupters. Both documents highlighted the relevance of developing and implementing screening strategies aimed at the rapid identification of xenoestrogens, in particular those of harm for human health (6). Given the large number of compounds that bind ERs, there is considerable interest in developing computational methods for the prediction of the affinity of compounds for the ER. Previous computational approaches were based mainly on regression techniques, such as quantitative structure—activity relationship (QSAR) and comparative molecular field analysis (CoMFA) (9, 10), and on molecular dynamics (MD) (11, 12).
An intrinsic complexity in the identification by computational methods of ligands to ERα is due to the flexibility of the binding site. Structural studies on the ligand binding domain (LBD) of ERα indicate that agonists, selective estrogen receptor modulators (SERMs), such as tamoxifen and raloxifene, and full antagonists bind at the same site but induce different conformations of the carboxy-terminal helix-12 (H12). Agonists stabilize a receptor “closed” conformation with H12 packed against helices 3, 5, 6, and 11, thus sealing the binding site (Figure 1a), whereas SERMs and antagonists stabilize an “open” conformation, in which H12 occupies a portion of ERα coactivator binding groove (Figure 1b) (13, 14). In addition to the flexibility of H12, ERα shows a number of active site residues whose side chains display different orientations in various ERα complexes (Figure 2) (15-17).
Figure 1.
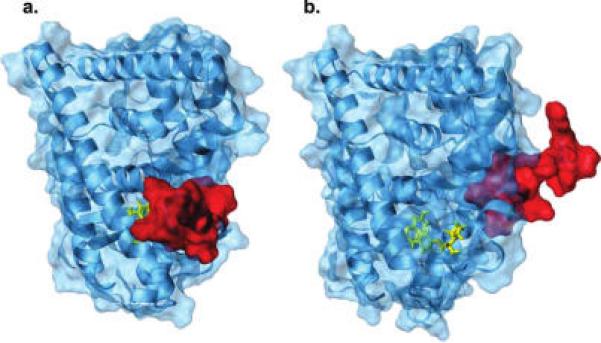
Ribbon and surface representation of ERα in complex with the agonist diethylstilbestrol (a) (PDB code: 3ERD), closed conformation, and with a dihydrobenzoxathiin SERM (b) (PDB code: 1XPC), open conformation. H12 (red) exhibits different orientations in the two complexes.
Figure 2.
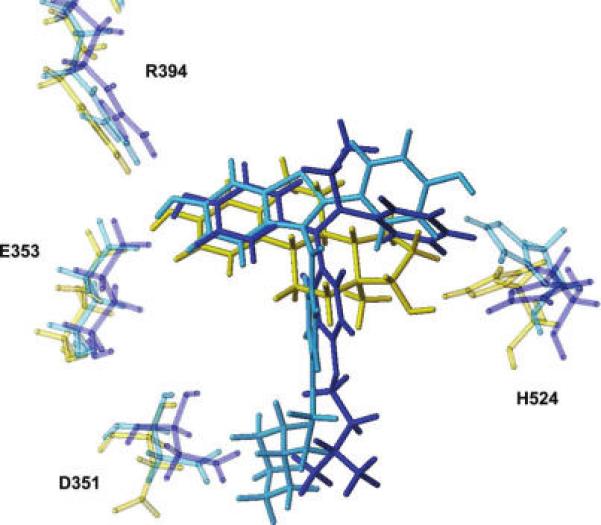
Overlay of the ERα binding site complexed with 17β-estradiol (PDB code: 1ERE, yellow), 4-hydroxytamoxifen (PDB code: 3ERT, blue), and raloxifene (PDB code: 1ERR, light-blue). Only the ligands and the key interacting residues are represented in capped sticks. Glu353 and Arg394, which interact with the estradiol A ring, show a very similar behavior in the different complexes. On the contrary, His524, which interacts with the estradiol D ring, displays different orientations in different complexes, reflecting the higher degree of freedom of ligand molecules in this portion of the binding pocket. The side chain of Asp351 shows two distinct orientations, depending on the presence (SERMs) or absence (agonist) of a bulky side chain able to interact with this residue.
As changes in the receptor conformation might have a dramatic impact in docking results (18-20), protein flexibility should be taken into account in searching for ERα ligands by computational approaches. The methods so far proposed may (21-24, 25) be classified in two main groups: those exploring the receptor conformations during the docking procedure (induced-fit approach) (17, 26-28) and those that dock ligands against multiple rigid protein conformations, obtained from X-ray crystallography (29, 30), NMR spectroscopy (31-33), or generated by computational routines (16, 34-36), mainly MD. Attempts to combine the two approaches have also been reported (37). In the present work, we applied an in silico screening for the identification of xenoestrogens among a library of commonly used food additives. This was achieved by combining the docking program GOLD (38) and the scoring function HINT (39, 40), a method that has been proved to be very powerful in the evaluation of the binding free energy between proteins and ligands, proteins and water molecules, and proteins and DNA (41-47). The H12 flexibility, which cannot be reproduced with the current docking protocols, was taken into account by carrying out the screening procedure on both the open and the closed receptor conformation. The computationally identified xenoestrogens were then assayed in vitro.
Experimental Procedures
Molecular Modeling Studies
The program Sybyl version 7.0 (Tripos, Inc., St. Louis, MO; www.tripos.com), used for this work, was installed on a FUEL Silicon Graphics workstation running o.s. IRIX 6.5. The program HINT (39, 40) 3.11 β test version (eduSoft, LC, Ashland, VA; www.edusoft-lc.com) was used as an add-on module within Sybyl. The program GOLD (38) version 3.1 (CCDC, Cambridge, United Kingdom; www.ccdc.cam.ac.uk) was installed on a dual Pentium processor, running operative system Linux Red Hat Enterprise 3.0.
Protein and Ligand Structure Preparation
The three-dimensional coordinates of protein—ligand complexes were retrieved from PDB (48) (www.rcsb.org) and imported into the molecular modeling program Sybyl. All structures were checked for chemical consistency of atom and bond type assignment. Amino-terminal and carboxyl-terminal groups were set to be protonated and deprotonated, respectively. Hydrogen atoms, not present in the PDB files, were added using Sybyl Biopolymer and Build/Edit menu tools. To avoid steric clashes, added hydrogen atoms were energy minimized using the Powell algorithm, with a convergence gradient of 0.5 kcal (mol Å)−1 for 1500 cycles. This procedure affected only hydrogen atoms. In the absence of available three-dimensional coordinates, ligands were built using the Sybyl Build/Edit menu tools and then energy minimized with a convergence gradient of 0.05 kcal (mol Å)−1 for 100 cycles.
The experimental value of binding affinity for the ERα receptor ligands contained in our data set was determined as IC50. In our analysis, these values were treated as proportional to Ki (49). Our data set did not contain compounds for which affinity was expressed as EC50.
Selection of the Database
The two major freely available food additive databases are the EAFUS (Everything Added to Food in the United States) database maintained by the U.S. Food and Drug Administration and the Combined Compendium of Food Additive Specifications of the Joint FAO/WHO Expert Committee on Food Additives (JECFA) (www.fao.org/ag/agn/jecfa-additives). More than 3000 substances are listed in EAFUS, but the chemical structures of various compounds are not comprised within the database, making it not usable for a screening procedure. The JECFA compendium includes about 1500 additives and is divided in two sections: One contains food additives used for purposes other than as flavoring agents, and the other contains flavoring agents. Because only the former includes the chemical structures of all of the listed compounds, we focused on this database containing 495 food additives. Because the chemical structures are not provided using 2D or 3D file formats, they were manually built.
Gold
The protein target and the ligands were prepared for docking using Sybyl (see above). Water molecules were removed from the PDB structure of the target protein. The input files were generated as.mol2. A radius of 20 Å, from the center of the active site, was used to direct site location. For each of the genetic algorithm run, a maximum number of 100000 operations were performed on a population of 100 individuals with a selection pressure of 1.1. Operator weights for crossover, mutation, and migration were set to 95, 95, and 10, respectively. The number of islands was set to 5, and the niche size was set to 2. Fifty genetic algorithm runs were carried out in each docking experiment. The default GOLDScore fitness function (38) was utilized for performing the energetic evaluations. The distance for hydrogen bonding was set to 2.5 Å, and the cutoff value for van der Waals calculation was set to 4.0 Å.
Water Molecules in ERα Complexes
A water molecule was present within the cavity of several ER complexes, predominantly interacting with Arg394 and Glu353. The energetic contribution of the water molecule to the binding free energy was analyzed for 24 ligand—ERα complexes. We found that the water—ligand HINT score (HS) ranged between 7 and 85 units, which was a negligible value with respect to the total HS for ligand—ERα complexes found to be in the order of 1500−3000. In agreement with this analysis, by applying the Rank method implemented in HINT software (41), the water molecule was predicted as “high probably conserved”, indicating that it cannot form other strong hydrogen bonds with ligands (unpublished data). Thus, in our in silico analysis, this water molecule was neglected.
Hydropathic Analysis
The software HINT (Hydrophatic Interactions) was used as a postdocking processor tool (45, 50). All of the 50 solutions proposed by GOLD for each ligand were rescored with HINT (the higher the HS the lower the predicted negative ΔG°) to predict the best binding mode. HINT first calculated LogPo/w for each component (protein and ligand) of the complexes. A partial LogPo/w value ai and a solvent-accessible surface area Si were assigned to each interacting atom. For the protein, the partition methods were dictionary, where HINT used a lookup table of parameters based on residue type and solvent condition. The “neutral” option was chosen as the solvent condition for protein partitioning (lysine and arginine side chains were protonated, while glutamic acid and aspartic acid side chains were deprotonated). A new HINT option that corrected the Si terms for backbone amide hydrogens by adding a 20 Å2 (51) was used in this study. This correction improved the relative energetics of inter- and intramolecular hydrogen bonds involving backbone amides, which were deemphasized in previous versions of HINT. For the ligands, the partitions were performed using the calculate method, an adaptation of the CLOG-P method of Leo (52). For both protein and ligand, a new “semiessential hydrogens” partition mode that treats polar hydrogens and hydrogens bonded to unsaturated carbons and carbons α to heteroatoms explicitly was used. In addition, hydrogens bonded to unsaturated carbons were, along with polar hydrogens, allowed to act as hydrogen bond donors. This was in accordance with several recent observations suggesting that some C—H···O hydrogen bonds were possible (53, 54).
After LogPo/w calculations, HINT provided a quantitative evaluation of the association process, as a sum of all single atom—atom interactions, using the following equation:
where bij is the interaction score between atom i and j, a is the hydrophobic atomic constant, S is the solvent accessibile surface area, Tij is a logic function assuming +1 or −1 values, depending on the nature of interacting atoms, and Rij and rij are functions of the distance between atoms i and j (40).
Postdocking Local Optimization
Genetic algorithms are not always suited for local optimization (55). Therefore, to allow a more accurate evaluation of the binding free energy, a local optimization, based on the HS, of the ligand rotatable bonds was performed after docking. This optimization generally affected only hydroxyl groups on the ligands.
Experimental Measurements
Unless otherwise specified, chemicals were purchased from Merck (Germany), culture media and additives were from Invitrogen Corp. (Scotland, United Kingdom), steroids were from Sigma Chemical Co. (MO), and food additives (curcumin, capsaicin, propyl gallate, octyl gallate, delphinidine, peonidine, malvidine, erythrosine B, and 4-hexylresorcinol) were from Sigma-Aldrich (Italy).
Cell Culture and Transactivation Studies
All studies were carried out using the B17 clone of MCF-7 obtained in our laboratory by stable transfection of a plasmid containing the luciferase gene under the control of an estrogen responsive promoter (56). Cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (Euroclone, United Kingdom), 50 U/mL penicillin G, 50 μg/mL streptomycin sulfate, 2 g/L sodium carbonate, and 0.11 g/L sodium pyruvate at 37 °C at 99% humidity and 5% CO2. Cells were split twice a week by seeding 2 × 106 cells in 100 mm diameter Petri (Corning, MA) dishes.
For transactivation studies, 105 cells/well were seeded in 24 well plate in phenol red-free RPMI 1640 medium (Sigma-Aldrich, MO) supplemented with 10% dextran-coated charcoal-stripped FBS, 1% essential amino acid, 1% vitamin mixture, 50 U/mL penicillin G, 50 μg/mL streptomycin sulfate, 2 g/L sodium carbonate, and 0.11 g/L sodium pyruvate and kept at 37 °C in a humidified incubator for 24 h. The culture medium was replaced with RPMI 1640 with 1% stripped FBS, and cells were incubated for a minimum of 4 h before adding 17β-estradiol or additives (at concentrations between 1 pM and 10 μM). After 24 h, cells were rinsed once with PBS before preparing the protein extract for the determination of luciferase content as previously described (56). Each experiment was carried out in triplicate. As a control, all compounds were run in parallel with 17β-estradiol. The average EC50 measured for 17β-estradiol was 0.02 nM, in good agreement with literature values.
Binding Studies
For binding analysis, 105 MCF-7 cells/well were seeded in 24 well plate in phenol red-free RPMI 1640 medium supplemented with 10% charcoal-stripped FBS and incubated at 37 °C in a humidified incubator for 24 h. The medium was replaced with fresh medium and 0.5 nM [2,4,6,7 3H] estradiol (Amersham, NJ) and increasing concentrations of cold competitors (additives or 17β-estradiol at concentrations of 1 nM to 0.25 μM). Nonspecific binding was assessed in the presence of 1 μM 17β-estradiol. Cells were incubated at 37 °C in a humidified incubator for 2 h to reach equilibrium of the binding reaction. Cells were then rapidly rinsed with cold PBS three times to eliminate unbound ligand, to separate bound from free radioligand, and then, the radiolabeled 17β-estradiol was extracted by treating the cells with 0.5 M NaOH for 30 min. Radioactivity in 450 μL of cellular extract was quantified by addition 4 mL of scintillation fluid (high flash point LSC-cocktail, PerkinElmer, MA) in the Liquid Scintillation Analyzer (Tri-Carb 1600 TR, Packard, MA). Data were analyzed by “plot sigmoidal one site competition curve” using a dedicated software (Prism5—GraphPad Software Inc., CA). Each experiment was carried out in triplicate. The binding affinity of 17β-estradiol was Ki = 0.03 nM. This value was in agreement with the values found in the literature. Analysis of binding data was performed by a nonlinear least-squares fitting using PRISM5 software (GraphPad Software Inc.) implemented with LIGAND program equations, to provide the basic molecular and cellular parameters for each ligand studied. Evaluation of the statistical significance of the parameter difference was based on the F test for the extra sum of squares principle (57).
Results
In Silico Screening Procedure. Prediction of Ligand Binding Modes to ERα
Prior to performing the screening on food additives to identify potential xenoestrogens, the computational procedure was validated by applying it to ligands for which ERα—ligand complexes are crystallographically determined. Thirty nonredundant X-ray structures of ERα—ligand complexes are deposited in the Protein Data Bank (PDB). For each of these complexes, the ligand was extracted and then redocked into the ERα binding site, using the GOLD docking package (38) to generate a set of 50 plausible ligand poses. The HINT program (39, 40) was used as a postdocking processor tool to select the best ligand conformation (see the Experimental Procedures and ref 45 for a detailed description of the procedure). The accuracy of this computational approach in reproducing the crystallographically detected conformation was estimated on the basis of the root-mean-square-deviation (rmsd) between the ligand coordinates in the HINT top docking pose and in the crystal structure. As shown in Table 1, the predicted binding modes reproduced very well the crystallographically determined ligand conformations. The best docking poses selected by HINT for 29 out of 30 compounds show rmsd values lower than 1.50 Å from the crystallographic binding mode, with a mean rmsd of 0.80 Å. For a single complex (PDB code: 2IOK), a rmsd of 2.55 Å was obtained. However, even in this case, the key interactions undertaken by the compound with ER were correctly predicted.
Table 1.
Results of Docking Predictions on ERα–Ligand Complexes
| PDB code | res. (Å) | ref | ligand | rmsd (Å) |
|---|---|---|---|---|
| 1ERE | 3.10 | (13) | 17β-estradiol | 0.51 |
| 1X7R | 2.00 | (78) | genistein | 0.31 |
| 3ERD | 2.03 | (14) | diethylstilbestrol | 1.10 |
| 3ERT | 1.90 | (14) | 4-hydroxytamoxifen | 1.42 |
| 1ERR | 2.60 | (13) | raloxifene | 0.49 |
| 1SJ0 | 1.90 | (79) | dihydrobenzoxathiin derivative | 0.69 |
| 1XP1 | 1.80 | (80) | dihydrobenzoxathiin derivative | 0.19 |
| 1XP6 | 1.70 | (80) | dihydrobenzoxathiin derivative | 0.18 |
| 1XP9 | 1.80 | (80) | dihydrobenzoxathiin derivative | 0.23 |
| 1XPC | 1.60 | (80) | dihydrobenzoxathiin derivative | 0.45 |
| 1YIN | 2.20 | (81) | chromane derivative | 0.62 |
| 1YIM | 1.90 | (81) | chromane derivative | 0.18 |
| 1X7E | 2.80 | (82) | WAY-244 | 1.01 |
| 1UOM | 2.28 | (83) | tetrahydroisoquinoline derivative | 1.18 |
| 1XQC | 2.05 | (83) | tetrahydroisoquinoline derivative | 1.23 |
| 1L2I | 1.95 | (84) | tetrahydrochrysene derivative | 0.63 |
| 2B1V | 1.80 | (85) | OBCP-1M | 1.34 |
| 2FAI | 2.10 | (85) | OBCP-2M | 1.23 |
| 1ZKY | 2.25 | (85) | OBCP-3M | 1.54 |
| 2B1Z | 1.78 | a | 17-methyl-17α-dihydroequilenin | 0.33 |
| 2G5O | 2.30 | a | 2-(but-1-enyl)-17β-estradiol | 0.55 |
| 2I0J | 2.90 | (86) | hexahydrocyclopenta[c]chromene derivative | 0.58 |
| 1R5K | 2.70 | (87) | GW5638 | 0.99 |
| 2G44 | 2.65 | a | OBCP-1M-G | 1.38 |
| 2Q70 | 1.95 | (88) | hexahydrocyclopenta[c]chromen-8-ole derivative | 0.31 |
| 2QE4 | 2.40 | (89) | benzopyran derivative | 0.38 |
| 2P15 | 1.94 | (90) | orthotrifluoro-methyl-phenyl-vinyl estradiol | 0.47 |
| 2AYR | 1.90 | (91) | naphthalen-2-ole derivative | 0.92 |
| 2IOG | 1.60 | (92) | 2-aryl indole derivative | 1.06 |
| 2IOK | 2.40 | (92) | 2-aryl indole derivative | 2.55 |
To be published.
Correlation between HS and Ligand Binding Affinity to ERα
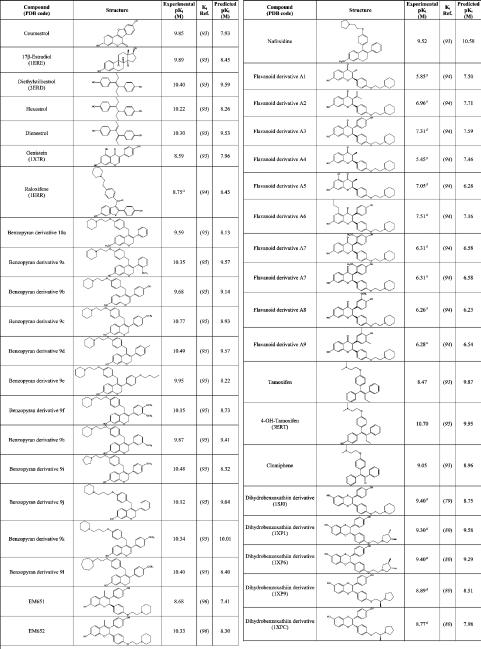
The next step of the procedure was to analyze a wider and more heterogeneous set of ligands with known affinity for ERα, with the aim of verifying the reliability of the procedure in the prediction of the ligand binding free energy, a key step in virtual screening investigations. The set included 15 of the crystallographic ligands, for which affinity values were available in literature, and another 42 compounds including drugs, natural hormone metabolites, phytoestrogens, and xenoestrogens for which the affinities to the ERα were known from literature. The whole data set comprising 57 ERα ligands is reported in Table 2. Because the backbone flexibility of ERα cannot be properly reproduced by the current docking programs, we carried out the docking analysis using two receptor structures, representative of the receptor “closed/agonist” and of the receptor “open/antagonist” conformations, respectively (see the Discussion). The two crystallographic structures that were selected among those available in PDB were the ERα-diethylstilbestrol complex (PDB code: 3ERD), representative of the “closed” conformation, and the complex between ERα and a dihydrobenzoxathiin derivative (PDB code: 1XPC), representative for the “open” conformation. This selection was dictated by the high quality of these crystallographic structures and by the low B-factors assigned to the binding site residues, mainly His524, in comparison with other structures of ERα complexes. Agonists were docked into the ERα “closed” binding site conformation, while antagonists/SERMs were docked in the open binding site conformation. The ligand pose showing the highest HS was considered as the most fitting candidate. The resulting correlation between the HINT scoring function and the experimentally determined binding affinities is shown in Figure 3. Data points were fitted to a linear regression (42, 44, 45):
| (1) |
with a r2 of 0.55 and a standard error of ± 1.18 pKi units, corresponding to 1.6 kcal mol−1. This calibration of the HS, specifically obtained for the ERα system (45), provides the basis for the affinity prediction during the following screening of food additives.
Table 2.
Structural and Experimental Affinity Data for the 57 ERα–Ligand Complexes
 |
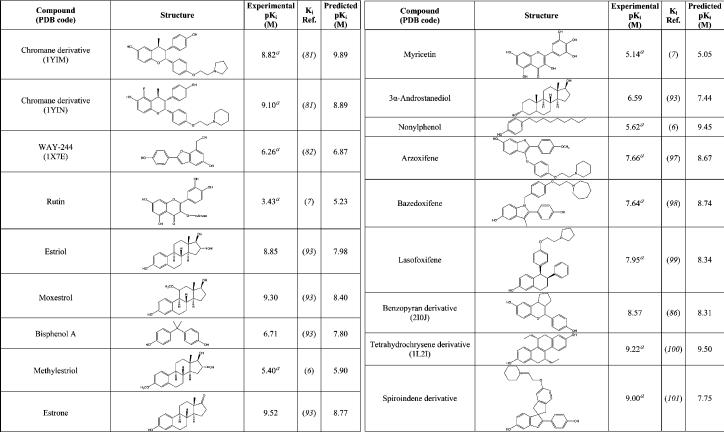 |
apIC50.
Figure 3.
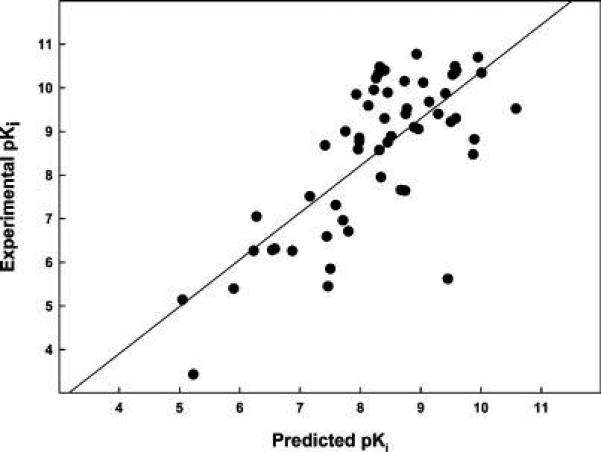
Correlation between the experimental pKi and the HINT-calculated pKi for the 57 ERα—ligand complexes.
Screening of Food Additives
Among the 495 food additives found in the JECFA database (see the Experimental Procedures), inorganic compounds, polymers, enzymes, polysaccharides, natural amino acids, sugars, fatty acids, nucleotides, vitamins, and redundant compounds (some entries report different salts of the same compound) were excluded. The remaining set includes a total of 112 additives that were examined using our docking/scoring procedure. Each additive was docked in both the closed/agonist and the open/antagonist conformation of ERα. The score of the best pose selected by HINT for each ligand was used to predict the binding affinity. Thirty-one potential ligands with a predicted pKi greater than 4.00 (Ki < 100 μM) were retained for the postdocking analysis. This analysis was based on (i) the evaluation of the ligand volume buried within the protein binding site (58) and (ii) the visual inspection of the docked complexes. Ligand volumes buried within the protein binding site were calculated using the software GRASP (59). Ligands with a buried volume lower than 150 Å3 were excluded from further analyses, as explained in the Discussion. The visual inspection of the retrieved docked candidates was carried out to evaluate (i) the chemical-geometric compatibility of conformation of the ligand in the binding site, (ii) the absence of charged groups adjacent to hydrophobic groups, and (iii) the presence of at least one hydrogen bond between protein and ligand (60, 61). By applying these criteria, 13 out of 31 potential ERα ligands were retained (Table 3). Among these 13 potential ligands, nine compounds were selected for experimental testing, since for four compounds data were already present in the literature assessing their binding capability to ERα. In particular, nordihydroguaiaretic acid, propyl p-hydroxybenzoate, and butyl p-hydroxybenzoate are known to bind to ER (6), thus indicating that the in silico screening procedure correctly identified them as xenoestrogens.
Table 3.
List of the 13 Food Additives Identified as Potential ERα Ligands by Virtual Screening
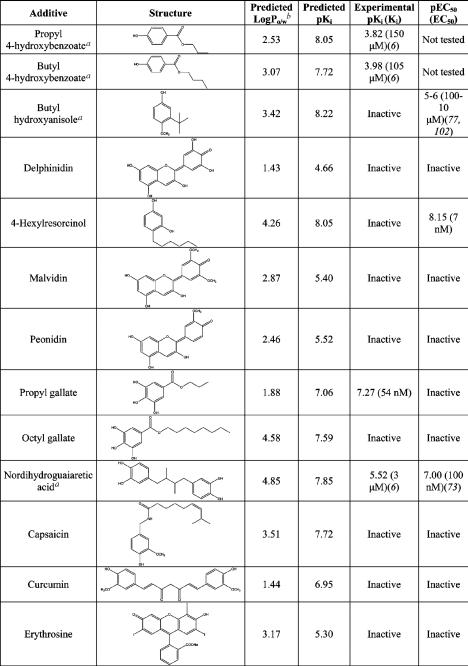 |
aAdditives for which experimental data were already available in the literature.
bLogPo/w calculated by HINT.
In Vitro Determination of Ligand Binding Affinity to ERα and Transactivation Potency
The nine compounds that were identified by in silico screening as potential ERα ligands were evaluated experimentally to determine their binding affinity to the receptor and their ability to elicit a ERα-dependent biological activity, that is, the transcription of genes that are under the control of the ERα response element, requiring a fully competent transcriptosome. The nine compounds exhibit LogP values between 1.43 and 4.85 (Table 3), thus ensuring easy diffusion through the cell membrane to reach the target receptor. The representative binding titration of propyl gallate to the ERα, carried out using the competition with radioactive-labeled estradiol (Figure 4a), can be fitted with a binding constant of 54 nM, in excellent agreement with the predicted value (Table 3). The corresponding transactivation potency assay (Figure 4b) indicates that propyl gallate is inactive, suggesting that it may act as a pure antagonist. Indeed, in the transactivation assay, propyl gallate blocked 17β-estradiol activity at a concentration compatible with its affinity: Figure 4f shows that propyl gallate at 10 and 100 nM was able to antagonize a 10-fold higher concentration of 17β-estradiol activity by 33 and 40%, respectively. In the case of 4-hexylresorcinol, the reverse behavior was found (Figure 4c,d). No binding to the receptor was detected in the competition assay (Figure 4c), but a very high potency was observed with a binding constant of 7 nM (Figure 4d) (see the Discussion). The comparison between predicted and experimental pKi, reported in Table 3, shows that in the case of octyl gallate, curcumine, and capsaicin, there is a large discrepancy between predicted and experimental behavior. The results can be explained by inspecting the predicted binding modes for these additives that are shown in Figure 5 (see the Discussion). Overall, the in silico screening procedure was able to identify food additives showing estrogen activity in the nanomolar range with a success rate of 23%.
Figure 4.
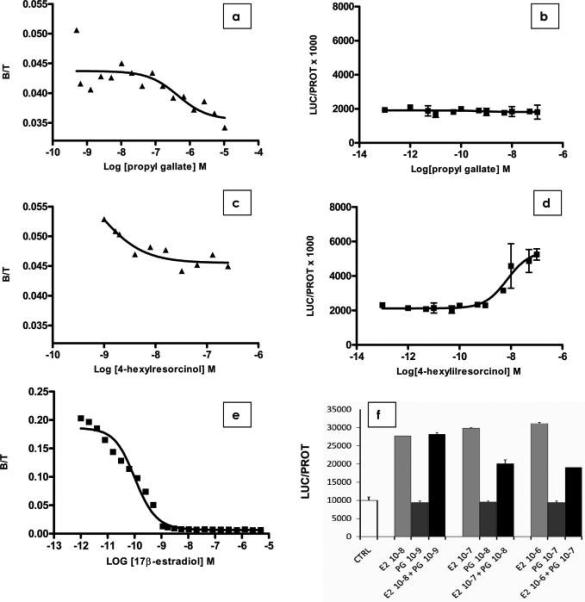
Binding titrations for propyl gallate (a) and 4-hexylresorcinol (c) and transactivation assays for propyl gallate (b), 4-hexylresorcinol (d), and 17β-estradiol (e). The antagonist activity of propyl gallate was tested in a transactivation assay (f). Each experiment was carried out in triplicate, according to the procedure reported in the Experimental Procedures.
Figure 5.
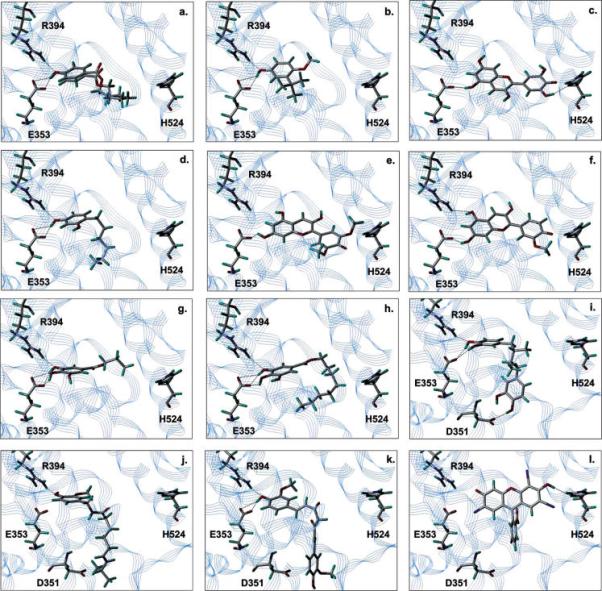
Predicted binding modes for the 13 food additives identified as potential ERα ligands by virtual screening. (a) Propyl 4-hydroxybenzoate and butyl 4-hydroxybenzoate, (b) butyl hydroxyanisole, (c) delphinidin, (d) 4-hexylresorcinol, (e) malvidin, (f) peonidin, (g) propyl gallate, (h) octyl gallate, (i) nordihydroguaiaretic acid, (j) capsaicin, (k) curcumin, and (l) erythrosine.
Discussion
The application of docking-based virtual screening to drug discovery speeds up the identification of potential ligands for a given receptor when screening large amounts of compounds. The main issue for in silico screening analysis lies in the difficulty of correctly ranking different compounds, that is, in correctly evaluating the relative free energy of binding. In this work, we have used the HINT force field that was shown to be a reliable tool for predictions of binding affinity in several studies of protein—ligand as well as protein—DNA interactions (42, 44-46, 62). Moreover, the results obtained correlating the HS and the experimentally determined binding affinities of crystallographically and noncystallographially ligand—ERα complexes (Figure 3) were remarkably good, indicating the robustness of HINT as postdocking scoring function. However, when examining a wide number of ligands characterized by very different chemical structures and unknown activity, failures of the scoring functions in discriminating between true ligands and nonbinders are common, and several studies report a large number of false positives among the predicted binders (63-66). Therefore, a careful analysis on the results generated from docking runs is mandatory (58, 67-69). We have adopted two filtering modes: (i) calculation of the ligand volume buried within the protein binding site using the software GRASP (59) and (ii) visual inspection of ligand binding mode generated through docking. The first approach, proposed as a good filter method by Stahl and Bohm (58), allows the identification of ligands leaving a large amount of empty space within the active site, which has to be filled by water molecules with an entropic penalty, particularly relevant for a closed hydrophobic cavity like the ERα binding pocket. None of the known ERα ligands exhibits a volume lower than about 200 Å3, and many of them (such as tamoxifen and raloxifene) display buried volumes around 300 Å3. The nonstringent buried volume cut off of 150 Å3, below which ligands were discarded, generated a selection among the potential binders, decreasing its number from 31 to 25. The visual inspection of the retrieved candidates was carried out evaluating the following compound features: (i) chemically plausible conformation of the bound ligand, (ii) the absence of charged groups adjacent to hydrophobic groups, and (iii) the presence of at least one hydrogen bond between protein and ligand. The two latter criteria are not particularly stringent, as often made in virtual screening studies (60, 70-72). This choice was made, even at the cost of a possible higher number of false positives, in order not to preclude the docking routine from identifying genuine ligands of ERα that bind with unusual modes. The inspection of the structures of the 13 compounds that were identified as potential ligands (Figure 5) indicates that capsaicin and erythrosine are placed within the active site with a binding mode that was not previously observed for estrogen ligands, that is, without hydrogen bond formation with Glu353. Indeed, none of these ligands resulted active in in vitro assays, suggesting that it is unlikely to observe estrogen ligands with binding modes different from the classic one.
Four among the 13 food additives identified as potentially estrogenic were predicted to preferentially bind to ERα in the “open” (SERM) conformation (Figure 5i—l). They are nordihydroguaiaretic acid, capsaicin, curcumin, and erythrosine. The other nine additives were predicted to bind to the ERα “closed” (agonist) conformation. The binding mode predicted for nordihydroguaiaretic acid (Figure 4i) is particularly interesting, because this compound is already known as a ERα ligand, but its activity as agonist or SERM has not yet been completely characterized (73). Our computational analysis may suggest a possible SERM-like nature of this compound that might deserve further investigation. The predicted binding modes of almost all of the food additives show a hydrogen bond between a phenolic group on the ligand and the carboxylate of Glu353 within the ERα binding site. Only capsaicin (Figure 5j) and erythrosine (Figure 5l) do not display hydrogen bonds with this glutamate residue, but both of them resulted inactive during in vitro assays. This is in line with previous reports indicating that hydrogen bonding with Glu353 is an essential feature for binding to ERα (13, 74). Furthermore, almost all known binders show a phenolic group, which mimics that of the 17β-estradiol A ring, acting as a hydrogen bond donor to the carboxylate of Glu353 and as a hydrogen bond acceptor from the guanidinium group of Arg394. Eight of the proposed hits possess this feature: propyl and butyl p-hydroxybenzoate (Figure 5a), butyl hydroxyanisole (Figure 5b), propyl gallate (Figure 5g), octyl gallate (Figure 5h), curcumin (Figure 5k), and nordihydroguaiaretic acid (Figure 5i). Among these compounds, we found the most active ligands (propyl gallate, 4-hexylresorcinol, and nordihydroguaiaretic acid) and also the two weak binders propyl and butyl p-hydroxybenzoates (Table 3). Most likely, the use of more stringent criteria during the visual inspection of the docking hits would have avoided some of the false positives collected at the end of the screening. It can also be noted that a number of ligands do not fully occupy the ERα binding cleft. This condition is energetically unfavorable because it causes the formation of lipophilic cavities (ERα binding site is mainly hydrophobic) that have to be filled by water molecules. However, 4-hexylresorcinol and propyl gallate, even if they fall in this category of ligands (Figure 5d,g, respectively) resulted experimentally strong xenoestrogens. This finding supports the choice of being relatively permissive in terms of buried ligand volume in the postdocking filtering.
The different activity profiles (Table 3) of propyl gallate and octyl gallate are somehow surprising. The number and the geometrical quality of the hydrogen bonds formed by the two additives with Glu353 and Arg394 are the same. Octyl gallate fills the binding pocket better than propyl gallate (ligand buried volume of 249 vs 176 Å3). Nonetheless, only the propyl gallate resulted active during experimental testing (Table 3). A possible explanation resides in the high flexibility of the octyl gallate long alkyl chain, whose confinement within the binding site may give rise to an entropic cost. Such negative contribution cannot be revealed by the scoring function. Similarly, curcumin, where the long alkyl chain that acts as a spacer between the two phenyl rings might increase ligand flexibility, was found to lack binding activity. Furthermore, this long chain does not allow the interaction of curcumin with the closed conformation of the ERα binding pocket (the molecule is too long).
The identification of novel molecules that bind to ERα in the micromolar range is of value both in the perspective of ligand optimization, the step following screening practices in drug discovery campaign, and in the search of food additives with estrogenic activity. However, it should be considered that xenoestrogens endowed with micromolar binding affinity for ERα might not be relevant because only ligands with a binding affinity in the nanomolar or in the low micromolar range might have the capability of interfering with endogenous ligands such as estradiol. In any event, three among the 13 potential xenoestrogens, nordihydroguaiaretic acid, propyl gallate, and 4-hexylresorcinol, exhibit a nanomolar binding affinity/potency, thus able to significantly compete with estradiol. This is a hit rate of 23%, well within the success rate of screening with heterogeneous compound libraries, usually of the order of 10−30% (64, 75). Furthermore, success rates reported in the literature are usually built considering as hits compounds that bind in the micromolar range (76) (or even in the hundreds of micromolar range). Applying this criterion to our results, we would obtain a hit rate of 38% (5/13). It should be also pointed out that four of the tested additives (delphinidin, malvidin, peonidin, and erythrosine) were predicted to be weak binders (Table 3). Considering only the predicted top binders (nine compounds), the hit rate of the computational protocol would rise to 56%.
Nordihydroguaiaretic acid was already known as an ERα binder (6), while for propyl gallate, only limited data showing transactivation activity in the high micromolar range were reported (77). To our knowledge, the 7.39 pKi (54 nM) that we found for propyl gallate in the present study identifies this compound as the strongest ligand among xenochemicals currently known as ERα binders. Propyl gallate showed another interesting feature. The compound was able to bind to ERα in the nanomolar range but did not show any transactivation activity at the concentrations used in the biological assay. Indeed, in a competition study, propyl gallate proved to be able to antagonize 17β-estradiol transactivation ability. On the other hand, 4-hexylesorcinol is very active in the transactivation assay but does not exhibit a direct binding. It is well-known that ligand—receptor interaction is necessary but not sufficient to activate the transcription machinery, and coregulators are attracted by the receptor to the promoter, and these molecules are responsible for the final activity of the receptor on transcription. Thus, the lack of correlation between receptor binding activity and transcriptional ability of the two compounds above suggests that each of them induces a different receptor conformation, attracting corepressors or coactivators able to stabilize the ER-DNA binding, resulting in transcription initiation. Alternatively, in the case of hexylresorcinol, it may be hypothesized that this compound has an indirect effect and facilitates the interaction between unliganded ER and coactivators, inducing the transcription of the reporter. As propyl gallate is a widely used antioxidant with an acceptable daily intake established by JECFA of 0−1.4 mg/kg (www.fao.org/ag/agn/jecfa-additives), some caution note might be issued for this additive.
Conclusions
The identification of xenoestrogens is of great interest, given the increasing concern regarding the possible adverse health effects of these compounds. The application of an integrated in silico and in vitro approach allows us to increase the speed in the analysis of food additives databases for the identification of potential xenoestrogens. The developed protocol was validated and allowed us to identify two new xenoestrogens, propyl gallate and 4-hexylresorcinol, exhibiting activity in the nanomolar range.
Acknowledgment
This work was carried out with the support of Regione-Emilia Romagna in the frame of the SIQUAL project (A.M. and P.C.) and by the European Union within the projects (Strep EWA, LSHM-CT-2005-518245) and NIH (R01AG027713-02) (to A.M.). We thank Prof. Glen E. Kellogg and Donald J. Abraham for the HINT software and Prof. Simone Ottonello and Dr. Roberta Ruotolo for helpful discussions.
Footnotes
Abbreviations: CoMFA, comparative molecular field analysis; EAFUS, Everything Added to Food in the United States; ER, estrogen receptor; ERE, estrogen response elements; FBS, fetal bovine serum; H12, helix-12; HS, HINT score; JECFA, Joint FAO/WHO Expert Committee on Food Additives; LBD, ligand binding domain; MD, molecular dynamic; PDB, protein data bank; QSAR, quantitative structure—activity relationship; SERM, selective estrogen receptor modulator.
References
- 1.Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 2006;58:773–781. doi: 10.1124/pr.58.4.8. [DOI] [PubMed] [Google Scholar]
- 2.Pozzi S, Benedusi V, Maggi A, Vegeto E. Estrogen action in neuroprotection and brain inflammation. Ann. N. Y. Acad. Sci. 2006;1089:302–323. doi: 10.1196/annals.1386.035. [DOI] [PubMed] [Google Scholar]
- 3.Gustafsson JA. Estrogen receptor beta—A new dimension in estrogen mechanism of action. J. Endocrinol. 1999;163:379–383. doi: 10.1677/joe.0.1630379. [DOI] [PubMed] [Google Scholar]
- 4.Osborne CK, Zhao H, Fuqua SA. Selective estrogen receptor modulators: Structure, function, and clinical use. J. Clin. Oncol. 2000;18:3172–3186. doi: 10.1200/JCO.2000.18.17.3172. [DOI] [PubMed] [Google Scholar]
- 5.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 6.Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, Tong W, Shi L, Perkins R, Sheehan DM. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000;54:138–153. doi: 10.1093/toxsci/54.1.138. [DOI] [PubMed] [Google Scholar]
- 7.Fang H, Tong W, Shi LM, Blair R, Perkins R, Branham W, Hass BS, Xie Q, Dial SL, Moland CL, Sheehan DM. Structure–activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001;14:280–294. doi: 10.1021/tx000208y. [DOI] [PubMed] [Google Scholar]
- 8.Mueller SO. Xenoestrogens: Mechanisms of action and detection methods. Anal. Bioanal. Chem. 2004;378:582–587. doi: 10.1007/s00216-003-2238-x. [DOI] [PubMed] [Google Scholar]
- 9.Gantchev TG, Ali H, van Lier JE. Quantitative structure-activity relationships/comparative molecular field analysis (QSAR/CoMFA) for receptor-binding properties of halogenated estradiol derivatives. J. Med. Chem. 1994;37:4164–4176. doi: 10.1021/jm00050a013. [DOI] [PubMed] [Google Scholar]
- 10.Tong W, Perkins R, Xing L, Welsh WJ, Sheehan DM. QSAR models for binding of estrogenic compounds to estrogen receptor alpha and beta subtypes. Endocrinology. 1997;138:4022–4025. doi: 10.1210/endo.138.9.5487. [DOI] [PubMed] [Google Scholar]
- 11.Oostenbrink BC, Pitera JW, van Lipzig MM, Meerman JH, van Gunsteren WF. Simulations of the estrogen receptor ligand-binding domain: Affinity of natural ligands and xenoestrogens. J. Med. Chem. 2000;43:4594–4605. doi: 10.1021/jm001045d. [DOI] [PubMed] [Google Scholar]
- 12.van Lipzig MM, ter Laak AM, Jongejan A, Vermeulen NP, Wamelink M, Geerke D, Meerman JH. Prediction of ligand binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear interaction energy method. J. Med. Chem. 2004;47:1018–1030. doi: 10.1021/jm0309607. [DOI] [PubMed] [Google Scholar]
- 13.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 14.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 15.Cozzini P, Dottorini T. Is it possible docking and scoring new ligands with few experimental data? Preliminary results on estrogen receptor as a case study. Eur. J. Med. Chem. 2004;39:601–609. doi: 10.1016/j.ejmech.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Sivanesan D, Rajnarayanan RV, Doherty J, Pattabiraman N. In-silico screening using flexible ligand binding pockets: A molecular dynamics-based approach. J. Comput.-Aided Mol. Des. 2005;19:213–228. doi: 10.1007/s10822-005-4788-9. [DOI] [PubMed] [Google Scholar]
- 17.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 18.Erickson JA, Jalaie M, Robertson DH, Lewis RA, Vieth M. Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 2004;47:45–55. doi: 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- 19.Murray CW, Baxter CA, Frenkel AD. The sensitivity of the results of molecular docking to induced fit effects: Application to thrombin, thermolysin and neuraminidase. J. Comput.-Aided Mol. Des. 1999;13:547–562. doi: 10.1023/a:1008015827877. [DOI] [PubMed] [Google Scholar]
- 20.Birch L, Murray CW, Hartshorn MJ, Tickle IJ, Verdonk ML. Sensitivity of molecular docking to induced fit effects in influenza virus neuraminidase. J. Comput.-Aided Mol. Des. 2002;16:855–869. doi: 10.1023/a:1023844626572. [DOI] [PubMed] [Google Scholar]
- 21.Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 22.Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol. Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- 23.Teague SJ. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discovery. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 24.Alonso H, Bliznyuk AA, Gready JE. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006;26:531–568. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 25.Jiang F, Kim SH. “Soft docking”: Matching of molecular surface cubes. J. Mol. Biol. 1991;219:79–102. doi: 10.1016/0022-2836(91)90859-5. [DOI] [PubMed] [Google Scholar]
- 26.Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J. Mol. Biol. 1994;235:345–356. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- 27.Schaffer L, Verkhivker GM. Predicting structural effects in HIV-1 protease mutant complexes with flexible ligand docking and protein side-chain optimization. Proteins. 1998;33:295–310. doi: 10.1002/(sici)1097-0134(19981101)33:2<295::aid-prot12>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 28.Moitessier N, Therrien E, Hanessian S. A method for induced-fit docking, scoring, and ranking of flexible ligands. Application to peptidic and pseudopeptidic beta-secretase (BACE 1) inhibitors. J. Med. Chem. 2006;49:5885–5894. doi: 10.1021/jm050138y. [DOI] [PubMed] [Google Scholar]
- 29.Claussen H, Buning C, Rarey M, Lengauer T. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 2001;308:377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 30.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 31.Huang SY, Zou X. Efficient molecular docking of NMR structures: Application to HIV-1 protease. Protein Sci. 2007;16:43–51. doi: 10.1110/ps.062501507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang SY, Zou X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 33.Knegtel RM, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J. Mol. Biol. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 34.Broughton HB. A method for including protein flexibility in protein-ligand docking: improving tools for database mining and virtual screening. J. Mol. Graphics Modell. 2000;18:247–257. 302–244. doi: 10.1016/s1093-3263(00)00036-x. [DOI] [PubMed] [Google Scholar]
- 35.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 36.Zavodszky MI, Lei M, Thorpe MF, Day AR, Kuhn LA. Modeling correlated main-chain motions in proteins for flexible molecular recognition. Proteins. 2004;57:243–261. doi: 10.1002/prot.20179. [DOI] [PubMed] [Google Scholar]
- 37.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 38.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 39.Kellogg GE, Abraham DJ. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur. J. Med. Chem. 2000;35:651–661. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 40.Kellogg GE, Burnett JC, Abraham DJ. Very empirical treatment of solvation and entropy: A force field derived from log Po/w. J. Comput.-Aided Mol. Des. 2001;15:381–393. doi: 10.1023/a:1011136228678. [DOI] [PubMed] [Google Scholar]
- 41.Amadasi A, Spyrakis F, Cozzini P, Abraham DJ, Kellogg GE, Mozzarelli A. Mapping the energetics of water-protein and water-ligand interactions with the “natural” HINT forcefield: Predictive tools for characterizing the roles of water in biomolecules. J. Mol. Biol. 2006;358:289–309. doi: 10.1016/j.jmb.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 42.Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, Mozzarelli A. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J. Med. Chem. 2002;45:2469–2483. doi: 10.1021/jm0200299. [DOI] [PubMed] [Google Scholar]
- 43.Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, Mozzarelli A. Free energy of ligand binding to protein: evaluation of the contribution of water molecules by computational methods. Curr. Med. Chem. 2004;11:3093–3118. doi: 10.2174/0929867043363929. [DOI] [PubMed] [Google Scholar]
- 44.Fornabaio M, Spyrakis F, Mozzarelli A, Cozzini P, Abraham DJ, Kellogg GE. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 3. The free energy contribution of structural water molecules in HIV-1 protease complexes. J. Med. Chem. 2004;47:4507–4516. doi: 10.1021/jm030596b. [DOI] [PubMed] [Google Scholar]
- 45.Spyrakis F, Amadasi A, Fornabaio M, Abraham DJ, Mozzarelli A, Kellogg GE, Cozzini P. The consequences of scoring docked ligand conformations using free energy correlations. Eur. J. Med. Chem. 2007;42:921–933. doi: 10.1016/j.ejmech.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 46.Spyrakis F, Cozzini P, Bertoli C, Marabotti A, Kellogg GE, Mozzarelli A. Energetics of the protein-DNA-water interaction. BMC Struct. Biol. 2007;7:4. doi: 10.1186/1472-6807-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spyrakis F, Fornabaio M, Cozzini P, Mozzarelli A, Abraham DJ, Kellogg GE. Computational titration analysis of a multiprotic HIV-1 protease-ligand complex. J. Am. Chem. Soc. 2004;126:11764–11765. doi: 10.1021/ja0465754. [DOI] [PubMed] [Google Scholar]
- 48.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng Y, Prusoff W. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 50.Amadasi A, Cozzini P, Incerti M, Duce E, Fisicaro E, Vicini P. Molecular modeling of binding between amidinobenzisothiazoles, with antidegenerative activity on cartilage, and matrix metalloproteinase-3. Bioorg. Med. Chem. 2007;15:1420–1429. doi: 10.1016/j.bmc.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Porotto M, Fornabaio M, Greengard O, Murrell MT, Kellogg GE, Moscona A. Paramyxovirus receptor-binding molecules: Engagement of one site on the hemagglutinin-neuraminidase protein modulates activity at the second site. J. Virol. 2006;80:1204–1213. doi: 10.1128/JVI.80.3.1204-1213.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansch C, Leo AJ. Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley and Sons Inc.; New York: 1979. [DOI] [PubMed] [Google Scholar]
- 53.Derewenda ZS, Lee L, Derewenda U. The occurrence of C-H···O hydrogen bonds in proteins. J. Mol. Biol. 1995;252:248–262. doi: 10.1006/jmbi.1995.0492. [DOI] [PubMed] [Google Scholar]
- 54.Vargas R, Garza J, Dixon DA, Hay BP. How strong is the C-H···OdC hydrogen bond? J. Am. Chem. Soc. 2000;122:4750–4755. [Google Scholar]
- 55.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 56.Ciana P, Di Luccio G, Belcredito S, Pollio G, Vegeto E, Tatangelo L, Tiveron C, Maggi A. Engineering of a mouse for the in vivo profiling of estrogen receptor activity. Mol. Endocrinol. 2001;15:1104–1113. doi: 10.1210/mend.15.7.0658. [DOI] [PubMed] [Google Scholar]
- 57.Capra V, Veltri A, Foglia C, Crimaldi L, Habib A, Parenti M, Rovati G. Mutational analysis of the highly conserved ERY motif of the thromboxane A2 receptor: Alternative role in G protein-coupled receptor signaling. Mol. Pharmacol. 2004;66:880–889. doi: 10.1124/mol.104.001487. [DOI] [PubMed] [Google Scholar]
- 58.Stahl M, Bohm HJ. Development of filter functions for protein-ligand docking. J. Mol. Graphics Modell. 1998;16:121–132. doi: 10.1016/s1093-3263(98)00018-7. [DOI] [PubMed] [Google Scholar]
- 59.Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 60.Gruneberg S, Stubbs MT, Klebe G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: Strategy and experimental confirmation. J. Med. Chem. 2002;45:3588–3602. doi: 10.1021/jm011112j. [DOI] [PubMed] [Google Scholar]
- 61.Lyne PD. Structure-based virtual screening: an overview. Drug Discovery Today. 2002;7:1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 62.Burnett JC, Botti P, Abraham DJ, Kellogg GE. Computationally accessible method for estimating free energy changes resulting from site-specific mutations of biomolecules: Systematic model building and structural/hydropathic analysis of deoxy and oxy hemoglobins. Proteins. 2001;42:355–377. doi: 10.1002/1097-0134(20010215)42:3<355::aid-prot60>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 63.Cummings MD, DesJarlais RL, Gibbs AC, Mohan V, Jaeger EP. Comparison of automated docking programs as virtual screening tools. J. Med. Chem. 2005;48:962–976. doi: 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- 64.Graves AP, Brenk R, Shoichet BK. Decoys for docking. J. Med. Chem. 2005;48:3714–3728. doi: 10.1021/jm0491187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J. Med. Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 66.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 67.Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 68.Feher M. Consensus scoring for protein-ligand interactions. Drug Discovery Today. 2006;11:421–428. doi: 10.1016/j.drudis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 69.Hoffmann D, Kramer B, Washio T, Steinmetzer T, Rarey M, Lengauer T. Two-stage method for protein-ligand docking. J. Med. Chem. 1999;42:4422–4433. doi: 10.1021/jm991090p. [DOI] [PubMed] [Google Scholar]
- 70.Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J. Med. Chem. 2004;47:5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 71.Kellenberger E, Springael JY, Parmentier M, Hachet-Haas M, Galzi JL, Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J. Med. Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 72.Lyne PD, Kenny PW, Cosgrove DA, Deng C, Zabludoff S, Wendoloski JJ, Ashwell S. Identification of compounds with nanomolar binding affinity for checkpoint kinase-1 using knowledge-based virtual screening. J. Med. Chem. 2004;47:1962–1968. doi: 10.1021/jm030504i. [DOI] [PubMed] [Google Scholar]
- 73.Fujimoto N, Kohta R, Kitamura S, Honda H. Estrogenic activity of an antioxidant, nordihydroguaiaretic acid (NDGA). Life Sci. 2004;74:1417–1425. doi: 10.1016/j.lfs.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 74.Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. 1997;62:268–303. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 75.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.ter Veld MG, Schouten B, Louisse J, van Es DS, van der Saag PT, Rietjens IM, Murk AJ. Estrogenic potency of food-packaging-associated plasticizers and antioxidants as detected in ERα and ERβ reporter gene cell lines. J. Agric. Food Chem. 2006;54:4407–4416. doi: 10.1021/jf052864f. [DOI] [PubMed] [Google Scholar]
- 78.Manas ES, Xu ZB, Unwalla RJ, Somers WS. Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure. 2004;12:2197–2207. doi: 10.1016/j.str.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 79.Kim S, Wu JY, Birzin ET, Frisch K, Chan W, Pai LY, Yang YT, Mosley RT, Fitzgerald PM, Sharma N, Dahllund J, Thorsell AG, DiNinno F, Rohrer SP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen receptor alpha modulators. J. Med. Chem. 2004;47:2171–2175. doi: 10.1021/jm034243o. [DOI] [PubMed] [Google Scholar]
- 80.Blizzard TA, Dininno F, Morgan JD, 2nd, Chen HY, Wu JY, Kim S, Chan W, Birzin ET, Yang YT, Pai LY, Fitzgerald PM, Sharma N, Li Y, Zhang Z, Hayes EC, Dasilva CA, Tang W, Rohrer SP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side chains and linkers. Bioorg. Med. Chem. Lett. 2005;15:107–113. doi: 10.1016/j.bmcl.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 81.Tan Q, Blizzard TA, Morgan JD, 2nd, Birzin ET, Chan W, Yang YT, Pai LY, Hayes EC, DaSilva CA, Warrier S, Yudkovitz J, Wilkinson HA, Sharma N, Fitzgerald PM, Li S, Colwell L, Fisher JE, Adamski S, Reszka AA, Kimmel D, DiNinno F, Rohrer SP, Freedman LP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. Part 10: Chromanes: Old scaffolds for new SERAMs. Bioorg. Med. Chem. Lett. 2005;15:1675–1681. doi: 10.1016/j.bmcl.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 82.Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, Harris HA, Hsiao C, Akopian T, Hum WT, Malakian K, Wolfrom S, Bapat A, Bhat RA, Stahl ML, Somers WS, Alvarez JC. Structure-based design of estrogen receptor-beta selective ligands. J. Am. Chem. Soc. 2004;126:15106–15119. doi: 10.1021/ja047633o. [DOI] [PubMed] [Google Scholar]
- 83.Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, Geiser M, Halleux C, Kallen J, Keller H, Ramage P. Selective estrogen receptor modulators with conformationally restricted side chains. Synthesis and structure-activity relationship of ERalpha-selective tetrahydroisoquinoline ligands. J. Med. Chem. 2005;48:364–379. doi: 10.1021/jm040858p. [DOI] [PubMed] [Google Scholar]
- 84.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 85.Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, Mrksich M, Greene GL. Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J. Biol. Chem. 2006;281:17909–17919. doi: 10.1074/jbc.M513684200. [DOI] [PubMed] [Google Scholar]
- 86.Norman BH, Dodge JA, Richardson TI, Borromeo PS, Lugar CW, Jones SA, Chen K, Wang Y, Durst GL, Barr RJ, Montrose-Rafizadeh C, Osborne HE, Amos RM, Guo S, Boodhoo A, Krishnan V. Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J. Med. Chem. 2006;49:6155–6157. doi: 10.1021/jm060491j. [DOI] [PubMed] [Google Scholar]
- 87.Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 88.Richardson TI, Dodge JA, Durst GL, Pfeifer LA, Shah J, Wang Y, Durbin JD, Krishnan V, Norman BH. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 3: Synthesis of cyclopentanone and cyclohexanone intermediates for C-ring modification. Bioorg. Med. Chem. Lett. 2007;17:4824–4828. doi: 10.1016/j.bmcl.2007.06.052. [DOI] [PubMed] [Google Scholar]
- 89.Norman BH, Richardson TI, Dodge JA, Pfeifer LA, Durst GL, Wang Y, Durbin JD, Krishnan V, Dinn SR, Liu S, Reilly JE, Ryter KT. Benzopyrans as selective estrogen receptor beta agonists (SERBAs). Part 4: Functionalization of the benzopyran A-ring. Bioorg. Med. Chem. Lett. 2007;17:5082–5085. doi: 10.1016/j.bmcl.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 90.Nettles KW, Bruning JB, Gil G, O'Neill EE, Nowak J, Guo Y, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hummel CW, Geiser AG, Bryant HU, Cohen IR, Dally RD, Fong KC, Frank SA, Hinklin R, Jones SA, Lewis G, McCann DJ, Rudmann DG, Shepherd TA, Tian H, Wallace OB, Wang M, Wang Y, Dodge JA. A selective estrogen receptor modulator designed for the treatment of uterine leiomyoma with unique tissue specificity for uterus and ovaries in rats. J. Med. Chem. 2005;48:6772–6775. doi: 10.1021/jm050723z. [DOI] [PubMed] [Google Scholar]
- 92.Dykstra KD, Guo L, Birzin ET, Chan W, Yang YT, Hayes EC, DaSilva CA, Pai LY, Mosley RT, Kraker B, Fitzgerald PM, DiNinno F, Rohrer SP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. Part 16: 2-Aryl indoles as highly subtype selective ligands for ERalpha. Bioorg. Med. Chem. Lett. 2007;17:2322–2328. doi: 10.1016/j.bmcl.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 93.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 94.Chen HY, Dykstra KD, Birzin ET, Frisch K, Chan W, Yang YT, Mosley RT, DiNinno F, Rohrer SP, Schaeffer JM, Hammond ML. Estrogen receptor ligands. Part 1: The discovery of flavanoids with subtype selectivity. Bioorg. Med. Chem. Lett. 2004;14:1417–1421. doi: 10.1016/j.bmcl.2004.01.031. [DOI] [PubMed] [Google Scholar]
- 95.Amari G, Armani E, Ghirardi S, Delcanale M, Civelli M, Caruso PL, Galbiati E, Lipreri M, Rivara S, Lodola A, Mor M. Synthesis, pharmacological evaluation, and structure-activity relationships of benzopyran derivatives with potent SERM activity. Bioorg. Med. Chem. 2004;12:3763–3782. doi: 10.1016/j.bmc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 96.Gauthier S, Caron B, Cloutier J, Dory YL, Favre A, Larouche D, Mailhot J, Ouellet C, Schwerdtfeger A, Leblanc G, Martel C, Simard J, Merand Y, Belanger A, Labrie C, Labrie F. (S)-(+)-4-[7-(2,2-Dimethyl-1-oxopropoxy)-4-methyl-2-[4-[2-(1-piperidinyl)-ethoxy]phenyl]-2H-1-benzopyran-3-yl]-phenyl 2,2-dimethylpropanoate (EM-800): A highly potent, specific, and orally active nonsteroidal antiestrogen. J. Med. Chem. 1997;40:2117–2122. doi: 10.1021/jm970095o. [DOI] [PubMed] [Google Scholar]
- 97.Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB. Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res. 2001;61:8412–8415. [PubMed] [Google Scholar]
- 98.Miller CP, Collini MD, Tran BD, Harris HA, Kharode YP, Marzolf JT, Moran RA, Henderson RA, Bender RH, Unwalla RJ, Greenberger LM, Yardley JP, Abou-Gharbia MA, Lyttle CR, Komm BS. Design, synthesis, and preclinical characterization of novel, highly selective indole estrogens. J. Med. Chem. 2001;44:1654–1657. doi: 10.1021/jm010086m. [DOI] [PubMed] [Google Scholar]
- 99.Rosati RL, Da Silva Jardine P, Cameron KO, Thompson DD, Ke HZ, Toler SM, Brown TA, Pan LC, Ebbinghaus CF, Reinhold AR, Elliott NC, Newhouse BN, Tjoa CM, Sweetnam PM, Cole MJ, Arriola MW, Gauthier JW, Crawford DT, Nickerson DF, Pirie CM, Qi H, Simmons HA, Tkalcevic GT. Discovery and preclinical pharmacology of a novel, potent, nonsteroidal estrogen receptor agonist/antagonist, CP-336156, a diaryltetrahydronaphthalene. J. Med. Chem. 1998;41:2928–2931. doi: 10.1021/jm980048b. [DOI] [PubMed] [Google Scholar]
- 100.Meyers MJ, Sun J, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor subtype-selective ligands: asymmetric synthesis and biological evaluation of cis- and trans-5,11-dialkyl-5,6,11,12-tetrahydrochrysenes. J. Med. Chem. 1999;42:2456–2468. doi: 10.1021/jm990101b. [DOI] [PubMed] [Google Scholar]
- 101.Blizzard TA, Morgan JD, 2nd, Mosley RT, Birzin ET, Frisch K, Rohrer SP, Hammond ML. 2-Phenylspiroindenes: A novel class of selective estrogen receptor modulators (SERMs). Bioorg. Med. Chem. Lett. 2003;13:479–483. doi: 10.1016/s0960-894x(02)00985-x. [DOI] [PubMed] [Google Scholar]
- 102.Jobling S, Reynolds T, White R, Parker MG, Sumpter JP. A variety of environmentally persistent chemicals, including some phthalate plasticizers, are weakly estrogenic. Environ. Health Perspect. 1995;103:582–587. doi: 10.1289/ehp.95103582. [DOI] [PMC free article] [PubMed] [Google Scholar]