

Abstract
Residual dipolar couplings provide complementary information to the nuclear Overhauser effect measurements that are traditionally used in biomolecular structure determination by NMR. In a de novo structure determination, however, lack of knowledge about the degree and orientation of molecular alignment complicates the analysis of dipolar coupling data. We present a probabilistic framework for analyzing residual dipolar couplings and demonstrate that it is possible to estimate the atomic coordinates, the complete molecular alignment tensor, and the error of the couplings simultaneously. As a by-product, we also obtain estimates of the uncertainty in the coordinates and the alignment tensor. We show that our approach encompasses existing methods for determining the alignment tensor as special cases, including least squares estimation, histogram fitting, and elimination of an explicit alignment tensor in the restraint energy.
Keywords: Protein structure, NMR structure determination, Residual dipolar couplings, Inferential structure determination, Markov chain Monte Carlo
Introduction
Residual dipolar coupling (RDC) measurements provide long-range orientational information for biomolecular structure determination by NMR (Prestegard 1998; Bax et al. 2001; Bax 2003; Lipsitz and Tjandra 2004; Bax and Grishaev 2005). Hence, dipolar couplings complement the nuclear Overhauser effect (NOE) data that are most commonly used in NMR structure determination. In favorable cases, orientational information may even be sufficient to determine the backbone conformation of a protein without any additional data. Using molecular fragment replacement, Delaglio et al. determined the backbone conformation of the protein ubiquitin to high accuracy from RDCs alone (2000).
In isotropic solution, dipolar couplings average to zero. Therefore, to observe dipolar couplings it is necessary to weakly align the molecule. This can be achieved by orienting the molecule in an external field (Tolman et al. 1995) or through interactions with an appropriate solvent medium such as liquid crystals (Tjandra and Bax 1997). The magnitude of a dipolar coupling depends on the degree of molecular alignment and on the average orientation of the internuclear vector relative to the external magnetic field. To calculate a dipolar coupling, knowledge of the degree of alignment and of the average orientation of the molecule is therefore required. This poses a problem in a de novo structure determination because the alignment tensor is a priori unknown. As a consequence the structure calculation requires estimates of the axial and rhombic component of the alignment tensor, in addition to the observed dipolar couplings.
Several ways of obtaining such estimates have been proposed. Losonczi et al. used singular value decomposition to calculate the alignment tensor from a known structure (1999). Clore et al. introduced a method which is based on an analysis of the histogram of all measured dipolar couplings and does not require any structural knowledge (1998b). Estimates of the axial and rhombic component are obtained by fitting the distribution of observed dipolar couplings with the analytical curve describing a chemical shift powder pattern. Once such estimates are available, the unknown orientation of the molecule relative to the alignment tensor is determined during the structure calculation (Tjandra et al. 1997). It has further been demonstrated that the axial and rhombic component can be optimized using a grid search (Clore et al. 1998a). Grzesiek and coworkers developed a restraint energy function that does not explicitly involve the alignment tensor (Moltke and Grzesiek 1999; Sass et al. 2001).
Each of these methods has its limitations. Tensor fitting by singular value decomposition requires a known structure and is therefore not applicable in a de novo structure determination. The histogram method provides only approximate estimates of the axial and rhombic components. These estimates very much depend on the smallest and the largest observed dipolar coupling and are therefore sensitive to noise. Furthermore, as the average orientation of the molecule cannot be derived by this method, it needs to be optimized during the structure calculation. Direct optimization of the axial and rhombic component by a grid search is calculation intensive. The tensor-free restraint energy is inflexible when it comes to the incorporation of a priori knowledge or the estimation of errors for individual data sets. A fundamental limitation common to all these methods is that they cannot assess the uncertainty in the alignment tensor and do not provide a generic way to take a priori knowledge into account.
Here we introduce a probabilistic model and estimation procedure for analyzing dipolar coupling data. Both integrate seamlessly with a probabilistic structure determination framework. Our approach builds on related work for three-bond scalar coupling constants (Habeck et al. 2005a). In this previous work, we showed that, using the Inferential Structure Determination (ISD) framework (Rieping et al. 2005a; Habeck et al. 2005b), it is straightforward to simultaneously estimate the molecular structure and the unknown coefficients of the Karplus curve. The same model can be applied to dipolar couplings, where the elements of the unknown alignment tensor play a role analogous to the Karplus parameters. By applying Bayes’ theorem we derive a joint posterior distribution for the atomic coordinates, the tensor elements, and the errors of the data sets. This probability distribution is uniquely determined by the observed dipolar couplings and the few basic assumptions required to model them. It quantifies the interdependence of the different groups of parameters and tells us how to simultaneously estimate all parameters from the data: All we need to do is find and explore the regions of high posterior probability by means of statistical sampling methods; additional heuristics are not required. A consequence of the probabilistic treatment is that we obtain precision estimates for all unknown parameters, most importantly for the tensor elements and the three-dimensional coordinates of the structure. A further advantage is that we can incorporate different kinds of a priori knowledge. We find that our probabilistic formulation contains the histogram method, singular value decomposition and a tensor-free restraint energy function as special cases and thus unifies these methods in a consistent way.
Theory
In the secular approximation, a dipolar coupling Dkl between two nuclear spins k and l with distance vector rkl has a magnitude of
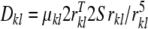 |
1 |
where S is the Saupe order matrix and
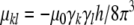 (Saupe and Englert 1963). This relation is strictly valid only if the molecule is rigid and undergoes rotational diffusion, which we will assume in the following. The Saupe tensor describes the average orientation of the molecule as well as the degree of alignment. It is determined by several factors such as the solvent medium, its concentration, the molecule’s shape and electrostatic properties (Zweckstetter and Bax 2000; Zweckstetter et al. 2004; Zweckstetter 2006). The alignment tensor is symmetric and traceless and can be parameterized with five independent elements s1,..., s5:
(Saupe and Englert 1963). This relation is strictly valid only if the molecule is rigid and undergoes rotational diffusion, which we will assume in the following. The Saupe tensor describes the average orientation of the molecule as well as the degree of alignment. It is determined by several factors such as the solvent medium, its concentration, the molecule’s shape and electrostatic properties (Zweckstetter and Bax 2000; Zweckstetter et al. 2004; Zweckstetter 2006). The alignment tensor is symmetric and traceless and can be parameterized with five independent elements s1,..., s5:
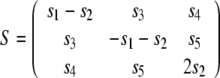 |
2 |
Using this parameterization, a dipolar coupling can be written as the scalar product between two five-dimensional vectors:
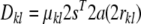 |
3 |
where
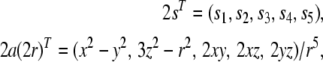 |
4 |
for an internuclear vector r with Cartesian coordinates x, y, z and length r. Equation (3) reveals that dipolar couplings depend linearly on the tensor elements, which allows us to treat them similarly to the Karplus parameters which also enter linearly into the Karplus relation (Karplus 1963).
In analogy to our treatment of scalar coupling constants (Habeck et al. 2005a), we model the observation of a single dipolar coupling with a Gaussian error distribution with an unknown error σ. The likelihood function, i.e. the probability of a data set comprising n measurements, is (Habeck et al. 2006)
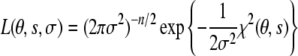 |
5 |
where θ are the conformational degrees of freedom of the molecule. The residual of the fit between observed and calculated dipolar couplings resulting from a Gaussian likelihood is
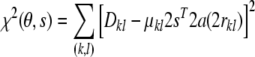 |
6 |
where the sum runs over all pairs of atoms for which a dipolar coupling has been measured. The likelihood function (5) is not a probability for θ, s, and σ in a strict sense, because it is normalized with respect to the data. However, similar to a probability the likelihood function quantifies how consistent settings for θ, s, and σ are with the observations and therefore ranks parameter values according to their ability to explain the data.
Most of the existing methods for structure calculation from dipolar couplings minimize the residual defined in Eq. (6) with respect to the conformational degrees of freedom; during this minimization, the alignment tensor remains fixed to some empirical estimate. Using our probabilistic framework, we are able to determine all unknowns simultaneously, including the conformational degrees of freedom, the five elements of the alignment tensor, and the error of the couplings. The estimation is based on the joint posterior probability distribution
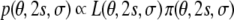 |
7 |
obtained from Bayes’ theorem (Jaynes 2003). Bayes’ theorem requires a prior probability π(θ,s,σ) that quantifies our background knowledge about the unknown parameters. In most situations we dispose of little a priori information about the tensor elements and therefore choose a uniform prior distribution for them.1 We also have little knowledge about the error, except that it is a scale parameter (Habeck et al. 2006) leading to π(σ) = 1/σ (Jeffreys 1946). The prior probability for the atomic coordinates is a canonical ensemble at inverse temperature β and is based on a standard molecular force field E(θ) (Rieping et al. 2005a; Habeck et al. 2005b).
Application of Bayes’ theorem results in the posterior distribution
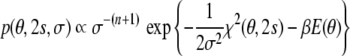 |
8 |
This distribution is a joint probability for all unknown parameters. We make practical use of the posterior distribution by generating a sequence of statistical samples from it. These samples approximate the posterior distribution and can be utilized to estimate θ, s, and σ or to compute an integral such as an expected value.
It is possible to eliminate uninteresting parameters by integrating them out (marginalization (Jaynes 2003; Habeck et al. 2005b)). If, for example, we are not interested in the alignment tensor we can use the marginal posterior distribution
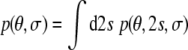 |
9 |
to determine the conformational degrees of freedom and the error of the measurements without explicit knowledge of the alignment tensor. In some cases it is possible to solve marginalization integrals analytically. In general, however, we need to integrate numerically using statistical sampling techniques.
A parameterization of the Saupe tensor in terms of five independent matrix elements exhibits several invariances that may complicate the parameter estimation. For example, a reflection of the coordinates along the x-axis can be compensated by changing the signs of s3 and s4. We can reparameterize the alignment tensor using its spectral decomposition
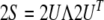 |
10 |
where U is a rotation matrix and Λ the diagonal matrix of eigenvalues λi which are numbered such that |λ1| < |λ2| < |λ3|; because S is traceless, λ1 + λ2 + λ3 = 0. The rotation matrix U describes the average orientation of the molecule. We define the magnitude A and the rhombicity R of the alignment tensor as
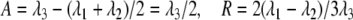 |
11 |
That is, A is related to the size of the largest principal axis and R measures the asymmetry of the alignment tensor along this axis. The strength of a dipolar coupling in the molecular reference frame defined by U is:
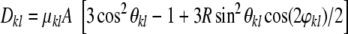 |
12 |
where φkl and θkl are the azimuthal and the polar angle of the internuclear vector rkl in the principal axis system.
If we describe U with Euler angles α, β, γ and replace the tensor elements s1,..., s5 with the new parameters A, R, α, β, γ, we obtain posterior probabilities for the new parameters. The new parameterization has the advantage that it is less degenerate, but the reparameterized posterior distributions become more complicated: R is confined to values between 0 and 2/3, the distribution of the Euler angles is not of a standard form. We therefore use the parametrization based on s1,..., s5.
Results
We applied the outlined formalism to data measured on the protein ubiquitin (Cornilescu et al. 1998). The data comprise 11 RDC sets that were recorded in two different liquid crystalline phases. For both phases, five different coupling types defining the orientation of the peptide planes (N–H, C′–N, C′–H, Cα–C′, Cα–Hα) are available; for the first phase, an additional set of Cα–Cβ couplings was also measured. To describe dipolar coupling data that were recorded in the same liquid crystalline phase, we use a single alignment tensor. However, each data set has its own error parameter σ. Thus the total number of unknowns describing the dipolar couplings is 21: 10 parameters for the two alignment tensors and 11 errors. In addition, we use the 2727 NOE-based distances that are also listed in the restraint file (PDB code 1D3Z). The likelihood function of the distance measurements is described in Rieping et al. (2005b).
Simultaneous estimation of structure and alignment tensor
In a de novo structure determination, all parameters, θ, s and σ, are unknown and need to be estimated from the data. The calculations were carried out with the software ISD (Rieping, Nilges, and Habeck submitted; the software can be downloaded from http://www.bioc.cam.ac.uk/isd and comes with a free academic license). ISD uses Gibbs sampling to break down the task of sampling from a high dimensional probability distribution into less complex steps that update a single parameter set at a time only (Geman and Geman 1984; Rieping et al. 2005a). The conditional posterior probabilities required for this are a five-dimensional Gaussian distribution for the tensor elements [cf. Eq. (16) below] and a gamma distribution for the inverse quadratic error (Habeck et al. 2006). The Gibbs sampler is embedded in a replica-exchange Monte Carlo scheme as described in (Habeck et al. 2005b, c).
Figure 1 shows the posterior histograms for the tensor elements obtained with our sampling algorithm. As a matter of principle, the tensors can be determined to a certain precision only. The precision is reflected in the width of the histograms and depends on various factors such as the availability of additional data (e.g. NOE data), the quality of the dipolar coupling measurements, the consistency of the data sets, and caveats in the relation used to calculate the dipolar couplings [(Eq. 1)]. The distribution of the tensor elements also reflects the uncertainty in the coordinates, because the joint posterior probability (8) couples all parameters and quantifies their degree of correlation. The distributions obtained from the Monte Carlo samples are marginal posterior distributions for the tensor elements, i.e. the variability in the coordinates is fully taken into account; the quantification of the influence of structural uncertainty on the precision of the alignment tensor (Losonczi et al. 1999; Zweckstetter and Bax 2002) is built-in to our framework.
Fig. 1.

Posterior histograms of the tensor elements. Upper panels show the elements si describing the alignment in the first liquid crystal phase. Lower panels show the si histograms for the second phase
Conditional and marginal posterior probabilities
The relationship between protein structure and alignment tensor can be further elucidated on the basis of the conditional and marginal posterior probabilities. To derive marginal posterior probabilities as, for example, Eq. (9), we rewrite the least squares residual, Eq. (6), in matrix notation:
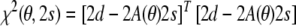 |
13 |
The n dimensional data vector d comprises the dipolar couplings; A is a conformation dependent n × 5 matrix whose rows are the vectors a(rkl) defined in Eq. (4). The residual can be written as the sum of two other strictly nonnegative residuals: 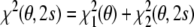 . The first residual depends only on the conformational degrees of freedom:
. The first residual depends only on the conformational degrees of freedom:
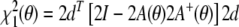 |
14 |
This term is minimal if the structure fully explains the data, i.e. if the data vector lies in the space spanned by the columns of A(θ). The second residual depends both on the structure and on the tensor elements:
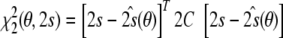 |
15 |
For any given structure, this term has its minimum at 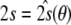 . In the above expressions, we introduced the 5 × 5 matrix
. In the above expressions, we introduced the 5 × 5 matrix 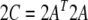 and the vector
and the vector 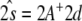 where
where 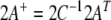 is the generalized inverse (Press et al. 1989) of A. If the same tensor describes multiple data sets, each having its own error, these expressions become more complicated but still only involve standard linear algebra.
is the generalized inverse (Press et al. 1989) of A. If the same tensor describes multiple data sets, each having its own error, these expressions become more complicated but still only involve standard linear algebra.
Distribution of tensor elements for a given structure
Consider now the case that the protein structure is known. For example, a crystal structure of the molecule may be available or the structure of a homologuous protein. Minimization of the least squares residual 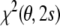 with respect to the tensor elements, while fixing the conformational degrees of freedom to the known structure, then yields an estimate of the Saupe tensor. Using the decomposition of the residual, Eqs. (14) and (15), this minimum can be calculated analytically. Only the second term,
with respect to the tensor elements, while fixing the conformational degrees of freedom to the known structure, then yields an estimate of the Saupe tensor. Using the decomposition of the residual, Eqs. (14) and (15), this minimum can be calculated analytically. Only the second term, 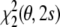 , depends on the tensor elements. Therefore, the optimal tensor for a given structure is the least squares solution
, depends on the tensor elements. Therefore, the optimal tensor for a given structure is the least squares solution 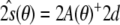 .
.
A convenient way to calculate the generalized inverse 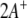 is singular value decomposition (Press et al. 1989), which in the context of dipolar coupling analysis has been proposed first by Losonczi et al. (1999).
is singular value decomposition (Press et al. 1989), which in the context of dipolar coupling analysis has been proposed first by Losonczi et al. (1999).
This rule follows directly from our model. If we fix the coordinates and the error in the joint posterior distribution (8), we obtain a five-dimensional Gaussian distribution for the tensor elements:
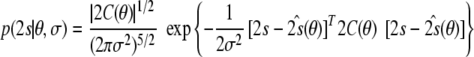 |
16 |
The conditional posterior probability, Eq. (16), is centered at the least squares estimate 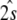 with covariance matrix
with covariance matrix 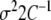 . It reaches its maximum at the least squares solution
. It reaches its maximum at the least squares solution 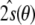 of Losonczi et al. In addition, we are able to make statements about the precision and correlation of the tensor elements, whereas methods without a firm probabilistic basis have to rely on some heuristic to compute an error estimate. Losonczi et al. proposed to add Gaussian noise to the measurements and then estimate the tensor elements by applying singular value decomposition to many realizations of such simulated data sets (1999). In this way, a distribution of possible tensor elements is obtained. However, it is not clear how much noise should be added, nor is there a sound theoretical basis for this procedure. In contrast, the conditional posterior distribution of the tensor elements in Eq. (16) follows unambiguously from the basic rules of probability theory.
of Losonczi et al. In addition, we are able to make statements about the precision and correlation of the tensor elements, whereas methods without a firm probabilistic basis have to rely on some heuristic to compute an error estimate. Losonczi et al. proposed to add Gaussian noise to the measurements and then estimate the tensor elements by applying singular value decomposition to many realizations of such simulated data sets (1999). In this way, a distribution of possible tensor elements is obtained. However, it is not clear how much noise should be added, nor is there a sound theoretical basis for this procedure. In contrast, the conditional posterior distribution of the tensor elements in Eq. (16) follows unambiguously from the basic rules of probability theory.
We generated alignment tensors from the conditional posterior probability (16) with coordinates set to those of the NMR structure 1D3Z (Cornilescu et al. 1998) and to those of the crystal structure 1UBQ (Vijay-Kumar et al. 1987). Figure 2 shows the distribution of the tensors. The finite widths of the posterior distributions again indicate that also for fixed coordinates the alignment tensor will remain imprecise to a degree depending on the quality of the data, their number and consistency, as well as the validity of the theoretical model (1).
Fig. 2.

Posterior histograms for the elements calculated from the X-ray structure 1UBQ (panel A) and from the NMR structure 1D3Z (panel B). In both panels, the upper row shows the tensor elements describing the first liquid crystal phase. The lower row shows the corresponding histograms for the second phase. The distribution of the tensor elements estimated along with the molecular coordinates (shown in Fig. 1 are plotted in grey for comparison)
Figure 3 illustrates the variability in the tensor elements in a different way. The molecular reference frames where reconstructed by spectral decomposition from the sampled alignment tensors (shown in Figs. 1 and 2). Structure ensembles were constructed by orienting the average structures into the sampled reference frames. The superposition of structures is not obtained by minimizing the coordinate RMSD; in this case the ensembles would be much tighter (cf. Fig. 7). The ensembles reflect the overall orientational ambiguity due to limitations of the dipolar coupling data. As already apparent from the posterior distributions of the tensor elements (Figs. 1 and 2), the variability is largest for the full simulation and smallest for 1D3Z, which is evident since 1D3Z was directly refined against the dipolar coupling data. Figure 4 shows a representation of the alignment tensors in the principal axis system. The agreement in the rhombic component is quite high, especially for the first phase. Whereas the distributions of the axial component show some differences, which is in accord with the observation that in the presence of “structural noise” the estimation of the axial component becomes problematic (Zweckstetter and Bax 2002).
Fig. 3.

Ensembles generated by applying the sampled rotations to the average structure of the full simulation (A), the crystal structure 1UBQ (B) and the NMR structure 1D3Z (C). The last three residues are poorly structured and therefore not shown for clarity
Fig. 7.

Structure ensembles calculated by statistical sampling from the full posterior distribution (left) and from the marginal posterior distribution (right)
Fig. 4.

Distributions of the axial and rhombic component for both liquid crystal phases. a, b: A and R distributions for the first phase, c, d: A and R distributions for the second phase. The filled histograms result from sampling the joint posterior probability and correspond to the distributions shown in Fig. 1. Distributions obtained from the crystal structure 1UBQ and from the NMR structure 1D3Z are shown as dashed and solid lines, respectively
Histogram method
Maximization of the conditional posterior probability of the Saupe tensor requires approximate knowledge of the coordinates and is therefore not applicable to a de novo structure determination. A way to deal with this situation is provided by the histogram method (Clore et al. 1998b) or its variants (Warren and Moore 2001). The histogram method estimates the axial and rhombic component of the Saupe tensor from dipolar couplings alone, without knowledge of a structure. The histogram method builds on the fact that for isotropically oriented bond vectors, the expected distribution of dipolar couplings has the same analytical form as a chemical shift powder pattern. The extrema and the maximum of the histogram of measured dipolar couplings therefore provide estimates of the axial and rhombic component. Considering the powder pattern as the probability for observing a dipolar coupling, Warren and Moore (2001) proposed a maximum likelihood version of this approach.
The maximum likelihood version of the histogram method follows from our model. The powder pattern is the distribution of dipolar couplings if the conformational degrees of freedom are averaged out. The assumption that the internuclear vectors are isotropically distributed is necessary to keep the averaging over conformational degrees of freedom analytically tractable. The Bayesian analog is the marginal posterior distribution
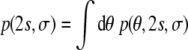 |
17 |
in which we integrate out the unknown conformational degrees of freedom. It can be proved that for the special case of neglected structural prior knowledge (β = 0 in Eq. 8) and perfect data (σ = 0) the marginal posterior is identical to the powder pattern. For real data, however, one would want to account for measurement errors as well as incorporate prior structural knowledge. The integration above is then no longer analytically tractable. A major advantage of Monte Carlo sampling over analytical marginalization is that we can relax the assumptions made by the histogram method.
This is illustrated in Fig. 5 showing the distribution of dipolar couplings for different combinations of prior knowledge and data. Also shown are the histogram of observed normalized dipolar couplings and the powder pattern obtained by maximum likelihood analysis. In the latter case, the axial and rhombic component are adapted such that the powder pattern exactly covers the observed range. Therefore, effectively only two data points, the extrema of the empirical histogram, determine the estimates of R and A. This can lead to unstable estimates in case of sparse and/or noisy data. The estimates calculated by Monte Carlo sampling are stable and capture the empirical RDC distribution better. The algorithm adapts the tensors and the data errors such that the simulated histogram maximally overlaps with the empirical distribution. The Bayesian histograms are smeared out at the limits and therefore not so much dependent on the exact values of the minimum and maximum observed coupling, because errors in the observed couplings are directly taken into account.
Fig. 5.

Comparison between the analytical powder pattern optimized via maximum likelihood (dashed line), the histogram of the normalized observed data (grey), and dipolar coupling distributions calculated by averaging over the conformational degrees of freedom using Monte Carlo sampling (solid line). Top row (a–c): analysis of the N–H couplings, only the distributions of these couplings are shown, middle (d–f): analysis of all couplings in the first liquid crystal phase, bottom row (g–i): analysis based on couplings from both phases, both data and histogram are shown for the first phase only. From left to right the following additional information is used in the simulation: left column (a, d, g): no additional data, no force field taken into account, middle column (b, e, h): force field used in the simulation, right column (c, f, i): force field and NOEs taken into account
Figure 6 shows the posterior probability of the axial and rhombic component for the different scenarios. The position of the posterior mode mainly changes in the axial component. This again is consistent with the observation that the axial component tends to be underestimated if variations in the coordinates are taken into account (Zweckstetter and Bax 2002). The rhombicity estimates are quite similar for the different settings and agree well with the estimates obtained from the crystal and the NMR structure. The shape of the posterior ellipsoids shows that with increasing number of data and prior knowledge also the tensor estimates become more precise. In almost all cases, the Bayesian posterior probabilities locate their main bulk of probability mass in the vicinity of the estimate obtained from the crystal structure 1UBQ. Only when NOEs are also taken into account, the posterior modes move towards the estimate obtained from the NMR structure 1D3Z. Maximum likelihood yields A = 11.7 Hz and R = 0.20 for all couplings in the first liquid crystal phase and A = 8.5 Hz and R = 0.26, which differs significantly from the values for the NMR and the crystal structure.
Fig. 6.

1σ-Ellipsoids of the posterior distributions for the axial and rhombic component in the first liquid crystal phase. The panels correspond to those shown in Fig. 5 The black ellipsoids indicate the 1σ-region of a two-dimensional Gaussian distribution that was fitted to the samples. Also shown as big grey dots are the axial and rhombic component obtained by maximum likelihood for all couplings observed in the first phase (ML) and for the N–H couplings only (ML) and by fitting the couplings to the NMR (1D3Z) and to the crystal structure (1UBQ)
Elimination of the alignment tensor
It is possible to eliminate the alignment tensor in the restraint energy and refine protein structures directly against observed dipolar couplings without any preanalysis (Moltke and Grzesiek 1999; Sass et al. 2001). To derive such a restraint energy, one minimizes the full residual, 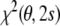 [Eq. (6)], with respect to both the conformational degrees of freedom and the tensor elements. The latter minimization can be done analytically, because
[Eq. (6)], with respect to both the conformational degrees of freedom and the tensor elements. The latter minimization can be done analytically, because 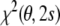 is quadratic in the tensor elements. The optimal conformation-dependent tensor is:
is quadratic in the tensor elements. The optimal conformation-dependent tensor is: 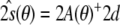 . After substituting this tensor into the full residual, one obtains the target function,
. After substituting this tensor into the full residual, one obtains the target function, 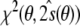 , which is equal to the residual
, which is equal to the residual 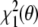 , Eq. (13), because
, Eq. (13), because 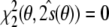 . That is, minimization of
. That is, minimization of 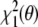 in conformation space will give the same results as minimization of
in conformation space will give the same results as minimization of 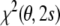 in joint structure-tensor space. The tensor-free target function has the advantage that it does not require knowledge of the Saupe tensor. The downside is that the calculation of the restraint energy and its gradient is quite involved and that analytical elimination of the alignment tensor may be impossible when additional prior knowledge is included. We point out that direct minimization of
in joint structure-tensor space. The tensor-free target function has the advantage that it does not require knowledge of the Saupe tensor. The downside is that the calculation of the restraint energy and its gradient is quite involved and that analytical elimination of the alignment tensor may be impossible when additional prior knowledge is included. We point out that direct minimization of 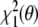 and iterative minimization of
and iterative minimization of 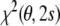 by repeated structure calculation and tensor fitting is equivalent—our Gibbs sampling algorithm is a probabilistic version of such an iterative scheme.
by repeated structure calculation and tensor fitting is equivalent—our Gibbs sampling algorithm is a probabilistic version of such an iterative scheme.
The probabilistic counterpart of Moltke’s and Grzesiek’s argument (1999) is to eliminate the tensor elements by integrating them out in the joint posterior probability. This integration can be done analytically, because the posterior probability is Gaussian in the tensor elements. The marginal posterior probability is
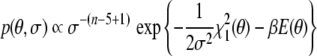 |
18 |
Note that the implicit estimation of the tensor elements consumes five data points, which is reflected in the reduced number of data in the exponent of σ when compared to Eq. (8). The maximum posterior estimate for θ is obtained by minimizing the negative logarithm of (18), which is identical to the target function proposed in (Moltke and Grzesiek 1999).
To demonstrate that both the full and the marginal posterior distribution [Eqs. (8) and (18)] convey the same information with regard to the protein structure, we also generated structures from p(θ,σ). Figure 7 displays structure ensembles from both simulations. The ensembles are virtually identical, and there is a high agreement in terms of accuracy, precision and quality.
Conclusions
We introduced a Bayesian probabilistic model to analyze RDC measurements. From a Bayesian perspective, existing heuristics to determine the alignment tensor, such as tensor fitting, the histogram method and tensor elimination all come under the same umbrella as special cases and approximations of a fully probabilistic treatment.
A fully probabilistic approach has the advantage of allowing for the incorporation of prior information (such as, for example, models described in Almond and Axelsen (2002) and Azurmendi and Bush (2002)), and the estimation of the reliability of the measured dipolar couplings as well as the assessment of the uncertainty of the alignment tensor and the coordinates. This is made possible through the simultaneous estimation of the protein structure and “nuisance parameters”, which in the present context are the tensor elements and the errors of the data sets. Although it is possible to eliminate these parameters analytically, we advice against doing so for several reasons: First, full flexibility with regard to the incorporation of prior knowledge is only maintained when the parameters are not eliminated analytically. For more advanced models, the marginalization integral may even not be analytically solvable. Second, to our experience, the joint posterior probability has better convergence properties. Third, the additional cost of estimating the nuisance parameters is negligible in comparison with updating the conformational degrees of freedom.
To apply our probabilistic model, it is not necessary to calculate structures by posterior sampling if the main interest is not so much the quantification of uncertainties. Hence, traditional structure determination based on restraint energy minimization can benefit from the insights presented here. A direct analog of the Gibbs sampling scheme would be an iterative maximization of the joint posterior probability (8). The individual updates maximize the conditional posterior probabilities, which can often be done analytically: The method of Losonczi et al. (1999) provides an update rule if the coordinates and the data error are given, how to estimate the error from a structural model is discussed in Habeck et al. (2006). Except for the treatment of the error parameters, such an iterative algorithm would be similar to the SCULPTOR approach (Hus et al. 2000).
Simultaneous optimization of all tensor elements is not routinely done during structure calculation. Often either the axial and rhombic components or the orientation of the alignment tensor is updated. Computationally, it is much simpler to estimate the full alignment tensor. But also in terms of quality, the structures seem to improve if the tensor is estimated during the structure calculation (Hus et al. 2000, 2001). Hus and Blackledge showed that the effects on the quality of the structure can be dramatic in the case of few measurements. A probabilistic approach has the additional advantage of providing error estimates and allowing for adaptive weighting of the RDC sets. We expect that, as in the case of NOE data (Rieping et al. 2005a), these features will help to improve structures calculated from sparse RDC data.
Acknowledgements
Wolfgang Rieping thanks the European Molecular Biology Organisation for financial support.
Footnotes
Because the tensor elements 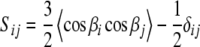 are directly related to the variance and correlation of the direction cosines,
are directly related to the variance and correlation of the direction cosines, 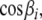 between the axes of the molecular reference frame and the static magnetic field (Bax et al. 2001), they are in principle restricted to certain physically reasonable ranges
between the axes of the molecular reference frame and the static magnetic field (Bax et al. 2001), they are in principle restricted to certain physically reasonable ranges 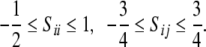 However, mainly for mathematical convenience we will work with an (improper) uniform prior for the tensor elements defined over the entire real axis.
However, mainly for mathematical convenience we will work with an (improper) uniform prior for the tensor elements defined over the entire real axis.
Contributor Information
Michael Habeck, Email: michael.habeck@tuebingen.mpg.de.
Wolfgang Rieping, Email: wolfgang.rieping@bioc.cam.ac.uk.
References
- Almond A, Axelsen JB (2002) Physical interpretation of residual dipolar couplings in neutral aligned media. J Am Chem Soc 124(34):9986–9987 [DOI] [PubMed]
- Azurmendi HF, Bush CA (2002) Tracking alignment from the moment of inertia tensor (TRAMITE) of biomolecules in neutral dilute liquid crystal solutions. J Am Chem Soc 124(11):2426–2427 [DOI] [PubMed]
- Bax A (2003) Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci 12:1–16 [DOI] [PMC free article] [PubMed]
- Bax A, Grishaev A (2005) Weak alignment NMR: a hawk-eyed view on biomolecular structure. Curr Opin Struct Biol 15:563–570 [DOI] [PubMed]
- Bax A, Kontaxis G, Tjandra N (2001) Dipolar couplings in macromolecular structure determination. Methods Enzymol 339:127–174 [DOI] [PubMed]
- Clore GM, Gronenborn AM, Tjandra N (1998a) Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J Magn Reson 131:159–162 [DOI] [PubMed]
- Clore GM, Bax A, Gronenborn AM (1998b) A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J Magn Reson 133:216–221 [DOI] [PubMed]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A (1998) Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc 120:6836–6837 [DOI]
- Delaglio F, Kontaxis G, Bax A (2000) Protein structure determination using molecular fragment replacement and NMR dipolar couplings. J Am Chem Soc 122:2142–2143 [DOI]
- Geman S, Geman D (1984) Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans PAMI 6(6):721–741 [DOI] [PubMed]
- Habeck M, Rieping W, Nilges M (2005a) Bayesian estimation of Karplus parameters and torsion angles from three-bond scalar coupling constants. J Magn Reson 177:160–165 [DOI] [PubMed]
- Habeck M, Nilges M, Rieping W (2005b) Bayesian inference applied to macromolecular structure determination. Phys Rev E 72:031912 [DOI] [PubMed]
- Habeck M, Nilges M, Rieping W (2005c) Replica-exchange Monte Carlo scheme for Bayesian data analysis. Phys Rev Lett 94:0181051–0181054 [DOI] [PubMed]
- Habeck M, Rieping W, Nilges M (2006) Weighting of experimental evidence in macromolecular structure determination. Proc Natl Acad Sci USA 103:1756–1761 [DOI] [PMC free article] [PubMed]
- Hus J-C, Marion D, Blackledge M (2000) De novo determination of protein structure by NMR using orientational and long-range order restraints. J Mol Biol 298:927–936 [DOI] [PubMed]
- Hus J-C, Marion D, Blackledge M (2001) Determination of protein backbone structure using only residual dipolar couplings. J Am Chem Soc 123:1541–1542 [DOI] [PubMed]
- Jaynes ET (2003) Probability theory: the logic of science. Cambridge University Press, Cambridge
- Jeffreys H (1946) An invariant form for the prior probability in estimation problems. Proc R Soc A 186:453–461 [DOI] [PubMed]
- Karplus M (1963) Vicinal proton coupling in nuclear magnetic resonance. J Am Chem Soc 85:2870–2871 [DOI]
- Lipsitz RS, Tjandra N (2004) Residual dipolar couplings in NMR structure analysis. Ann Rev Biophys Biomol Struct 33:387–412 [DOI] [PubMed]
- Losonczi JA, Andrec M, Fischer MWF, Prestegard JH (1999) Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson 138:334–342 [DOI] [PubMed]
- Moltke S, Grzesiek S (1999) Structural constraints from residual tensorial couplings in high resolution NMR without an explicit term for the alignment tensor. J Biomol NMR 15:77–82 [DOI] [PubMed]
- Press WH, Flannery BP, Teukolsky SA, Vetterling WT (1989) Numerical recipes: the art of scientific computing. Cambridge University Press, Cambridge
- Prestegard J (1998) New techniques in structural NMR—anisotropic interactions. Nat Struct Biol 5(Suppl):517–522 [DOI] [PubMed]
- Rieping W, Habeck M, Nilges M (2005a) Inferential structure determination. Science 309:303–306 [DOI] [PubMed]
- Rieping W, Habeck M, Nilges M (2005b) Modeling errors in NOE data with a lognormal distribution improves the quality of NMR structures. J Am Chem Soc 27:16026–16027 [DOI] [PubMed]
- Sass H-J, Musco G, Stahl SJ, Wingfield PT, Grzesiek S (2001) An easy way to include weak alignment constraints into NMR structure calculations. J Biomol NMR 21:275–280 [DOI] [PubMed]
- Saupe A, Englert G (1963) High-resolution nuclear magnetic resonance spectra of orientated molecules. Phys Rev Lett 11:462–464 [DOI]
- Tjandra N, Bax A (1997) Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278:1111–1114 [DOI] [PubMed]
- Tjandra N, Omichinski JG, Gronenborn AM, Clore GM, Bax A (1997) Use of dipolar H1-N15 and H1-C13 couplings in the structure determination of magnetically oriented macromolecules in solution. Nat Struct Biol 4:732–738 [DOI] [PubMed]
- Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH (1995) Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci USA 92:9279–9283 [DOI] [PMC free article] [PubMed]
- Vijay-Kumar S, Bugg CE, Cook WJ (1987) Structure of ubiquitin refined at 1.8 Å resolution. J Mol Biol 194(3):531–544 [DOI] [PubMed]
- Warren JJ, Moore PB (2001) A maximum likelihood method for determining and R for sets of dipolar coupling data. J Magn Reson 149:271–275 [DOI] [PubMed]
- Zweckstetter M (2006) Prediction of charge-induced molecular alignment: residual dipolar couplings at pH 3 and alignment in surfactant liquid crystalline phases. Eur Biophys J 35:170–180 [DOI] [PubMed]
- Zweckstetter M, Bax AJ (2000) Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc 122:3791–3792 [DOI]
- Zweckstetter M, Bax A (2002) Evaluation of uncertainty in alignment tensors obtained from dipolar couplings. J Biomol NMR 23:127–137 [DOI] [PubMed]
- Zweckstetter M, Hummer G, Bax A (2004) Prediction of charge-induced alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys J 86:3444–3460 [DOI] [PMC free article] [PubMed]