

Abstract
The present study was undertaken to shed light on the mechanism of the epimerization of cis-1,2,3-trisubstituted tetrahydro-β-carbolines into the trans isomers via a potential carbocationic intermediate at C(1). In order to study the pathway involved in C(1)–N(2) bond cleavage, the electronic character of the carbon atom at C-1 was altered by substitution of electron-rich and electron-poor phenyl rings at this position. This provided direct evidence of the effects of charge at the proposed site of the carbocationic intermediate. In this regard, a diverse set of 1-(phenyl substituted)-2-benzyl-3-ethoxycarbonyl-1,2,3,4-tetrahydro-β-carbolines has been synthesized via the Pictet–Spengler reaction by condensation of L-tryptophan derivatives with electron-poor and electron-rich aromatic aldehydes. The epimers involved in the isomerization mechanism were investigated by dynamic 1H and 13C NMR spectroscopic and X-ray crystallographic analyses. The kinetic studies, which involved conversion of cis diastereomers into their trans counterparts, were carried out in dilute TFA/CH2Cl2. The 1-(4-methoxyphenyl) cis diastereomer epimerized at a much faster rate into the corresponding trans diastereomer than the related 1-(4-nitrophenyl) cis diastereomer epimerized. These observations provide support for the carbocationic intermediate in the C(1)–N(2) scission process. The understanding of this epimerization process is of importance when Pictet–Spengler reactions are carried out under acidic conditions during the synthesis of indole alkaloids.
Introduction
The Pictet–Spengler reaction,1 which involves the cyclization of electron-rich aryl or heteroaryl groups onto imine or iminium ion electrophiles, has long been a standard method for the construction of both tetrahydroisoquinolines1 and tetrahydro-β-carbolines.2 These privileged scaffolds appear in a diverse array of biologically active compounds; both of these ring systems are popular choices for combinatorial libraries targeted at drug discovery.3 Recent advances in medicinal chemistry include potential implications for β-carbolines in mechanisms which operate in alcoholism.4,5 The enantiospecific Pictet–Spengler reaction, in particular, has stimulated widespread interest in both organic and medicinal chemistry6–18 and has resulted in the total synthesis of a number of natural products including (−)-alstophylline,19,20 (−)-macralstonine,21,22 ajmaline23 and (−)-raumacline.21,24 Ungemach et al.6 reported on the diastereospecific Pictet–Spengler reaction after condensation of Nb-benzyltryptophan methyl ester with aldehydes which resulted in the 100% stereoselective formation of trans-1,3-disubstituted diastereomers. Alternatively, several groups have extended the scope to examine the ratio of cis/trans isomers of the Pictet–Spengler reaction employing tryptophan derivatives with aldehydes.25,26 These studies conclusively indicated that the steric bulk of the incoming carbonyl compound, the substituents at the Nb-nitrogen (N2) position and the size of ester function play major roles in the diastereoselective formation of the trans isomer. Although progress has been made in regard to synthesis, interestingly, the mechanism of the cis to trans isomerization, in the presence of Brönsted acids, has not been fully defined. In efforts aimed at the investigation of the mechanism of the isomerization at C(1), a set of readily accessible electron-donating and electron-withdrawing aromatic aldehydes have been reacted with tryptophan ethyl ester to provide 1-phenyl substituted tetrahydro-β-carbolines (Scheme 1).
Scheme 1.

Synthesis of Cis and Trans Na-H, Nb-H Tetrahydro-β-carbolines via the Pictet–Spengler Reaction
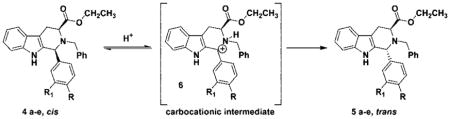
It is well documented that Na-methyl, Nb-benzyl tryptophan alkyl esters undergo the Pictet–Spengler reaction in nonacidic aprotic media with aldehydes of varied steric bulk to furnish the trans and cis diastereomers in an approximate ratio of 4:1, with the trans isomer predominating.25 Furthermore, the diastereomeric ratio changes to approximately 3:2 for Na-H, Nb-H tryptophan alkyl esters in both nonacidic and acidic media. Aldehydes as small as acetaldehyde provide the cis diastereomer in excess.25 These diastereomers are usually separated via silica gel chromatography; however, the separation remained difficult in many cases due to their close retention times. This is especially important in large scale reactions. On the other hand, the thermodynamically more stable trans isomer, an important chiral intermediate in the total synthesis of macroline/sarpagine/ajmaline indole alkaloids,27–31 could be exclusively obtained by epimerization of the cis stereoisomer or a mixture of the cis and trans isomers of the Nb-benzylated moieties under acidic conditions.3,32–35 On the basis of the elegant work of Joule et al. on reserpine,36 three potential intermediates, depicted in Figure 1, were considered for this epimerization process at C(1).
Figure 1.

Proposed intermediates in the cis to trans epimerization.
Among the three potential intermediates (6, 7, 8) depicted in Figure 1, the cis to trans epimerization process, which proceeds by protonation of the Nb-nitrogen atom with concomitant cleavage of the C(1)–N(2) bond followed by reclosure to the trans isomer, cation 6 is the most consistent with recent evidence.25,34,35,37 Importantly, intermediate 7 has been excluded on the basis of epimerizations carried out in CF3COOD.25 The carbocation 6, so generated, after rotation of the C-1/C-9a carbon–carbon bond, could recyclize to provide the more thermodynamically stable trans diastereomer.34,35 Recent results during the synthesis of alkoxy substituted indole alkaloids indicated that such an intermediate might be involved especially in the case of indoles substituted at C(4) or C(6) with methoxyl groups.31,38–41 This epimerization afforded 100% stereoselective formation of the trans diastereomer, specifically at C-1, under standard acidic conditions of TFA/CH2Cl2.25 Similar findings have been observed by Han et al.37 during a conformational study of cis and trans Na-methyl and Na-H substituted Nb-benzyl diesters. Evidence from simple kinetic studies is in agreement with the presence of the carbocationic intermediate 6 in these isomerizations. Furthermore, analysis of the conformations of the isomers by NMR spectroscopy provided a structural basis for the generation of carbocationic intermediate, 6, in the cis to trans isomerization (at C-1).37 It was felt that preparation of 1-phenyl substituted tetrahydro-β-carbolines from substituted benzaldehydes and tryptophan ethyl ester would provide a means to study the C(1)–N(2) scission process by varying the electronic character at C(1), the direct site of the proposed carbocation (see 6). Examination of the results described herein via kinetic data is in agreement with the generation of the carbocationic intermediate 6 as a key intermediate in this cis to trans epimerization process. This is, presumably, important with regard to Pictet–Spengler reactions carried out under acidic conditions in both the tetrahydro-β-carboline and tetrahydroisoquinoline series.
Results and Discussion
It is well accepted that the Pictet–Spengler condensation of Na-H, Nb-H tryptophan alkyl esters provides a mixture of cis and trans isomers.25,26 It is clear that the presence of a benzyl group on the Nb-nitrogen atom affects the diastereochemical outcome of the Pictet–Spengler reaction to provide the corresponding trans diastereomer, often stereospecifically under acidic conditions. In this regard, systematic variation of substituents which contain electron donating or electron withdrawing groups at C-1 should effect the stabilization of a carbocationic intermediate, consequently, affecting the rate of the epimerization process. In this vein, three experiments were devised which employed a Pictet–Spengler cyclization, followed by Nb-benzylation and then kinetic experiments on the cis to trans epimerization. The synthetic process employed to access the cis and trans isomers is depicted in Schemes 1, 2, and 3. The pure cis and trans isomers represented by 2 and 3, respectively, were prepared analogous to the method employed by Zhang20,21,24,42 for related Na-methyl tetrahydro-β-carbolines. First, the Pictet–Spengler cyclization of L-tryptophan ethyl ester (1) with substituted aryl aldehydes provided the optically active Na-H, Nb-H-1,2,3,4-tetrahydro-β-carbolines. However, in many cases the Pictet–Spengler reaction was carried out in benzene/TFA to prevent epimerization at C-3 when electron-rich aldehydes were employed.25,26
Scheme 2.

Nb-Benzylation of the Cis Diastereomers
Scheme 3.

Nb-Benzylation of the Trans Diastereomers
A number of aldehydes including 4-nitrobenzaldehyde, 3-nitrobenzaldehyde, 4-chlorobenzaldehyde, 4-tolualdehyde and 4-methoxybenzaldehyde were reacted with the free base of tryptophan ethyl ester. This afforded a series of 1-aryl substituted-3-ethoxycarbonyl-tetrahydro-β-carbolines. As expected, the process tolerated a diverse set of aromatic aldehydes. The Pictet–Spengler cyclization resulted in a mixture of cis and trans isomers which were then separated by careful flash column chromatography. Excellent yields of tetrahydro-β-carbolines were obtained upon stirring for 30 h with 4-nitro- and 3-nitrobenzaldehydes in nonacidic, aprotic media, providing a cis: trans ratio of 2:3. On the other hand, nonacidic aprotic media was less effective when electron-rich aldehydes were employed (Table 1). Very poor yields of tetrahydro-β-carbolines were observed and oftentimes epimerization occurred at the tryptophan chiral center.25,43 These reactions did not go to completion in 30 h and yields were less than 40% (Table 1). The yields could be improved on prolonged heating; however, the optical purity of the products was low (2e, −6.0) under these conditions. In order to effect cyclization, it was necessary to add TFA to the reaction medium to increase the rate of the Pictet–Spengler cyclization and to prevent epimerization of the chiral center in the intermediate imine.25 Each of these individual Pictet–Spengler reactions were repeated in the presence of 1–2 equiv of TFA which produced the tetrahydro-β-carbolines in 4–7 h in high optical purity. Indeed, the reactions cleanly provided the cis and trans diastereomers (2c–2e, cis; 3c–3e, trans) in reasonable yields. The cis (2c–2e) and trans (3c–3e) diastereomers prepared under acidic conditions were isolated, and the ratio of cis to trans diastereomer remained approximately 2:3, respectively. An overall increase in optical purity (2e, −14.5) was observed under acidic conditions. The successful formation of the desired tetrahydro-β-carbolines was established by 1H NMR, 13C NMR, and mass spectrometry. The stereochemical configuration of cis-1-(4-methoxylphenyl)-1,2,3,4-tetrahydro-β-carboline (2e) and trans-1-(4-methoxyphenyl)-1,2,3,4-teterahydro-β-carboline (3e) was confirmed as cis and trans by single crystal X-ray crystallography, respectively (Figures S1 and S2, Supporting Information).
Table 1.
Ratio of Cis and Trans Diastereomers of 1,3-Disubstituted Tetrahydro-β-carbolines
| diastereomer | R | R1 | nonacidic media cis:trans ratio | acidic media cis:trans ratio | yield [%]a cis + trans |
|---|---|---|---|---|---|
| 2a, 3a | NO2 | H | 48:52 | NA | 88b |
| 2b, 3b | H | NO2 | 43:57 | NA | 84b |
| 2c, 3c | Cl | H | 30:70 | 45:55 | 76c |
| 2d, 3d | CH3 | H | 38:62 | 40:60 | 78c |
| 2e, 3e | OCH3 | H | 33:67 | 45:55 | 82c |
Isolated yield.
Determined under aprotic conditions.
Determined under acidic conditions.
The Nb-alkylation of the Nb-H tetrahydro-β-carbolines (cis-2; trans-3) was achieved by stirring either the cis or trans diastereomer (individually) in the presence of Hunig’s base (N,N-diisopropylethylamine, DIPEA) and benzyl bromide in dry acetonitrile. These mixtures were heated at reflux for 8–24 h under an inert atmosphere (Schemes 3 and 4). Purification by rapid flash column chromatography gave the Na-H, Nb-benzyl-1,2,3-trisubstituted tetrahydro-β-carbolines (cis-4; trans-5). Epimerization was not observed in the alkylation. The benzylation step was also successful if a cis and trans mixture was employed as the starting material; however, separation of the diastereomers remained tedious due to the close Rf values (i.e., for 4a and 5a, TLC, silica gel, cis 0.50; trans 0.45, EtOAc/hexane). Therefore, the cis and trans isomers (2 and 3) were separated and individually alkylated with benzyl bromide. Attempts to achieve Nb-benzylation using bases other than Hunig’s base, such as triethylamine, pyridine, and Na2CO3, as well as in different solvents (DMSO, CH2Cl2, or dioxane) afforded only a trace of the desired tetrahydro-β-carbolines. Among the various solvents employed, CH3CN was the most effective. Assignment of the proton and carbon resonances of the cis (4) and trans (5) isomers was carried out on the basis of 1D and 2D NMR (COSY, HSQC, HMBC) experiments. The data are presented in Tables S1 and S2, respectively, and the spectra are provided in the Supporting Information. Inspection of the proton and carbon spectra of the individual diastereomers clearly demonstrated a marked difference between the cis and trans isomers, in particular, the proton resonances between δ 3.30 and 3.50 were quite distinct. Examination of the 1H NMR spectrum of the cis isomer (see 4a) indicated the presence of two distinct signals for the two nonequivalent, diastereotopic methylene protons of H4ax (δ 3.18, ddd, J = 15.6, 5.1, 1.0 Hz) and H4eq (δ 3.44, ddd, J = 15.6, 6.3, 1.3 Hz). Both protons appear as a significant doublet of doublets with a common geminal coupling constant of 15.6 Hz and an additional vicinal coupling constant of 6.3 Hz (3J H3–H4ax) and 5.1 Hz (3J H3–H4eq), respectively. The structures of the Nb-benzyl cis and trans diastereomers (4a and 5a) were confirmed by X-ray analysis (Figures S3 and S4, Supporting Information).
Scheme 4.

Retro Pictet–Spengler Process
The effect of the 4-substituted phenyl substituent at C(1) on the rate of the cis to trans isomerization in the Nb-benzyl series was studied, analogous to the method of Han.37 The pure Na-H, Nb-benzyl cis diastereomers, 4a–e were dissolved in dilute TFA in CH2Cl2 and allowed to stir at room temperature (Table 2). At 30 min intervals, an aliquot of the reaction mixture was removed and quenched with cold, dilute, mild base (Na2CO3). The concentrated sample was then taken up in CDCl3. The progress of the reaction was monitored by 1H NMR spectroscopy and the ratio of cis:trans diastereomers was determined by integration of the proton spectrum of the crude material. Analysis of the proton NMR spectra showed the collapse of individual doublets (H4a and H4b) in the cis compound with emergence of a doublet centered between the individual doublets for the trans isomer. The initial progress of the reaction, as indicated by TLC, illustrated the epimerization of the entire series had been completed in less than 4 h (Table 2). Analysis of the data in Table 2 indicated the 4-methoxy substituted cis isomer 4e epimerized at a more rapid rate than the corresponding 4-nitro cis analog (4a). More importantly, analysis of the optical rotations indicated that the optical activity had been retained.
Table 2.
Acid-Mediated Epimerization of Cis Diastereomers (4) into Trans 0Diastereomers (5)
|
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| approximate ratio of cis:trans isomersa |
|||||||||
| entry | R | R1 | epimerization process | initial | 0.5 h | 1 h | 1.5 h | 3 h | |
| 4a (cis) | NO2 | H | 4a → 5a | 100:0 | 78:22 | 35:55 | 14:86 | 6:94 | |
| 4b (cis) | H | NO2 | 4b → 5b | 100:0 | 70:30 | 40:60 | 25:75 | 0:100 | |
| 4c (cis) | Cl | H | 4c → 5c | 100:0 | 58:52 | 19:81 | 0:100 | ||
| 4d (cis) | CH3 | H | 4d → 5db | 100:0 | 2:98 | 0:100 | |||
| 4e (cis) | OCH3 | H | 4e → 5eb | 100:0 | 0:100 | ||||
| 5a (trans) | NO2 | H | 5a → 4ac | 0:100 | 0:100 | ||||
| 5e (trans) | OCH3 | H | 5e → 4ec | 0:100 | 0:100 | ||||
The cis to trans ratio was determined by analysis of the proton NMR spectrum of the crude reaction mixture.
The epimerization of 4d and 4e containing electron-donating substituents on the aromatic ring was completed in 30 min.
The trans to cis conversion was not observed in the last two reactions.
The cis isomer, 4a, −57.2 (c 1.50, CHCl3), epimerized to the trans diastereomer, 5a, with 100% conversion and a rotation of −73.6 (c 1.50, CHCl3). When the above rotation of the trans isomer from the epimerization process was compared to the trans isomer 5a originally obtained from the Nb-benzylation of 3a → 5a (Scheme 3), they were found to be identical. Analysis of this data further supported the proposed mechanism of C(1)–N(2) scission via the carbocationic intermediate 6. In addition, the optical rotation data eliminated the possibility of epimerization at C(3). Had epimerization occurred at C(3), the enantiomer of 5a would have been formed and the rotation would have been equal but opposite in sign, namely +73.6. In addition, comparison of the NMR and mass spectral data for the two trans isomers 5a confirmed they were identical. With the intent of determining the final equilibrium ratio of this epimerization, the trans diastereomer (5a) was heated in excess acid (at length). At no time was any of the cis isomer (4a) observed nor the corresponding Nb-benzyl tryptophan ethyl ester, 9, which would have arisen from a retro Pictet–Spengler/hydrolysis process (Scheme 4).35 In all of the epimerization experiments, 9 was not detected by TLC or NMR spectroscopy, arguing against a retro Pictet–Spengler mechanism.35 To investigate this trend further, the 3-nitro-, 4-chloro-and 4-methylphenyl substituted cis-tetrahydro-β-carbolines (4b, 4c, 4d) were employed in the kinetic study. Similar trends were observed in the above series (Table 2), and the conversions yielded optically active trans diastereomers.
The rate of epimerization was related to the amount of electron density on the 1-phenyl substituent. Examination of the data clearly indicated the 4-nitro- (4a), 3-nitro- (4b), and 4-chlorophenyl (4c) cis-Nb-benzyl substituted isomers epimerized at a slower rate than the corresponding 4-methyl- (4d) and 4-methoxyphenyl (4e) substituted analogs. Donation of electrons from the methoxyl group, presumably, stabilized the carbocationic intermediate at C(1) in 6 (see Figure 1), thereby stabilizing intermediate 10 via an oxonium ion contributor 11, as well as iminium ion form 12 (see Figure 2). Previous results with 6-methoxy indoles, pioneered by Zhang and Cox et al., observed stabilization of the carbocationic pathway (6, Figure 1) by electron donation of the methoxyl substituent on ring A.20,26,33,37 In the case of the 4-methylphenyl substituted tetrahydro-β-carboline 5d, the rate of isomerization of cis 4d to trans 5d also increased when compared to that of 4a → 5a. The 4-nitro group effectively retarded the acid induced epimerization of the cis isomer into the trans diastereomer as the nitro and chloro groups withdrew electron density from the phenyl group and destabilized the carbocationic intermediate (see Figure 3) which would result from C(1)–N(2) cleavage. This trend, the rate enhancement in the case of 4d and 4e as compared to the rate decrease with 4a and 4b, is in agreement with the mechanism of C(1)–N(2) scission via a carbocationic intermediate 6 (Scheme 5). A similar trend might be expected for the retro-Pictet–Spengler process as electron donation would stabilize iminium ion 8. However, if protonation at the indole-2 position was the rate determining step, an electron withdrawing group (i.e., NO2) would presumably increase the rate of protonation at the indole-2 position and the 4-methoxyphenyl substituent would decrease the rate (Scheme 4). Therefore, the rate of epimerization would have increased. This is the exact opposite of what was observed here. Further experimentation is in progress to address this issue.
Figure 2.

Stabilization of the carbocation intermediate via electron donating effects.
Figure 3.

Effect of the p-nitro group on the stability of the carbocationic intermediate.
Scheme 5.

Proposed Mechanism for Epimerization of the Cis Diastereomers into the Trans Isomers via Carbocationic Intermediate 6
Conclusion
The Pictet–Spengler reaction of L-tryptophan ethyl ester with aromatic aldehydes provided a mixture of the cis (2) and trans (3) diastereomers in approximately a 2:3 ratio, as expected.6,25,26 Chromatographic separation and Nb-benzylation of the pure cis and trans isomers, respectively, gave the required cis-Nb-benzyl tetrahydro-β-carbolines (see 4) and trans-Nb-benzyl diastereomers (see 5) for the study of the C(1)–N(2) scission process. This study focused on the stabilization of the carbocationic intermediate (see 6) in a more direct fashion than previously possible.35,37 It is clear that the 4-methoxyphenyl C(1) substituted cis isomer 4e isomerized at a more rapid rate than the corresponding isomerization of the 4-nitrophenyl analog cis-4a to trans-5a (see Table 2) under acidic conditions. The rate of isomerization of the cis-4-nitrophenyl isomer and the 4-methoxyphenyl cis diastereomer were in agreement with the ability to stabilize the incipient carbocation at C(1). During the epimerization experiments, 9 was not observed, which would be formed by a retro Pictet–Spengler/hydrolysis process of iminium intermediate 8.35 These results lend more direct support for the involvment of a carbocationic intermediate at C(1) in the C(1)–N(2) scission process, however, similar results might be observed for the retro Pictet–Spengler mechanism, depending on the rate determining step. Therefore, intermediate 8 cannot be entirely excluded. If protonation at C-2 in the retro Pictet–Spengler mechanism is the rate determining step, the 4-nitrophenyl substituent at C-1 would increase the rate of epimerization whereas the 4-methoxyphenyl substituent would decrease the rate. This is opposite to what was observed and argues against the retro Pictet–Spengler mechanism. Further work is in progress to better distinguish between these two reaction mechanisms. These results have implications with regard to the use of asymmetric Pictet–Spengler reactions in acidic media in the synthesis of natural products.12–14,19–22,24
Experimental Section
General Procedure for the Preparation of Both Cis and Trans Diastereomers (2a, 2b, 3a, 3b) via the Pictet–Spengler Reaction of Aromatic Aldehydes under Nonacidic Conditions
L-Tryptophan ethyl ester (1, 13.5 g, 0.058 mol) was added to a 500 mL, three-neck, round-bottom flask containing dry benzene (300 mL). The flask was attached to a Dean–Stark trap topped by a reflux condenser for azeotropic removal of water. The corresponding aldehyde (0.070 mol, 1.2 equiv) was added to the reaction mixture and was then heated to reflux for 18–24 h under argon, until all of the tryptophan ethyl ester, 1, was consumed as indicated by TLC (silica gel, hexanes:EtOAc, 3:1). The reaction mixture was cooled to rt, and the solvent was removed under reduced pressure. Examination of the TLC (silica gel, hexanes:EtOAc, 3:1) of the crude mixture indicated only two major spots along with some unreacted aldehyde. The residue was chromatographed on silica gel (gradient elution, EtOAc: hexane = 1:10, 2:10, 3:10, 4:10) to provide pure cis and trans diastereomers, 2a–b and 3a–b, respectively.
General Procedure for the Preparation of Both Cis and TransDiastereomers(2c,2d,2e,3c,3d,3e)viathePictet–Spengler Reaction of Aromatic Aldehydes under Acidic Conditions
L-Tryptophan ethyl ester (13.5 g, 0.058 mol) was added to a 500 mL, three-neck, round-bottom flask containing dry benzene (300 mL). The flask was attached to a Dean–Stark trap topped by a reflux condenser for azeotropic removal of water during the course of the process. To the reaction mixture was added trifluoroacetic acid (0.116 mol, 2.0 equiv), and this was followed by addition of the aldehyde in dry benzene (0.070 mol, 1.2 equiv). The mixture was heated to reflux for 4–8 h under argon, until all of the tryptophan ethyl ester, 1, was consumed as indicated by TLC (silica gel, hexanes:EtOAc, 3:1). The reaction mixture was then cooled to rt, and the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (300 mL) and washed with cold, saturated NaHCO3 (3 × 300 mL) to remove TFA. The organic layer was separated and dried (Na2SO4). Analysis by TLC (silica gel, hexanes:EtOAc, 3:1) of the crude oil indicated the presence of two major components along with some unreacted aldehyde. The residue was chromatographed on silica gel (gradient elution, EtOAc: hexane = 1:10, 2:10, 3:10, 4:10) to provide the pure cis and trans diastereoisomers, 2c–e and 3c–e, respectively.
General Procedure for the Alkylation of the Nb-H Tetrahydro-β-carbolines (4a–e, 5a–e)
To a solution of tetrahydro-β-carboline 2 or 3 (5.8 mmol, 1 equiv) in dry acetonitrile (10 mL) were added Hunig’s base (N,N-diisopropylethylamine, DIPEA) (8.7 mmol, 1.5 equiv) and benzyl bromide (7.0 mmol, 1.2 equiv). This mixture was heated to reflux for 8–24 h under argon. The solvent was removed under reduced pressure, and the residue was dissolved in cold EtOAc (10 mL) to remove the salt of the Hunig’s base which had precipitated. The solution was filtered, and the filtrate was dried (K2CO3) and concentrated in vacuo to yield a brown oil. Analysis of the crude material indicated the presence of a major component along with some unreacted benzyl bromide and starting material. The residue was purified on a silica gel column via flash chromatography (hexane:EtOAc 7:1) to yield the pure isomer 4 or 5, respectively, as an oil which was then triturated to form a pale yellow or white solid.
General Procedure for the Kinetic Epimerization Experiments (4a–e → 5a–e)
To a solution of pure diastereomer 4 (0.015 mol) in dry CH2Cl2 (5 mL) was added dilute TFA (a solution of 0.2 mL of TFA dissolved in 10 mL of CH2Cl2 was prepared; from this dilution only 0.2 mL was employed). The mixture was allowed to stir at rt and at 30 min intervals (as indicated in Table 1); 1 mL of reaction solution was removed and diluted with CH2Cl2 (25 mL) after which it was then quenched with cold aq 0.1 N NH4OH (2 × 50 mL). The organic layer was separated and dried (K2CO3). The solvent was removed under reduced pressure, and the residue was taken up in CDCl3 to determine the ratio of the two diastereoisomers by integration of the 1H NMR spectrum (see Table 2).
L-Tryptophan Ethyl Ester (1)
To a saturated solution of 100% ethyl alcohol (6 L) and HCl (anhydrous) was added L-tryptophan (500 g, 2.45 mol). The reaction vessel was brought to reflux overnight and then cooled to 5 °C after all the starting material was consumed as indicated by TLC (NH4OH, hexanes:EtOAc, 2:1, silica gel). A white precipitate formed which was filtered in vacuo and dried. Multiple crystallizations were performed on the filtrate, leaving the HCl salt of 1 (690 g, 2.44 mol, 99%). This material was extracted with cold, saturated NaHCO3 and CH2Cl2 (3 × 300 mL), when required, for the Pictet–Spengler reactions. 1H NMR (500 MHz, CDCl3): δ 7.63 (d, J = 7.8 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.19 (t, J = 7.3 Hz, 1H), 7.12 (t, J = 7.7 Hz, 1H), 7.05 (d, J = 2.2 Hz, 1H), 4.21–4.14 (m, 2H), 3.83 (dd, J = 7.6 Hz, 5.0 Hz, 1H), 3.30 (dd, J = 14.3 Hz, 4.9 Hz, 1H), 3.07 (dd, J = 14.0 Hz, 7.6 Hz, 1H), 1.26 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 175.6, 137.0, 128.5, 123.4, 122.4, 119.8, 119.2, 111.7, 111.5, 61.1, 55.2, 31.4, 15.0.
cis-1-(4-Nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (2a)
The general procedure was followed. L-Tryptophan ethyl ester (13.5 g, 0.058 mol) and 4-nitrobenzaldehyde (11.8 g, 0.078 mol) were used to obtain the product. Mp: 164 °C. : −74.5 (c 6.40, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.27 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 7.3 Hz, 1H), 7.47 (s, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.26–7.19 (m, 2H), 5.42 (s, 1H), 4.39–4.30 (m, 2H), 4.02 (dd, J = 10.9 Hz, 4.1 Hz, 1H), 3.32 (ddd, J = 14.9 Hz, 4.0 Hz, 1.9 Hz, 1H), 3.11–3.06 (m, 1H), 2.30 (br s, 1H), 1.42 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 172.7, 148.5, 148.3, 136.8, 133.5, 130.2, 129.7, 127.4, 123.0, 120.6, 119.1, 111.3, 109.9, 61.9, 58.6, 57.2, 25.9, 14.9. MS (EI) m/e (rel intensity): 365 (M+, 93), 292 (100), 264 (65), 243 (40), 217 (85), 169 (41), 144 (50). HRMS calcd for C20H19N3O4 (M + H)+ 365.1376; found, 365.1305. The product was used directly in a following step.
trans-1-(4-Nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (3a)
The general procedure was followed. L-Tryptophan ethyl ester (13.5 g, 0.058 mol) and 4-nitrobenzaldehyde (11.8 g, 0.078 mol) were used to obtain the product. Mp: 119 °C. : −86.4 (c 6.40, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.01 (d, J = 7.6 Hz, 2H), 7.46 (d, J = 7.0 Hz, 2H), 7.40 (d, J = 7.6 Hz, 2H), 7.18 (s, 1H), 7.14–7.07 (m, 2H), 5.67 (s, 1H), 4.48–4.40 (m, 2H), 4.20 (t, J = 5.4 Hz, 1H), 3.64 (dd, J = 5.7 Hz, 2.4 Hz, 1H), 3.52 (dd, J = 6.8 Hz, 2.9 Hz, 1H) 2.28 (br s, 1H), 1.84 (t, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 174.0, 149.8, 148.2, 136.7, 132.2, 129.8, 127.3, 124.4, 123.0, 120.3, 118.9, 111.4, 109.5, 61.7, 54.7, 53.1, 25.1, 14.6. MS (EI) m/e (rel intensity): 365 (M+, 100), 292 (57), 243 (33), 217 (73), 169 (32), 144 (33). HRMS calcd for C20H19N3O4 (M + H)+ 365.1376; found, 365.1394. The product was used directly in a following step.
cis-2-Benzyl-1-(4-nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (4a)
The general procedure was followed. 2a (2.0 g, 5.5 mmol), DIPEA (1.1 g, 8.2 mmol), and benzyl bromide (1.1 g, 6.4 mmol) were used to obtain the product. Mp: 166–170 °C. : −57.2 (c 1.50, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.17 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 7.0, 2H), 7.42 (s, 1H), 7.38–7.34 (m, 4H), 7.32–7.27 (m, 2H), 7.24–7.18 (m, 2H), 5.10 (s, 1H), 4.08 (d, J = 14.5 Hz, 1H), 4.01 (d, J = 14.5 Hz, 1H), 3.98–3.93 (m, 2H), 3.80–3.76 (m, 1H), 3.42 (dd, J = 6.4, 1.4 Hz, 1H), 3.19 (dd, J = 8.4, 3.8 Hz, 1H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.3, 148.7, 147.9, 138.0, 137.0, 131.8, 130.4, 129.7, 128.8, 127.9, 127.1, 123.9, 122.8, 120.3, 119.0, 111.4, 108.4, 61.2, 61.14, 61.09, 58.5, 23.9, 14.4. MS (EI) m/e (rel intensity): 455 (M+, 26), 382 (100), 265 (15), 217 (35). Anal. Calcd for C27H25N3O4: C, 71.19; H, 5.53; N, 9.22. Found: C, 70.91; H, 5.75; N, 9.25.
trans-2-Benzyl-1-(4-nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (5a)
The general procedure was followed. 3a (2.1 g, 5.8 mmol), DIPEA (1.1 g, 8.5 mmol), and benzyl bromide (1.2 g, 7.0 mmol) were used to obtain the product. Mp: 81 °C. : −73.6 (c 1.50, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.26 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H), 7.59 (d, J = 7.6 Hz, 1H), 7.41 (s, 1H), 7.36 (d, J = 6.6 Hz, 2H), 7.33 (d, J = 3.9 Hz, 3H), 7.25 (d, J = 8.0 Hz, 1H), 7.21–7.16 (m, 2H), 5.62 (s, 1H), 4.22–4.19 (m, 1H), 4.14–4.11 (m, 1H), 4.05 (d, J = 13.8 Hz, 1H), 3.98 (t, J = 4.8 Hz, 1H), 3.80 (d, J = 13.8 Hz, 1H), 3.30–3.28 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.2, 150.5, 148.3, 139.1, 137.1, 133.5, 130.1, 129.0, 127.9, 127.3, 124.5, 122.7, 120.2, 118.9, 111.4, 107.9, 60.9, 56.6, 55.2, 24.7, 14.7. MS (EI) m/e (rel intensity): 455 (M+, 25), 382 (57), 364 (100), 217 (42). HRMS calcd for C27H25N3O4 (M + H)+ 455.1845; found, 455.1784.
cis-1-(3-Nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (2b)
The general procedure was followed. L-Tryptophan ethyl ester (17.1 g, 0.074 mol) and 3-nitrobenzaldehyde (12.9 g, 0.085 mol) were used to obtain the product. Mp: 185 °C. : +77.06 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.36 (s, 1H), 8.29 (d, J = 8.2 Hz, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.64–7.60 (m, 2H), 7.41 (s, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.25–7.19 (m, 2H), 5.46 (s, 1H), 4.39–4.33 (m, 2H), 4.03 (dd, J = 11.2, 4.2 Hz, 1H), 3.32 (dd, J = 15.2, 4.0 Hz, 1H), 3.12–3.07 (m, 1H), 2.68 (br s, 1H), 1.42 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 172.9, 149.0, 143.8, 136.8, 135.3, 133.5, 130.4, 127.4, 124.1, 124.0, 122.9, 120.4, 118.9, 111.4, 110.2, 61.8, 58.6, 57.2, 25.9, 14.7. MS (EI) m/e (rel intensity): 365 (M+, 89), 292 (100), 264 (66), 243 (46), 217 (80), 169 (49), 144 (44). Anal. Calcd for C20H19N3O4: C, 65.74; H, 5.24; N, 11.50. Found: C, 65.59; H, 5.04; N, 11.27.
trans-1-(3-Nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (3b)
The general procedure was followed. L-Tryptophan ethyl ester (17.1 g, 0.074 mol) and 3-nitrobenzaldehyde (12.9 g, 0.085 mol) were used to obtain the product. Mp: 173 °C. : −71.27 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.29 (s, 1H), 8.23 (d, J = 8.2 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.64–7.60 (m, 2H), 7.57 (t, J = 8.0 Hz, 1H), 7.33–7.27 (m, 2H), 7.27–7.19 (m, 2H), 5.61 (s, 1H), 4.27–4.20 (m, 2H), 3.99 (t, J = 5.8 Hz, 1H), 3.35 (dd, J = 15.4, 5.6 Hz, 1H), 3.22 (dd, J = 17.0, 6.4 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 174.4, 149.1, 145.0, 136.8, 134.9, 132.2, 130.1, 127.4, 123.8, 123.6, 122.9, 120.3, 118.9, 111.4, 109.5, 61.6, 54.7, 53.2 25.0, 14.6. MS (EI) m/e (rel intensity): 365 (M+, 100), 336 (30), 292 (78), 243 (39), 217 (70), 169 (41), 144 (31). HRMS calcd for C20H19N3O4 (M + H)+ 365.1376; found, 365.1422. The product was used directly in a following step.
cis-2-Benzyl-1-(3-nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carbo-line-3-carboxylic Acid Ethyl Ester (4b)
The general procedure was followed. 2b (2.0 g, 5.5 mmol), DIPEA (1.1 g, 8.2 mmol), and benzyl bromide (1.1 g, 6.4 mmol) were used to obtain the product. Mp: 167–168 °C. : −48.2 (c 6.80, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.22 (s, 1H), 8.12 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.63 (d, J = 9.0 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.45 (s, 1H), 7.38 (d, J = 7.2 Hz, 2H), 7.35–7.32 (m, 2H), 7.29–7.26 (m, 2H), 7.25–7.20 (m, 2H), 5.11 (s, 1H), 4.06 (d, J = 14.7 Hz, 1H), 4.03 (d, J = 14.3 Hz, 1H), 3.99–3.93 (m, 2H), 3.82–3.78 (m, 1H), 3.45 (dd, J = 14.8, 6.6 Hz, 1H), 3.19 (dd, J = 15.2, 3.6 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.3, 148.6, 143.5, 138.1, 137.0, 135.7, 131.9, 129.8, 129.7, 128.8, 127.8, 127.1, 124.6, 123.3, 122.8, 120.3, 119.0, 111.4, 108.5, 61.5, 61.4, 61.3, 58.8, 24.0, 14.4. MS (EI) m/e (rel intensity): 455 (M+, 11), 382 (100), 352 (8), 264 (8), 217 (20). Anal. Calcd for C27H25N3O4: C, 71.19; H, 5.53; N 9.22. Found: C, 71.31; H, 5.62; N, 9.02.
trans-2-Benzyl-1-(3-nitrophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (5b)
The general procedure was followed. 3b (0.5 g, 1.4 mmol), DIPEA (0.27 g, 2.1 mmol), and benzyl bromide (0.28 g, 1.6 mmol) were used to obtain the product. Mp: 158–160 °C. : −54.0 (c 6.80, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.41 (s, 1H), 8.20 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.59 (d, J = 8.7 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 7.39–7.27 (m, 5H), 7.28 (d, J = 7.6 Hz, 1H), 7.23–7.18 (m, 2H), 5.64 (s, 1H), 4.23–4.18 (m, 1H), 4.15–4.12 (m, 1H), 4.05 (d, J = 13.8 Hz, 1H), 4.00 (t, J = 4.7 Hz, 1H), 3.83 (d, J = 13.8 Hz, 1H), 3.31 (d, J = 4.8 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 172.8, 149.1, 145.4, 139.2, 137.1, 135.2, 133.7, 130.2, 129.0, 127.8, 127.3, 124.1, 123.6, 122.6, 120.1, 118.9, 111.4, 107.7, 60.9, 56.8, 55.2, 24.8, 14.7. MS (EI) m/e (rel intensity): 455 (M+, 26), 382 (63), 364 (100), 217 (51). Anal. Calcd for C27H25N3O4: C, 71.19; H, 5.53; N, 9.22. Found: C, 71.16; H, 5.52; N, 8.83.
cis-1-(4-Chlorophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (2c)
The general procedure was followed. L-Tryptophan ethyl ester (13.5 g, 0.058 mol), 4-chlorobenzaldehyde (9.4 g, 0.067 mol), and TFA (13.2 g, 0.12 mol) were used to obtain the product. Mp: 140–142 °C. 1H NMR (500 MHz, CDCl3): δ 7.61 (d, J = 7.6 Hz, 1H), 7.42 (s, 1H), 7.41 (s, 3H), 7.32 (s, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.23–7.17 (m, 2H), 5.29 (s, 1H), 4.38–4.31 (m, 2H), 4.00 (dd, J = 7.1, 4.1 Hz, 1H), 3.28 (dd, J = 11.3, 3.8 Hz, 1H), 3.08–3.03 (m, 1H), 2.42 (br s, 1H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.1, 139.8, 136.7, 134.9, 134.6, 130.4, 129.6, 127.5, 122.6, 120.2, 118.8, 111.4, 109.7, 61.7, 58.5, 57.3, 26.1, 14.7. MS (EI) m/e (rel intensity): 354 (M+, 50), 281 (84), 253 (67), 218 (100), 169 (42), 144, (63). Anal. Calcd for C20H19ClN2O2: C, 67.70; H, 5.40; N, 7.89. Found: C, 67.75; H, 5.35; N, 7.75. HRMS calcd for C20H19ClN2O2 (M + H)+ 355.1213; found, 355.1217. The product was used directly in a following step.
trans-1-(4-Chlorophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (3c)
The general procedure was followed. L-Tryptophan ethyl ester (13.5 g, 0.058 mol), 4-chlorobenzaldehyde (9.4 g, 0.067 mol), and TFA (13.2 g, 0.12 mol) were used to obtain the product. Mp: 152 °C. 1H NMR (500 MHz, CDCl3): δ 7.73 (d, J = 5.0 Hz, 1H), 7.63 (d, J = 7.4 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.30–7.20 (m, 5H), 5.43 (s, 1H), 4.26–4.22 (m, 2H), 3.96 (t, J = 6.6 Hz, 1H), 3.25 (dd, J = 5.4, 1.0 Hz, 1H), 3.20–3.18 (m, 1H), 2.42 (br s, 1H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 174.0, 141.7, 136.7, 134.4, 133.2, 130.2, 129.3, 127.4, 122.6, 120.1, 118.8, 111.4, 109.1, 61.6, 54.7, 52.9, 25.1, 14.6. MS (EI) m/e (rel intensity): 354 (M+, 74), 281 (84), 253 (27), 217 (100), 169 (44), 144 (64). HRMS calcd for C20H19ClN2O2 (M + H)+ 355.1213; found, 355.1221. The product was used directly in a following step.
cis-2-Benzyl-1-(4-chlorophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (4c)
The general procedure was followed. 2c (1.5 g, 4.2 mmol), DIPEA (0.81 g, 6.2 mmol), and benzyl bromide (0.86 g, 5.0 mmol) were used to obtain the product. Mp: 156 °C. : +66.92 (c 1.04, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 8.2 Hz, 1H), 7.38–7.28 (m, 10H), 7.26 (d, J = 7.2 Hz, 1H), 7.22–7.16 (m, 2H), 4.98 (s, 1H), 4.10 (d, J = 14.8 Hz, 1H), 3.97 (d, J = 14.8 Hz, 1H), 3.92–3.88 (m, 2H), 3.82–3.78 (m, 1H), 3.42 (dd, J = 16.4, 3.0 Hz, 1H), 3.13 (dd, J = 16.0, 5.2 Hz, 1H), 1.18 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.6, 139.3, 138.4, 136.8, 134.2, 133.1, 131.1, 129.6, 129.0, 128.6, 127.6, 127.2, 122.5, 120.1, 118.9, 111.2, 108.0, 61.30, 61.28, 61.1, 57.6, 24.3, 14.3. MS (EI) m/e (rel intensity): 444 (M+, 38), 371 (100), 337 (23), 254 (40), 218 (53). Anal. Calcd for C27H25ClN2O2: C, 72.88; H, 5.66; N, 6.30. Found: C, 73.01; H, 5.80; N, 6.26. HRMS calcd for C27H25ClN2O2 (M + H)+ 455.1683; found, 445.1665.
trans-2-Benzyl-1-(4-chlorophenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (5c)
The general procedure was followed. 3c (0.63 g, 1.8 mmol), DIPEA (0.34 g, 2.7 mmol), and benzyl bromide (0.36 g, 2.1 mmol) were used to obtain the product. Mp: 172 °C. 1H NMR (500 MHz, CDCl3): δ 7.59 (d, J = 7.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.43 (s, 1H), 7.39–7.29 (m, 7H), 7.27 (d, J = 7.4 Hz, 1H), 7.21–7.15 (m, 2H), 5.51 (s, 1H), 4.22–4.17 (m, 1H), 4.14–4.11 (m, 1H), 4.00–3.98 (m, 2H), 3.87 (d, J = 13.8 Hz, 1H), 3.28–3.26 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.4, 141.3, 139.7, 137.0, 134.8, 134.2, 130.7, 129.4, 129.0, 128.9, 127.7, 127.5, 122.3, 119.9, 118.7, 111.3, 107.2, 60.8, 56.5, 54.8, 24.8, 14.7. MS (EI) m/e (rel intensity): 444 (M+, 63), 371, (85), 353 (100), 279 (34), 254 (49), 217 (91). HRMS calcd for C27H25ClN2O2 (M + H)+ 444.1605; found, 444.1509.
cis-1-(4-Methylphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (2d)
The general procedure was followed. L-Tryptophan ethyl ester (18.0 g, 0.077 mol), 4-methyl-benzaldehyde (10.7 g, 0.089 mol), and TFA (17.6 g, 0.15 mol) were used to obtain the product. Mp: 230–235 °C. 1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 8.2 Hz, 1H), 7.47 (s, 1H), 7.34–7.32 (m, 2H), 7.27–7.24 (m, 3H), 7.21–7.16 (m, 2H), 5.27 (s, 1H), 4.36–4.30 (m, 2H), 4.01 (dd, J = 11.0, 4.2 Hz, 1H), 3.28 (dd, J = 15.0, 4.2 Hz, 1H), 3.09–3.03 (m, 1H), 2.45 (s, 3H), 2.30 (br s, 1H), 1.40 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.2, 138.8, 138.2, 136.6, 135.4, 130.0, 129.0, 127.7, 122.3, 120.0, 118.7, 111.3, 109.3, 61.6, 58.7, 57.5, 26.2, 21.6, 14.7. MS (EI) m/e (rel intensity): 334 (M+, 100), 305 (26), 261 (93), 233 (74), 218 (88), 169 (36), 144 (41). Anal. Calcd for C21H22N2O2: C, 75.42; H, 6.63; N, 8.38. Found: C, 75.31; H, 6.52; N, 8.02. HRMS calcd for C21H22N2O2 (M + H)+ 335.1760; found, 335.1756. The product was used directly in a following step.
trans-1-(4-Methylphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (3d)
The general procedure was followed. L-Tryptophan ethyl ester (18.0 g, 0.077 mol), 4-methyl-benzaldehyde (10.7 g, 0.089 mol), and TFA (17.6 g, 0.15 mol) were used to obtain the product. Mp: 174 °C. : −48.19 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.62–7.59 (m, 2H), 7.29 (d, J = 7.6 Hz, 1H), 7.23–7.17 (m, 6H), 5.45 (s, 1H), 4.27–4.20 (m, 2H), 4.01 (t, J = 6.7 Hz, 1H), 3.32 (dd, J = 15.4, 4.8 Hz, 1H), 3.18 (dd, J = 15.4, 7.6 Hz, 1H), 2.58 (br s, 1H), 2.40 (s, 3H), 1.33 (t, J = 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 174.1, 139.6, 138.3, 136.6, 133.9, 129.8, 128.7, 127.6, 122.3, 119.9, 118.7, 111.3, 108.9, 61.4, 55.2, 53.0, 25.1, 21.5, 14.6. MS (EI) m/e (rel intensity): 334 (M+, 100), 305 (48), 261 (85), 246 (28), 232 (42), 218 (85), 169 (38), 144 (36). Anal. Calcd for C21H22N2O2: C, 75.42; H, 6.63; N, 8.83. Found: C, 75.21; H, 6.61; N, 8.17.
cis-2-Benzyl-1-(4-methylphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (4d)
The general procedure was followed. 2d (1.1 g, 3.3 mmol), DIPEA (0.6 g, 4.6 mmol), and benzyl bromide (0.66 g, 3.9 mmol) were used to obtain the product. Mp: 149–150 °C. 1H NMR (500 MHz, CDCl3): δ 7.58 (d, J = 7.4 Hz, 1H), 7.38–7.25 (m, 8H), 7.23 (d, J = 6.6 Hz, 1H), 7.20–7.14 (m, 4H), 4.97 (s, 1H), 4.13 (d, J = 14.9 Hz, 1H), 3.96 (d, J = 14.9 Hz, 1H), 3.89–3.81 (m, 3H), 3.42 (dd, J = 14.4, 7.6 Hz, 1H), 3.11 (dd, J = 15.2, 4.8 Hz, 1H), 2.40 (s, 3H), 1.17 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.7, 138.6, 138.2, 137.6, 136.8, 134.2, 129.7, 129.6, 128.4, 127.43, 127.36, 122.1, 119.9, 118.7, 111.2, 107.5, 62.1, 61.7, 61.0, 57.0, 24.9, 21.6, 14.3. MS (EI) m/e (rel intensity): 424 (M+, 26), 351 (100), 259 (21), 234 (47), 218 (57). HRMS calcd for C28H28N2O2 (M + H)+ 424.2151; found, 424.2149.
trans-2-Benzyl-1-(4-methylphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (5d)
The general procedure was followed. 3d (4.0 g, 0.012 mol), DIPEA (2.3 g, 0.018 mol), and benzyl bromide (2.4 g, 0.014 mol) were used to obtain the product. Mp: 174 °C. : −48.19 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.57 (d, J = 6.8 Hz, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.40 (s, 1H), 7.37–7.34 (m, 4H), 7.31–7.29 (m, 1H), 7.25–7.20 (m, 3H), 7.19–7.13 (m, 2H), 5.50 (s, 1H), 4.21–4.18 (m, 1H), 4.14–4.10 (m, 1H), 3.99 (t, J = 4.8 Hz, 1H), 3.97 (d, J = 13.8 Hz, 1H), 3.92 (d, J = 13.9 Hz, 1H), 3.27 (d, J = 6.4 Hz, 2H), 2.40 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.6, 140.1, 139.6, 138.2, 136.9, 135.7, 129.9, 129.3, 129.0, 128.8, 127.6, 127.5, 121.9, 119.7, 118.6, 111.2, 106.8, 61.1, 60.6, 56.5, 54.7, 24.9, 21.6, 14.7. MS (EI) m/e (rel intensity): 424 (M+, 78), 351 (93), 333 (100), 259 (41), 234 (36), 218 (67). Anal. Calcd for C28H28N2O2: C, 79.22; H, 6.65; N, 6.60. Found: C, 79.08; H, 6.83; N, 6.47.
cis-1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (2e)
The general procedure was followed. L-Tryptophan ethyl ester (18.5 g, 0.053 mol), 4-meth-oxybenzaldehyde (8.3 g, 0.061 mol), and TFA (12.0 g, 0.11 mol) were used to obtain the product. Mp: 134–138 °C. : −14.5 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 8.2 Hz, 1H), 7.52 (s, 1H), 7.36 (d, J = 8.6, 2H), 7.26 (d, J = 6.8, 1H), 7.22–7.16 (m, 2H), 6.95 (d, J = 8.6 Hz, 2H), 5.25 (s, 1H), 4.36–4.31 (m, 2H), 4.00 (dd, J = 11.2, 4.2 Hz, 1H), 3.87 (s, 3H), 3.28 (dd, J = 15.0, 4.2 Hz, 1H), 3.08–3.03 (m, 1H), 2.46 (br s, 1H), 1.40 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.3, 160.3, 136.6, 135.6, 133.3, 130.2, 127.7, 122.3, 120.0, 118.7, 114.7, 111.3, 109.4, 61.6, 58.5, 57.5, 55.8, 26.2, 14.7. MS (EI) m/e (rel intensity): 350 (M+, 100), 321 (29), 277 (80), 249 (71), 218 (62), 204 (37), 169 (39), 144 (39). Anal. Calcd for C21H22N2O3: C, 71.98; H, 6.33; N, 7.99. Found: C, 71.77; H, 6.37; N, 7.75.
trans-1-(4-Methoxyphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (3e)
The general procedure was followed. L-Tryptophan ethyl ester (18.5 g, 0.053 mol), 4-meth-oxybenzaldehyde (8.3 g, 0.061 mol), and TFA (12.0 g, 0.11 mol) were used to obtain the product. Mp: 190–192 °C. : −44.0 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.62 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 7.4 Hz, 1H), 7.25 (d, J = 8.6 Hz, 2H), 7.23–7.17 (m, 2H), 6.92 (d, J = 8.8 Hz, 2H), 5.43 (s, 1H), 4.28–4.20 (m, 2H), 4.00 (t, J = 6.1 Hz, 1H), 3.86 (s, 3H), 3.32 (dd, J = 15.4, 5.4 Hz, 1H), 3.17 (dd, J = 16.2, 7.0 Hz, 1H), 2.51 (br s, 1H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 174.2, 159.9, 136.6, 134.7, 134.1, 130.0, 127.6, 122.3, 119.9, 118.7, 114.5, 111.3, 108.9, 61.4, 55.8, 54.8, 53.0, 25.1, 14.6. MS (EI) m/e (rel intensity): 350 (M+, 100), 321 (55), 277 (71), 262 (33), 248 (50), 218 (55), 204 (43), 169 (44), 144 (36), 134 (26). Anal. Calcd for C21H22N2O3: C, 71.98; H, 6.33; N, 7.99. Found: C, 71.66; H, 6.46; N, 7.90.
cis-2-Benzyl-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (4e)
The general procedure was followed. 2e (1.5 g, 4.3 mmol), DIPEA (0.83 g, 6.2 mmol), and benzyl bromide (0.87 g, 5.1 mmol) were used to obtain the product. Mp: 131–133 °C. : −48.2 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.58 (d, J = 7.4 Hz, 1H), 7.37–7.30 (m, 7H), 7.28–7.24 (m, 2H), 7.20–7.14 (m, 2H), 6.90 (d, J = 9.0 Hz, 2H), 4.96 (s, 1H), 4.12 (d, J = 14.9 Hz, 1H), 3.95 (d, J = 14.9 Hz, 1H), 3.90–3.82 (m, 3H), 3.85 (s, 3H), 3.41 (dd, J = 14.4, 7.4 Hz, 1H), 3.10 (dd, J = 16.0, 5.0 Hz, 1H), 1.17 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.7, 159.8, 138.8, 136.8, 134.3, 132.6, 130.9, 129.6, 128.4, 127.39, 127.36, 122.2, 119.9, 118.8, 114.3, 111.2, 107.6, 61.8, 61.7, 61.0, 57.0, 55.8, 24.8, 14.3. MS (EI) m/e (rel intensity): 440 (M+, 37), 376 (100), 274 (22), 250 (42), 218 (37). Anal. Calcd for C28H28N2O3: C, 76.34; H, 6.41; N, 6.36. Found: C, 76.43; H, 6.46; N, 6.34.
trans-2-Benzyl-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic Acid Ethyl Ester (5e)
The general procedure was followed. 3e (1.5 g, 4.3 mmol), DIPEA (0.83 g, 6.2 mmol), and benzyl bromide (0.87 g, 5.1 mmol) were used to obtain the product. Mp: 150–151 °C. : −52.0 (c 2.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.57 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 11.4 Hz, 2H), 7.40 (s, 1H), 7.35 (d, J = 4.4 Hz, 4H), 7.31–7.28 (m, 1H), 7.25 (d, J = 7.0 Hz, 1H), 7.18–7.12 (m, 2H), 6.93 (d, J = 8.6 Hz, 2H), 5.48 (s, 1H), 4.21–4.16 (m, 1H), 4.13–4.10 (m, 1H), 3.97 (t, J = 4.9 Hz, 1H), 3.93 (d, J = 2.6 Hz, 2H), 3.85 (s, 3H), 3.25 (d, J = 5.2 Hz, 2H), 1.25 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 173.6, 159.9, 140.1, 136.9, 135.7, 134.6, 130.5, 129.0, 128.7, 127.6, 127.5, 121.9, 119.7, 118.6, 114.5, 111.2, 106.8, 60.64, 60.62, 56.5, 55.7, 54.6, 24.9, 14.7. MS (EI) m/e (rel intensity): 440 (M+, 73), 367 (88), 349 (100), 275 (23), 250 (39), 218 (37). HRMS calcd for C28H28N2O3 (M + H)+ 440.2100; found, 440.2091.
Supplementary Material
Acknowledgments
We thank Mr. Frank Laib for providing mass spectroscopic data and elemental analysis. X-ray crystallographic studies reported here were supported by NIDA under contract Y1-DA6002. We thank UW—Whitewater for providing the sabbatical support for H.J.K. and UW—Milwaukee and The Lynde and Harry Bradley Foundation for financial support. This paper is dedicated to Dr. Frank Ungemach and Mr. Joe Sandrin for their initial efforts in this area.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra of compounds 2a–e, 3a–e, 4a–e and 5a–e; COSY, HSQC, and HMBC 2D spectra for compounds 4a–e and 5a–e; X-ray crystallographic data and ORTEP plots for 2e, 3e, 4a, and 5a · HCl. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pictet A, Spengler T. Ber Dtsch Chem Ges. 1911;44:2030–2036. [Google Scholar]
- 2.Tatsui G. Yakugaku Zasshi. 1928;48:453–459. [Google Scholar]
- 3.Bunin BA, Dener JM, Kelly DE, Paras NA, Tario JD, Tushup SP. J Comb Chem. 2004;6:487–496. doi: 10.1021/cc0340776. [DOI] [PubMed] [Google Scholar]
- 4.Harvey SC, Foster KL, McKay PF, Carroll MR, Seyoum R, Woods JE, II, Grey C, Jones CM, McCane S, Cummings R, Mason D, Ma C, Cook JM, June HL. J Neurosci. 2002;22:3765–3775. doi: 10.1523/JNEUROSCI.22-09-03765.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster KL, McKay PF, Seyoum R, Milbourne D, Yin W, Sarma P, Cook JM, June HL. Neuropsychopharmacology. 2004;29:269–284. doi: 10.1038/sj.npp.1300306. [DOI] [PubMed] [Google Scholar]
- 6.Ungemach F, DiPierro M, Weber R, Cook JM. J Org Chem. 1981;46:164–168. [Google Scholar]
- 7.Czarnocki Z, Suh D, MacLean DB, Hultin PG, Szarek WA. Can J Chem. 1992;70:1555–1561. [Google Scholar]
- 8.Reddy MS, Cook JM. Tetrahedron Lett. 1994;35:5413–5416. [Google Scholar]
- 9.Waldmann H, Schmidt G, Henke H, Burkard M. Angew Chem, Int Ed Engl. 1995;34:2402–2403. [Google Scholar]
- 10.Yamada H, Kawate T, Matsumizu M, Nishida A, Yamaguchi K, Nakagawa M. J Org Chem. 1998;63:6348–6354. doi: 10.1021/jo980810h. [DOI] [PubMed] [Google Scholar]
- 11.Gremmen C, Willemse B, Wanner MJ, Koomen GJ. Org Lett. 2000;2:1955–1958. doi: 10.1021/ol006034t. [DOI] [PubMed] [Google Scholar]
- 12.Taylor MS, Jacobsen EN. J Am Chem Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]
- 13.Seayad J, Seayad AM, List B. J Am Chem Soc. 2006;128:1086–1087. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- 14.Wanner MJ, van der Haas RNS, de Cuba KR, van Maarseveen JH, Hiemstra H. Angew Chem, Int Ed Engl. 2007;46:7485–7487. doi: 10.1002/anie.200701808. [DOI] [PubMed] [Google Scholar]
- 15.Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klausen RS, Jacobsen EN. Org Lett. doi: 10.1021/ol802887h. [Online early access] Published Online: 2009. [DOI] [Google Scholar]
- 17.Sewgobind NV, Wanner MJ, Ingemann S, de Gelder R, van Maarseveen JH, Hiemstra H. J Org Chem. 2008;73:6405–6408. doi: 10.1021/jo8010478. [DOI] [PubMed] [Google Scholar]
- 18.Shi XX, Liu SL, Xu W, Xu YL. Tetrahedron: Asymmetry. 2008;19:435–442. [Google Scholar]
- 19.Liao X, Zhou H, Yu J, Cook JM. J Org Chem. 2006;71:8884–8890. doi: 10.1021/jo061652u. [DOI] [PubMed] [Google Scholar]
- 20.Zhang LH, Cook JM. J Am Chem Soc. 1990;112:4088–4090. [Google Scholar]
- 21.Fu X, Cook JM. J Am Chem Soc. 1992;114:6910–6912. [Google Scholar]
- 22.Trudell ML, Cook JM. J Am Chem Soc. 1989;111:7504–7507. [Google Scholar]
- 23.Li J, Wang T, Yu P, Peterson A, Weber R, Soerens D, Grubisha D, Bennett D, Cook JM. J Am Chem Soc. 1999;121:6998–7010. [Google Scholar]
- 24.Fu X, Cook JM. J Org Chem. 1993;58:661–672. [Google Scholar]
- 25.Czerwinski KM, Cook JM. In: Advances in Heterocyclic Natural Products Synthesis. Pearson W, editor. Vol. 3. JAI Press; Greenwich, CT: 1996. pp. 217–277. [Google Scholar]
- 26.Cox ED, Cook JM. Chem Rev. 1995;95:1797–1842. [Google Scholar]
- 27.Bi YZ, Zhang LH, Hamaker LK, Cook JM. J Am Chem Soc. 1994;116:9027–9041. [Google Scholar]
- 28.Yu P, Wang T, Li J, Cook JM. J Org Chem. 2000;65:3173–3191. doi: 10.1021/jo000126e. [DOI] [PubMed] [Google Scholar]
- 29.Wang T, Cook JM. Org Lett. 2000;2:2057–2059. doi: 10.1021/ol000095+. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Deschamps JR, Cook JM. Org Lett. 2002;4:3339–3342. doi: 10.1021/ol020101x. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Yin W, Zhou H, Cook JM. Org Lett. 2007;9:3491–3494. doi: 10.1021/ol071220l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen TE, Meldal M. J Org Chem. 2004;69:3765–3773. doi: 10.1021/jo049918p. [DOI] [PubMed] [Google Scholar]
- 33.Zhang LH, Gupta AK, Cook JM. J Org Chem. 1989;54:4708–4712. [Google Scholar]
- 34.Cox ED, Li J, Hamaker LK, Yu P, Cook JM. Chem Commun. 1996:2477–2478. [Google Scholar]
- 35.Cox ED, Hamaker LK, Li J, Yu P, Czerwinski KM, Deng L, Bennett DW, Cook JM, Watson WH, Krawiec M. J Org Chem. 1997;62:44–61. doi: 10.1021/jo951170a. [DOI] [PubMed] [Google Scholar]
- 36.Gaskell AJ, Joule JA. Tetrahedron. 1967;23:4053–4063. doi: 10.1016/s0040-4020(01)97916-5. [DOI] [PubMed] [Google Scholar]
- 37.Han D, Foersterling FH, Deschamps JR, Parrish D, Liu X, Yin W, Huang S, Cook JM. J Nat Prod. 2007;70:75–82. doi: 10.1021/np060391g. [DOI] [PubMed] [Google Scholar]
- 38.Liao X, Zhou H, Wearing X, Ma J, Cook JM. Org Lett. 2005;7:3501–3504. doi: 10.1021/ol051208y. [DOI] [PubMed] [Google Scholar]
- 39.Yu J, Wearing X, Cook JM. J Am Chem Soc. 2004;126:1358–1359. doi: 10.1021/ja039798n. [DOI] [PubMed] [Google Scholar]
- 40.Yu J, Wang T, Wearing X, Ma J, Cook JM. J Org Chem. 2003;68:5852–5859. doi: 10.1021/jo030116o. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Deschamps JR, Cook JM. Org Lett. 2002;4:3339–3342. doi: 10.1021/ol020101x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang LH, Bi Y, Yu F, Menzia G, Cook JM. Heterocycles. 1992;34:517–547. [Google Scholar]
- 43.Bailey PD. Tetrahedron Lett. 1987;28:5181–5184. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.