

Abstract
Monomethylarsonous acid (MMAIII), a trivalent metabolite of arsenic, is highly cytotoxic and recent cell culture studies suggest it might act as a carcinogen. The general consensus of studies indicates the cytoxicity of MMAIII is a result of increased levels of reactive oxygen species (ROS). A longstanding relationship between arsenic and selenium metabolism has led to the use of selenium as a supplement in arsenic exposed populations, however the impact of organic arsenicals (methylated metabolites) on selenium metabolism is still poorly understood. In this study we determined the impact of exposure to MMAIII on the regulation of expression of TrxR1 and its activity using a primary lung fibroblast line, WI-38. The promoter region of the gene encoding the selenoprotein thioredoxin reductase 1 (TrxR1) contains an antioxidant responsive element (ARE) that has been shown to be activated in the presence of electrophilic compounds. Results from radiolabeled selenoproteins indicate exposure to low concentrations of MMAIII resulted in increased synthesis of TrxR1 in WI-38 cells, and lower incorporation of selenium into other selenoproteins. MMAIII treatment led to increased mRNA encoding TrxR1 in WI-38 cells, while lower levels of mRNA coding for cellular glutathione peroxidase (cGpx) were detected in exposed cells. Luciferase activity of TrxR1 promoter fusions increased with addition of MMAIII, as did expression of a rat quinone reductase (QR) promoter fusion construct. However, MMAIII induction of the TRX1 promoter fusion was abrogated when the ARE was mutated, suggesting that this regulation is mediated via the ARE. These results indicate that MMAIII alters the expression of selenoproteins based on a selective induction of TrxR1, and this response to exposure to organic arsenicals requires the ARE element.
Keywords: Monomethylarsonous acid, thioredoxin reductase, antioxidant response element, arsenic, selenium, carcinogenesis
Introduction
Arsenic exposure is a worldwide public health concern. Mammals metabolize arsenic primarily in the liver by a series of methylation reactions (Styblo et al., 2000; Thomas et al., 2004; Li et al., 2005; Thomas et al., 2007). One of these methylated metabolites, MMAIII, has been shown to contribute to carcinogenesis, and exposure to MMAIII has led to mitogenic signaling critical to transformation (Bredfeldt et al., 2006; Eblin et al., 2007; Eblin et al., 2008). MMAIII has been shown to be the most cytotoxic of the trivalent arsenicals (Petrick et al., 2000), and has also been shown to cause lipid peroxidation, protein carbonylation, and oxidative DNA damage in cell culture studies (Wang et al., 2007a). MMAIII exposure to cells in culture induced hyperproliferation, anchorage-independent growth, and tumorigenicity in an immortalized human urothelial cell line (Bredfeldt et al., 2006). The results from this study led to a suggestion by the authors that MMAIII be classified as a carcinogen.
Selenocysteine is the 21st amino acid and is encoded by a stop codon, UGA. Selenoprotein synthesis is a complex process involving a myriad of proteins required for insertion of selenium into the final selenoprotein product (see (Papp et al., 2007) for a thorough review). One of the factors required for selenoprotein synthesis is the selenocysteine insertion sequence (SECIS), a stem-loop structure in the 3′ untranslated region of the mRNA (Berry et al., 1993). Another protein involved is SECIS binding protein 2 (SBP2). SBP2 is required for efficient selenoprotein synthesis, and may help to recruit the ribosome to the UGA codon (Copeland et al., 2000). Under selenium deficient conditions, some selenoproteins are preferentially produced over others in what is known as the selenoprotein hierarchy (Behne et al., 1988). At the top of this hierarchy is thioredoxin reductase 1 (TrxR1) and phospholipid hydroperoxide glutathione peroxidase (PHGPx). Other selenoproteins such as cellular glutathione peroxidase (cGPx) are produced in selenium replete conditions (Berry, 2005). The mechanism of this hierarchy is beginning to be understood at the molecular level. Because it uses a stop codon, selenoprotein-coding mRNAs are sensitive to nonsense-mediated decay (NMD) (de Jesus et al., 2006). NMD is in part determined by the introns present prior to the UGA codon. Another important factor is the efficiency of binding of SBP2 to the SECIS element. SBP2 has been shown to prefer certain SECIS elements over others (Low et al., 2000). Thus, SBP2 likely has a key role in the regulation of this hierarchy of expression of selenoproteins.
Thioredoxin reductase (TrxR) has three primary isoforms. Expression of cytosolic selenoprotein TrxR1 is regulated at the transcriptional level through an antioxidant response element (ARE) in its promoter (Hintze et al., 2003b). The ARE target sequence allows transcription of TrxR1 to be controlled by the Nrf2/Keap1 system. Many electrophilic compounds can induce the Nrf2/Keap1 response. In particular, it was found that sulforaphane induced transcription of the gene encoding TrxR1 through this system (Hintze et al., 2003b; Zhang et al., 2003). TrxR1 is expressed at higher levels in many tumors derived from breast, thyroid, prostate, liver, malignant melanoma, and colorectal tissue, as compared to normal tissues (Berggren et al., 1996; Gladyshev et al., 1998). Cytosolic thioredoxin (Trx), the primary substrate of TrxR1, is required for DNA synthesis, and is thus integral to cell proliferation (Laurent et al., 1964). Ras oncogene activation has recently been shown to trigger activation of elevated levels of TrxR1, leading to a proliferative phenotype (Yoo et al., 2007). It has been suggested that TrxR1 not only be a biomarker for cancer, but a potential target for cancer therapy (Arner and Holmgren, 2006). Several recent studies have shown that trivalent arsenicals are potent inhibitors of TrxR (Holmgren, 1977; Lin et al., 2001; Lu et al., 2007), and one of the most widely used therapies for acute promyelocytic leukemia is arsenic trioxide (ATO). ATO has been shown to act both as a potent inhibitor of TrxR1 (Lu et al., 2007) and to block selenium metabolism (Talbot et al., 2008), although it is not yet known whether these properties are underlying the molecular mechanism of the drugs efficacy for cancer treatment.
Inorganic arsenic and selenium are known to interact in what is known as the mutual sparing effect, first described by Moxon during studies of seleniforous grains (Moxon, 1938). This mutual sparing effect has also been demonstrated in animal and cell culture studies in which levels of inorganic selenium and arsenic were varied (Levander, 1977; Styblo and Thomas, 2001). A key finding in the interaction of selenium and arsenic was the discovery of the seleno-bis(S-glutathionyl) arsinium ion (Gailer et al., 2002a; Gailer et al., 2002b). This was shown to be excreted in the bile of rabbits treated with selenite and arsenate or arsenite (Gailer et al., 2002). Recently we analyzed the affect of trivalent arsenicals on selenoprotein synthesis using a keratinocyte cell culture model (Ganyc et al., 2007). It was found that arsenite decreased selenium incorporation into selenoproteins. In contrast, MMAIII induced selenium incorporation into TrxR1, while decreasing selenium incorporation into smaller selenoproteins, such as cGPx. Currently, there is no knowledge of a selenium and MMAIII complex formed, thus it appears that exposure to MMAIII leads to changes in the expression pattern of the genes encoding selenoproteins. To further understand the mechanism in this study we investigate the mechanism by which MMAIII affects selenoprotein synthesis using a primary lung fibroblast model (WI-38). The data reveal that exposure to MMAIII leads to significant increases in the mRNA encoding TrxR1 at the expense of cGpx. This increase expression requires the ARE element, suggesting that the differential expression of selenoproteins in response to MMAIII is due to activation of the Nrf2 pathway.
Materials and Methods
Materials
Sodium selenite was obtained from Acros Organics (Geel, Belgium). Radioisotope 75Se was purchased from University of Missouri Research Reactor (MURR, Columbia, MO). MMAIII was obtained from the laboratory of Dr. William Cullen (University of British Columbia, Vancouver, Canada).
Cell culture
To determine the response to exposure to MMAIII within a target tissue (lung) a normal lung fibroblast (WI-38) culture was obtained from the American Type Culture Collection (Manassas, VA). Cells were cultivated in Eagle’s Minimum Essential Medium (EMEM) with Earle’s balanced salt solution, non-essential amino acids, sodium pyruvate, 10% FBS, 50 μg/mL streptomycin, and 50 IU/mL penicillin. Culture monolayers were maintained at 37°C, in a humidified atmosphere containing 5% CO2. Population doublings of WI-38 cells were determined as previously described (Hayflick, 1985). No cultures beyond a population doubling of 34.0 were used in this study to avoid the potential impact of cellular senescence.
Cell viability assay
Cells were plated at a concentration of 5,000 per well in 96-well culture dishes, and treated in triplicate with varying concentrations of MMAIII; 0.2, 0.5, 1, 3, 6, 9, 12, 15, 18, 21, 24, 28 μM. After 24 hours of incubation, the tetrazolium dye, 3-(4,5 dimethylthiazol-2-yl)- 2,5-diphenyl tetrazolium bromide (MTT) (Amresco, Solon, Ohio) was added to a final concentration of 1.2 mM and cells were subsequently incubated for 4 hours at 37°C. The formazan product of the dye was solubilized by addition of SDS (10%) and HCl (5 mM) followed by overnight incubation at 37°C. Absorbance of the reduced MTT was measured at 570 nm using a SpectraMax 190 spectrophotometer (Molecular Devices, Sunnyvale, CA). Cell viability was determined by dividing the absorbance of treated samples to untreated controls and reported as a percentage with error being the standard deviation from triplicate wells.
Radioisotope labeling to follow selenoprotein synthesis
Cells were cultured in 6-well tissue culture plates and treated at a density of 70% confluence. WI-38 cells were treated with 0, 0.2, 1, or 2 μM MMAIII in triplicate wells for 24 hours. In all wells radioisotope selenium (2 μCi of 75Se), in the form of selenite (10 nM), was added to follow the metabolism of selenium.
Cells were harvested using trypsin-EDTA, collected by centrifugation, and subsequently washed with Dulbecco’s modified phosphate buffered saline (DPBS). Cell pellets were resuspended in 200 μL of lysis buffer (50 mM tricine, pH 8.0, 0.1 mM benzamidine, 0.5 mM EDTA, and 1 mM DTT) and lysed by sonication for 6–8 sec using a Model 100 sonic dismembrator (Thermo Fisher Scientific, Pittsburgh, PA) at a power setting of 4 watts. Crude cell lysates were obtained by centrifugation at 11,000 rpm for 7 minutes at 4°C. Radiolabeled selenium was analyzed in lysates using a Wallac Wizard Gamma Counter, Model 1470 (PerkinElmer, Wellesly, MA). Protein concentration was determined by Bradford assay (Bradford, 1976) with bovine serum albumin used to generate a standard curve. For visualization of labeled selenoproteins 25 μg of protein from crude cell extracts was separated by SDS-PAGE (15%). The gel was stained using Imperial Protein Stain (Pierce, Rockford, IL) and subsequently dried prior to exposure to a phosphor screen. Labeled proteins were analyzed using a Storm Molecular Dynamics phosphorimager (Sunnyvale, CA) and densitometry was carried out using ImageQuant software.
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR)
WI-38 cells were cultured in 25 cm2 flasks, and grown to approximately 70% confluence. Cells were treated with 0, 0.2, or 2 μM MMAIII. Cells were harvested as described above with the exception that DPBS was supplemented with diethylpyrocarbonate (DEPC, 0.1%). Cells were resuspended in DPBS containing DEPC and RNA was isolated using a ChargeSwitch total RNA cell kit (Invitrogen, Carlsbad, CA). RNA was quantified by UV-visible spectrophotometry at 260 nm using an Agilent 8453 UV-visible spectrophotometer (Agilent Technologies, Santa Clara, CA). Complementary DNA (cDNA) was synthesized from 0.5 μg of RNA using an iScript cDNA synthesis kit (BioRad, Hercules, CA).
Oligonucleotides utilized for real-time RT-PCR were as follows (listed forward then reverse): TrxR1; 5′-AGCTCAGTCCACCAATAGTGA-3′ and 5′-GGTATTT TTCCAGTCTTTTCAT-3′; HO-1; 5′-GTCTTCGCCCCTGTCTACTTC-3′ and 5′-CTGGGCAATCTTTTTGAGCAC-3′; cGPx; 5′-GGACTACACCCAGATGAA-3′ and 5′-CAAGGTGTTCCTCCCTCGTAG-3′; β-actin; 5′-CATGTACGTTGCTATCCA-3′ and 5′-CTCCTTAATGTCACGCACGAT-3′. Real time PCR reactions were carried out using a Bio-Rad I-Cycler (Bio-Rad). Each reaction was a total volume of 25 μL consisting of SYBRgreen supermix (Bio-Rad) with each oligonucleotide at a concentration of 0.2 mM, forward and reverse primers (at a concentration of 0.2 μM each), and 5 μL of 1:100 diluted cDNA. Reaction conditions were as follows; one cycle at 95.0°C for 3 minutes, followed by 40 cycles of 95.0°C for 10 sec, and 55.0°C for 45 sec. With the cGPx-specific primers the following reaction conditions were used; a single cycle at 94.0°C for 3 minutes, subsequent 40 cycles of 94.0°C for 30 sec, 55°C for 30 sec, and 70.0°C for 30 sec. Melt curves and agarose gel electrophoresis were used to verify formation of a single product. Efficiency of amplification of each primer pair was calculated using a 10-fold dilution series with untreated cDNA. The Pfaffl method (Pfaffl, 2001) was used to determine the relative expression (reported as fold increase or decrease) for each of the mRNAs of interest using β-actin as an internal standard.
TrxR Activity Assays
WI-38 cells were cultured in 75 cm2 flasks to generate sufficient material for TrxR enzyme assays. Cells were treated with 0, 0.2, or 2 μM MMAIII for 48 hours before harvesting as described above. Cells pellets were resuspended in 500 μL of lysis buffer (5 mM potassium phosphate pH 7.4, 0.5 mM EDTA). Cells were lysed by sonication as described above followed by centrifugation (10,000 × g) for 7 min at 4 °C to yield crude cell extracts. After protein determination, cells extracts were diluted in a buffer consisting of 10 mM potassium phosphate pH 7.4, 154 mM potassium chloride, and 1 mg/mL of bovine serum albumin (BSA) to a final concentration of 1 mg/mL. TrxR assays were carried out as previously described (Smith and Levander, 2002) using 50 μg of cell extract to start the reaction. Auranofin was used to treat cell extracts (20 minutes) to determine the gold-inhibited activity that is subtracted from the total reductase activity. Reduction of DTNB was followed at a wavelength of 412 nm at 37 °C for 3 minutes taking a reading every 15 seconds using a SpectraMax 190 spectrophotometer (Molecular Devices). Activity of TrxR is reported as nmol/min/mg of protein as previously described (Smith and Levander, 2002).
Promoter fusion luciferase assays
Luciferase promoter fusion constructs of human wild-type TrxR1, mutant (Nrf2 binding site mutant) TrxR1, and rat quinone reductase (QR) have been previously described (Hintze et al., 2003b). Cells were seeded in 24-well tissue culture plates at 60,000 per well in 500 μL of EMEM. Transfection complexes were prepared with either 1 μg (TrxR1 or TrxR1 mutant construct) or 2 μg (for QR construct) of plasmid DNA. For formation of transfection complexes 7.5 μL Superfect reagent (Qiagen, Valencia, CA) was added per μg of plasmid DNA, 540 μL Optipro serum free medium (Invitrogen), and 2.5 ng of a Renilla luciferase internal control plasmid pRL-SV40 (Promega, Madison, WI) were mixed, added to cells and incubated for 3 hours. The culture medium was then removed and MMAIII was added in the following concentrations in triplicate; 0, 0.2, or 2 μM. Cells were then incubated for 24 hours before assaying for luciferase activity. Luciferase assay was carried out using the dual luciferase reporter kit (Promega, Madison, WI) according to the manufacturer’s instructions with the exception that passive lysis buffer was used at 2X concentration and cells were manually scraped from the plate to enhance cell lysis. A Glomax luminometer (Promega) was used to measure luminescence. Luciferase activity is reported using Renilla luciferase as a baseline for transfection efficiency. The ratio of Firefly luciferase to Renilla luciferase in untreated cells is set to an arbitrary value of 1.0.
Statistical Analysis
All statistical analyses were performed using Microsoft Excel. Unpaired t-tests were performed to test for statistical significance.
Results
Cytotoxicity of MMAIII in WI-38 cells
WI-38 cells are a primary lung fibroblast, which behave accordingly to the Hayflick model, and have been used extensively for studies on cellular senescence (Hayflick and Moorhead, 1961; Place et al., 2005). Our goal in this study is to determine the effects of MMAIII on selenoproteins in a primary cell line, using a sub-toxic concentration of MMAIII. This cell line was chosen since the rigorous assessment of the affects to selenium metabolism by MMAIII are best determined with the use of primary cells. To determine a sub-toxic dose of MMAIII, we determined the cytotoxicity of MMAIII in WI-38 cells by MTT assay. A 50% decrease in cell viability was observed in cells treated with 6 μM MMAIII (Figure 1). Treatment of cells with concentrations at or below 2.5 μM MMAIII did not significantly reduce viability. Indeed low levels of MMAIII stimulated the reduction of MTT by these cells, approximately 20% above untreated controls. This stimulatory effect has been reported previously (Ganyc et al., 2007), yet the mechanism behind it remains elusive. Nonetheless for this study we evaluated the effects of MMAIII on selenium metabolism at concentrations at or below 2 μM in all subsequent experiments.
Figure 1. Cytotoxicity of MMAIII in WI-38 cells.
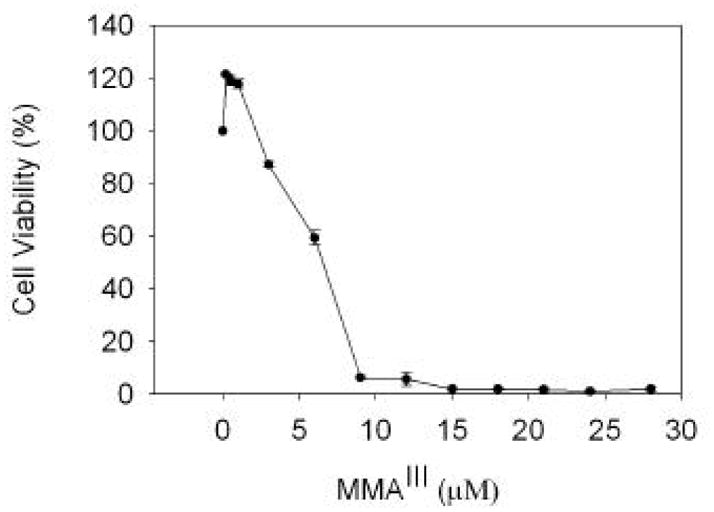
Cells were treated with MMAIII for 24 hours and cell viability is reported as a percentage of viable cells based on untreated controls. The mean value is reported and error shown is the standard deviation from at least three independent cultures.
Effect of MMAIII exposure on selenoprotein synthesis
To determine the effects of MMAIII on selenium metabolism and/or selenoprotein synthesis, cells were exposed to MMAIII and simultaneously labeled with 75Se (selenite) radioisotope. Increasing exposure to MMAIII led to an increase in a prevalent band of molecular weight near 60 KDa (Figure 2). Immunoblot analysis of cell extracts confirmed that this is TrxR1 (data not shown). Incorporation of selenium into other selenoproteins decreased as incorporation into TrxR1 increased. Further analysis of the densitometry of the TrxR1 band revealed a two-fold increase with exposure to 1 and 2 μM MMAIII compared to control (data not shown). The densitometry analysis of one of the smaller selenoproteins (denoted with an asterisk) showed a more than two-fold decease with increasing exposure to MMAIII. This pattern is similar to that seen when HaCat cells were exposed to MMAIII (Ganyc et al., 2007). Incorporation of selenium into newly synthesized TrxR1 in increased at the expense of other selenoproteins. To further analysis the mecahinsm for this phenotype, we determined whether this regulation occurred at the level of transcription.
Figure 2. MMAIII treatment of WI-38 cells leads to increased synthesis of TrxR1 at the expense of other selenoproteins.
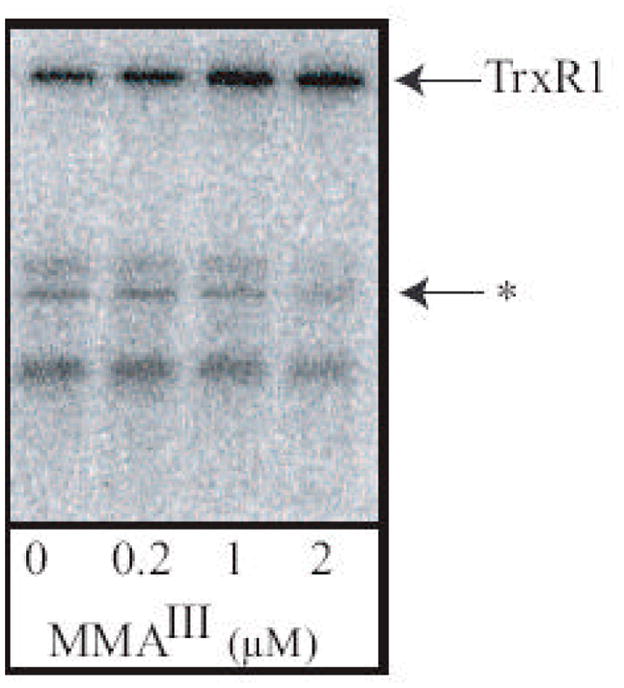
WI-38 cells were treated with MMAIII at the concentrations indicated in triplicate. During exposure to MMAIII, 75Se (selenite) was added to the culture medium and cells were incubated for 24 hours. Crude cell extracts (25 μg protein) were resolved by SDS-PAGE (15%) and labeled selenoproteins were visualized using phosphor screen. TrxR1 – cytosolic thioredoxin reductase; Arrow labeled with an asterisk indicates a selenoprotein used for analysis of selenium incorporation into proteins other than TrxR1.
MMAIII increases TrxR1 mRNA levels in WI-38 cells
We next we determined whether any changes occur to the mRNA encoding TrxR1 or cGPx (a selenoprotein known to have a lower hierarchy status) by using real time RT-PCR. A significant increase in mRNA levels of TrxR1 (*, p<0.05) (Figure 3A) was observed in WI-38 cells treated with 2 μM MMAIII. However, treatment of cells with 0.2 μM MMAIII did not elicit the same effect. This correlates well to the radiolabeling data in which exposure to 2 μM MMAIII resulted in increased levels of TrxR1.
Figure 3. Exposure of WI-38 cells to MMAIII results in increases in TrxR1 mRNA levels.

Cells were cultured with MMAIII for 24 hours after which cells were harvested for real-time RT-PCR analysis. β-actin was used as an internal standard. Relative expression plotted is a representative experiment (cultures in triplicate) with standard deviation as error. Statistical significance was determined by t-test. There is a significant difference in TrxR1 mRNA levels (*, p<0.05) in WI-38 cells when treated with 2 μM MMAIII (A). As TrxR1 mRNA levels are increasing with exposure to MMAIII, cGPx levels significantly decrease with treatment of 2 μM MMAIII (*, p<0.05) (B).
Likewise, treatment of WI-38 cells with MMAIII also caused mRNA levels of cGPx to decrease (Figure 3B). In cells exposed to 2 μM MMAIII, there was a significant decrease in cGPx mRNA (*, p<0.05). This also correlates with the selenium radiolabeling data in this cell line. Specifically, there was a decrease in selenium incorporation into selenoproteins other than TrxR1 with exposure to MMAIII (Figure 2).
Exposure to MMAIII reduces TrxR activity in WI-38 cells
It is well established that MMAIII and other trivalent arsenicals are potent inhibitors of TrxR when assayed using purified enzyme(Lin et al., 1999). In addition exposure to trivalent arsenicals led to decreases in TrxR activity in cultured hepatocytes(Lin et al., 2001) To determine whether MMAIII exposure is affecting TrxR activity, cells were cultured with MMAIII for 48 hours. Cells were subsequently harvested and TrxR activity was determined by DTNB assay (Smith and Levander, 2002; Hintze et al., 2003a). Treatment with 0.2 μM MMAIII led to an increase in the activity of TrxR above that of control (from 9.88 ± 0.290 to 14.7 ± 1.19 nmol/min/mg). In contrast, when cells were cultured in the presence of 2 μM MMAIII, TrxR activity decreased as compared to control (from 9.88 ± 0.290 to 6.33 ± 0.212 nmol/min/mg). Although there is increased incorporation of selenium into TrxR1, the enzyme’s activity has been reduced, likely mediated by direct inhibition with the arsenical as observed in a previous study (Lin et al., 1999).
MMAIII exposure results in activation of transcription of the TrxR1 promoter
The TrxR1 promoter contains an ARE along with binding sites for Oct-1, Sp1, and Sp3 (Rundlof et al., 2001; Hintze et al., 2003a). To test if MMAIII induces synthesis of TrxR1 through the ARE, promoter fusion luciferase constructs were transfected into WI-38 cells. This included a wild type TrxR1 promoter containing the ARE as well as a second construct with a mutated (Mut) ARE element. If exposure to MMAIII exerts any effects on the activation of the ARE of TrxR1 by Nrf2, the same effects should not be seen in the mutated construct.
WI-38 cells transfected with the wild-type promoter fusion construct displayed increased luciferase activity upon exposure to 2 μM MMAIII (*, p< 0.05) (Figure 4). There was a slight increase in activity in cells treated with 0.2 μM MMAIII, but not enough to be considered statistically significant. No significant change in luciferase activity was observed in WI-38 cells transfected with the mutant promoter fusion construct and exposed to 0.2 and 2 μM MMAIII, clearly indicating that exposure to MMAIII is leading to activation of the TrxR1 promoter via the ARE. Based on other studies, this activation is likely mediated through the Nrf2 transcriptional activator (Aono et al., 2003; Pi et al., 2003; Wang et al., 2007b).
Figure 4. MMAIII treatment activates transcription of TrxR1 promoter through the ARE element.
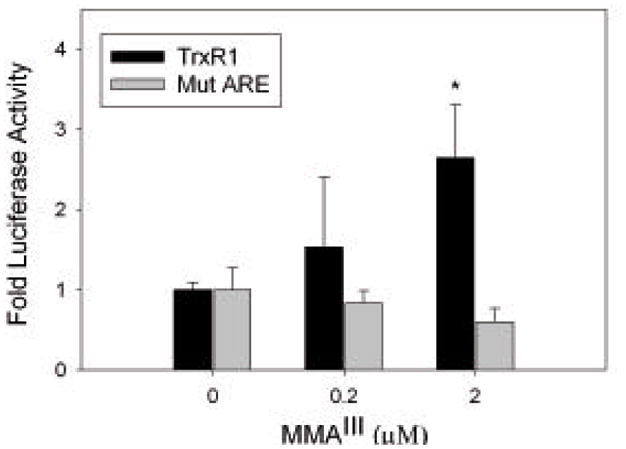
WI-38 cells were transiently transfected with TrxR1 and mutant TrxR1 (mutation in ARE) promoter fusion constructs that have been previously described (Hintze et al., 2003b). Cells were treated with MMAIII at the concentrations indicated in triplicate for each construct for 24 hours prior to assaying Firefly and Renilla luciferase activity. Fold luciferase activity is plotted and based on the ratio of Firefly luciferase to Renilla luciferase activity. The error bars represent standard deviation and student’s t-test was used for statistical analysis. In WI-38 cells transfected with the TrxR1 construct and exposed to 2 μM MMAIII there is a significant increase (*, p < 0.05) in luciferase activity. This increase in luciferase activity is not observed with the mutant ARE construct.
Activation of transcription of the promoter for quinone reductase in WI-38 cells by MMAIII
The gene encoding quinone reductase also contains an ARE in its promoter region, and this gene product is not known to be involved in selenium metabolism (Hintze et al., 2003b). WI-38 cells were transfected with a rat quinone reductase (QR) promoter fusion construct that contained the ARE. In WI-38 cells transfected with the QR construct and exposed to varying concentrations of MMAIII, an increase in QR promoter fusion activity was detected upon exposure to 2 μM MMAIII (Figure 5). There was also a slight increase in luciferase activity with treatment of cells with 0.2 μM MMAIII, again not enough to be considered significant.
Figure 5. Treatment of WI-38 cells with MMAIII activates quinone reductase promoter confirming ARE regulation.
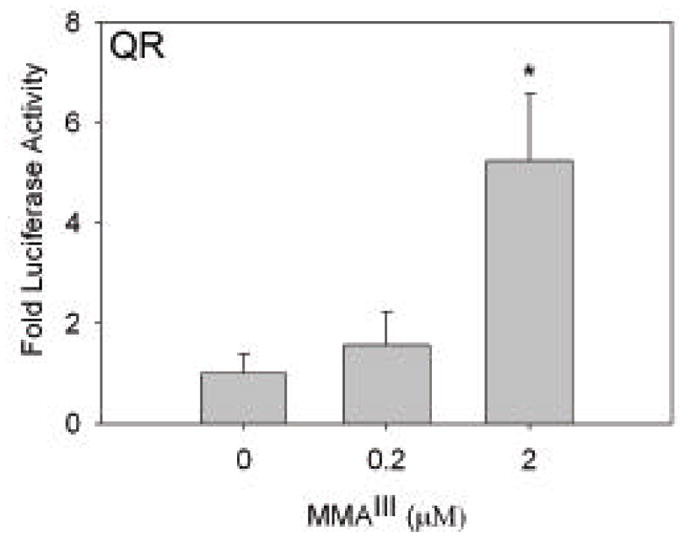
WI-38 cells were transiently transfected with rat quinone reductase (QR) promoter fusion construct (Hintze et al., 2003b) and exposed to MMAIII at the concentrations indicated. Similar to the results for TrxR1 construct, cells treated with 2 μM MMAIII increased activation of the promoter of QR (*, p<0.05).
In addition to the induction of QR, we also examined the level of mRNA encoding heme oxygenase 1 (HO-1), another gene whose promoter contains an ARE, during exposure to MMAIII. At treatment of cells with 2 μM MMAIII there was ten-fold induction of mRNA levels encoding HO-1 as determined by RT-PCR (data not shown). This provides further evidence that treatment of cells with MMAIII respond through the ARE and this response is very likely to be mediated by the Nrf2 response.
Discussion
MMAIII is a metabolite of arsenic produced during the methylation reactions carried out to facilitate excretion. It is not found in the natural environment, but is generated during this excretory pathway. It has been found to be the most cytotoxic of all arsenicals and could prove to be carcinogenic (Petrick et al., 2000; Bredfeldt et al., 2006). Human exposure to arsenic is primarily through inorganic forms, especially arsenate (Nickson et al., 1998). A recent study has shown that exposure to mice to arsenate (50 ppm in water) for 12 weeks led to an accumulation of MMAIII in lung tissue of 0.1 μg/gm of tissue (Kenyon et al., 2008). This level (0.1 ppm) is comparable to exposure to cells at 1 μM. Our experiment design, since it follows incorporation of radioisotope selenium, is limited to short term experiments. Nonetheless we see a significant shift in the synthesis of selenoprotein synthesis at exposure levels (2 μM) very close to those seen in animal studies (Kenyon et al., 2008). Since humans are usually exposed for far longer than 12 weeks, our results and experimental design are clearly relevant to human exposure. Furthermore many studies have utilized exposures to methylated arsenicals in the micromolar range to elicit cellular responses in the short term (Styblo et al., 2000; Lin et al., 2001; Liu et al., 2001). These studies also have shown that significant methylation of arsenicals is exhibited by hepatocytes and cells of the renal system, with little methylation of arsenic occurring in other cell types including fibroblasts (Wang and Lazarides, 1984; Styblo et al., 2000).
It has been shown that selenium and arsenic interact according to a mutual sparing effect (Moxon, 1938). Based on several studies it is well established that exposure to mammals to inorganic arsenic leads to decreased selenium levels (Levander and Argrett, 1969; Levander, 1977). If selenium levels were depleted, the cell will begin to preferentially make some selenoproteins over others (Low et al., 2000). TrxR1, because of its importance in maintaining the cellular redox environment is one of the selenoproteins at the top of the hierarchy and will likely be expressed under selenium limiting conditions. A recent study has demonstrated that administration of selenium to hairless mice significantly reduced the arsenic-enhanced carcinogeneis by UV radiation (Burns et al., 2008). Thus it is clear that a thorough understanding of the effects of arsenic exposure on selenium metabolism is justified. Moreover culture model systems are a more direct way to establish how cells react to any of the metabolic intermediates in a controlled manner. In this study we aimed to determine whether exposure to MMAIII revealed any changes in selenium metabolism and selenoprotein synthesis in a primary cell, as well as determine the mechanisms involved.
When WI-38 cells were exposed to MMAIII TrxR1 synthesis increased, while smaller selenoproteins decreased as determined by radiolabeling with 75Se. This phenotype with MMAIII exposure was also observed in a previous study with HaCat cells (human keratinocytes) (Ganyc et al., 2007) yet in this work we determined the mechanism by which the incorporation into this particular selenoprotein occurs. This phenotype (i.e. increased TrxR1 levels with lower levels of cGpx) was also observed in an animal study studying the differential regulation of selenoproteins in response to sulforaphane (Hintze et al., 2003a). This correlation suggested to us that the Nrf2 response was responsible for the differential regulation of expression of selenoproteins. In this study we demonstrate that MMAIII induces TrxR1 through the ARE, although it is not surprising since other trivalent arsenicals have been shown to induce an active Nrf2 response (Aono et al., 2003; He et al., 2006; Wang et al., 2007b). It should be noted that induction of heme oxygenase, known to be induced by Nrf2, has been used as biomarker for arsenic exposure (Falkner et al., 1993; Kenyon et al., 2005). In one study with inorganic arsenic exposures in mice of up to 100 μmol/kg, significant increases in heme oxygenase was detected in liver and kidney but not in lung (Kenyon et al., 2005). However a study using one dose of inorganic arsenic at 75 μmol/kg in guinea pigs did find an induction of heme oxygenase activity in lungs (Falkner et al., 1993)..
Based on the results presented in this and several previous studies one can begin to determine the combined effects of arsenic exposure at the molecular level with respect to selenium metabolism and selenoenzyme activity. Specifically, 1.) exposure to arsenite will lead to reduction of selenium levels in tissues, due to the Moxon effect (Gailer et al., 2002; Ganyc et al., 2007; Burns et al., 2008), 2.) Exposure to arsenite or MMAIII will lead to induction of the Nrf2 response (Aono et al., 2003; Pi et al., 2003; Wang et al., 2007b), 3.) Synthesis of TrxR1 mRNA will increase in tissues exposed to either arsenite or MMAIII, mediated through the ARE, 4.) Higher expression of TrxR1 mRNA will lead to preferential production of TrxR1 over cGPx and other selenoproteins due to non-sense mediated decay of selenoproteins at a lower level on the hierarchy (de Jesus et al., 2006; Small-Howard et al., 2006), and 5.) TrxR activity will be inhibited by direct binding of trivalent arsenicals to the active site CXU motif of TrxR1 (Lin et al., 1999; Lin et al., 2001). Figure 6 is overview describing these cumulative effects. This combination will reduce significantly the cell’s ability to defend against oxidative stress through decreases in Gpx isoenzymes and catalytic inactivation of the thioredoxin system. The thioredoxin system is needed not only for proliferation, but also as an electron donor for peroxiredoxins and methionine sulfoxide reductases (Moskovitz, 2005; Rhee et al., 2005). Future studies to examine whether this series of events are the basis for the carcinogenic potential of arsenic in animal models is thus warranted.
Figure 6. Overview of the impact of arsenite and MMAIII on selenoenzymes.

Arrows indicate positive or negative impact on several aspects of selenoenzymes and redox metabolism. Overview is based on data presented in this study and cumulative data from others (Lin et al., 1999; Gailer et al., 2000; Lin et al., 2001; Rundlof et al., 2001; Gailer et al., 2002; Moskovitz, 2005; Rhee et al., 2005; Ganyc et al., 2007; Talbot et al., 2008).
Acknowledgments
This research was supported by grants to WTS from the National institutes of Health (NIEHS ES01434) and the Florida Department of Health (05-NIR-10). We thank Dr. William Cullen (University of British Columbia) for supplying MMAIII.
Abbreviations
- MMAIII
Monomethylarsonous acid
- TrxR
thioredoxin reductase
- TrxR1
cytosolic thioredoxin reductase
- QR
quinone reductase
- ARE
antioxidant response element
- cGpx
cytosolic glutathione peroxidase
- PHGpx
phosphohydrolipid glutathione peroxidase
Footnotes
Conflict of Interest Statement
No conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aono J, Yanagawa T, Itoh K, Li B, Yoshida H, Kumagai Y, Yamamoto M, Ishii T. Activation of Nrf2 and accumulation of ubiquitinated A170 by arsenic in osteoblasts. Biochemical and biophysical research communications. 2003;305:271–277. doi: 10.1016/s0006-291x(03)00728-9. [DOI] [PubMed] [Google Scholar]
- Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16:420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Behne D, Hilmert H, Scheid S, Gessner H, Elger W. Evidence for specific selenium target tissues and new biologically important selenoproteins. Biochim Biophys Acta. 1988;966:12–21. doi: 10.1016/0304-4165(88)90123-7. [DOI] [PubMed] [Google Scholar]
- Berry MJ. Insights into the hierarchy of selenium incorporation. Nat Genet. 2005;37:1162–1163. doi: 10.1038/ng1105-1162. [DOI] [PubMed] [Google Scholar]
- Berry MJ, Banu L, Harney JW, Larsen PR. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. Embo J. 1993;12:3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Jagadish B, Eblin KE, Mash EA, Gandolfi AJ. Monomethylarsonous acid induces transformation of human bladder cells. Toxicology and applied pharmacology. 2006;216:69–79. doi: 10.1016/j.taap.2006.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns FJ, Rossman T, Vega K, Uddin A, Vogt S, Lai B, Reeder RJ. Mechanism of selenium-induced inhibition of arsenic-enhanced UVR carcinogenesis in mice. Environ Health Perspect. 2008;116:703–708. doi: 10.1289/ehp.10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus LA, Hoffmann PR, Michaud T, Forry EP, Small-Howard A, Stillwell RJ, Morozova N, Harney JW, Berry MJ. Nuclear assembly of UGA decoding complexes on selenoprotein mRNAs: a mechanism for eluding nonsense-mediated decay? Mol Cell Biol. 2006;26:1795–1805. doi: 10.1128/MCB.26.5.1795-1805.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Buffington S, Gandolfi AJ. Mitogenic signal transduction caused by monomethylarsonous acid in human bladder cells: role in arsenic-induced carcinogenesis. Toxicol Sci. 2007;95:321–330. doi: 10.1093/toxsci/kfl160. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Gandolfi AJ. Immortalized human urothelial cells as a model of arsenic-induced bladder cancer. Toxicology. 2008;248:67–76. doi: 10.1016/j.tox.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Falkner KC, McCallum GP, Cherian MG, Bend JR. Effects of acute sodium arsenite administration on the pulmonary chemical metabolizing enzymes, cytochrome P-450 monooxygenase, NAD(P)H:quinone acceptor oxidoreductase and glutathione S-transferase in guinea pig: comparison with effects in liver and kidney. Chem Biol Interact. 1993;86:51–68. doi: 10.1016/0009-2797(93)90111-b. [DOI] [PubMed] [Google Scholar]
- Gailer J, George GN, Pickering IJ, Madden S, Prince RC, Yu EY, Denton MB, Younis HS, Aposhian HV. Structural basis of the antagonism between inorganic mercury and selenium in mammals. Chem Res Toxicol. 2000;13:1135–1142. doi: 10.1021/tx000050h. [DOI] [PubMed] [Google Scholar]
- Gailer J, George GN, Pickering IJ, Prince RC, Younis HS, Winzerling JJ. Biliary excretion of [(GS)(2)AsSe](−) after intravenous injection of rabbits with arsenite and selenate. Chem Res Toxicol. 2002;15:1466–1471. doi: 10.1021/tx025538s. [DOI] [PubMed] [Google Scholar]
- Ganyc D, Talbot S, Konate F, Jackson S, Schanen B, Cullen W, Self WT. Impact of trivalent arsenicals on selenoprotein synthesis. Environ Health Perspect. 2007;115:346–353. doi: 10.1289/ehp.9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. The cell biology of aging. Clinics in geriatric medicine. 1985;1:15–27. [PubMed] [Google Scholar]
- He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 × Keap1 × Cul3 complex and recruiting Nrf2 × Maf to the antioxidant response element enhancer. The Journal of biological chemistry. 2006;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Keck AS, Finley JW, Jeffery EH. Induction of hepatic thioredoxin reductase activity by sulforaphane, both in Hepa1c1c7 cells and in male Fisher 344 rats. J Nutr Biochem. 2003a;14:173–179. doi: 10.1016/s0955-2863(02)00282-6. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Wald KA, Zeng H, Jeffery EH, Finley JW. Thioredoxin reductase in human hepatoma cells is transcriptionally regulated by sulforaphane and other electrophiles via an antioxidant response element. J Nutr. 2003b;133:2721–2727. doi: 10.1093/jn/133.9.2721. [DOI] [PubMed] [Google Scholar]
- Kenyon EM, Del Razo LM, Hughes MF, Kitchin KT. An integrated pharmacokinetic and pharmacodynamic study of arsenite action 2. Heme oxygenase induction in mice. Toxicology. 2005;206:389–401. doi: 10.1016/j.tox.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Kenyon EM, Hughes MF, Adair BM, Highfill JH, Crecelius EA, Clewell HJ, Yager JW. Tissue distribution and urinary excretion of inorganic arsenic and its methylated metabolites in C57BL6 mice following subchronic exposure to arsenate in drinking water. Toxicology and applied pharmacology. 2008;232:448–455. doi: 10.1016/j.taap.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Laurent TC, Moore EC, Reichard P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B. J Biol Chem. 1964;239:3436–3444. [PubMed] [Google Scholar]
- Li J, Waters SB, Drobna Z, Devesa V, Styblo M, Thomas DJ. Arsenic (+3 oxidation state) methyltransferase and the inorganic arsenic methylation phenotype. Toxicology and applied pharmacology. 2005;204:164–169. doi: 10.1016/j.taap.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lin S, Cullen WR, Thomas DJ. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem Res Toxicol. 1999;12:924–930. doi: 10.1021/tx9900775. [DOI] [PubMed] [Google Scholar]
- Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem Res Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci U S A. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Grundner-Culemann E, Harney JW, Berry MJ. SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. Embo J. 2000;19:6882–6890. doi: 10.1093/emboj/19.24.6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J. Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Current pharmaceutical design. 2005;11:1451–1457. doi: 10.2174/1381612053507846. [DOI] [PubMed] [Google Scholar]
- Moxon A. The influence of arsenic on the toxicity of seleniforous grains. Science. 1938:81. doi: 10.1126/science.88.2273.81. [DOI] [PubMed] [Google Scholar]
- Nickson R, McArthur J, Burgess W, Ahmed KM, Ravenscroft P, Rahman M. Arsenic poisoning of Bangladesh groundwater. Nature. 1998;395:338. doi: 10.1038/26387. [DOI] [PubMed] [Google Scholar]
- Papp LV, Lu J, Holmgren A, Khanna KK. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid Redox Signal. 2007;9:775–806. doi: 10.1089/ars.2007.1528. [DOI] [PubMed] [Google Scholar]
- Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Vasken Aposhian H. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicology and applied pharmacology. 2000;163:203–207. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free radical biology & medicine. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Rundlof AK, Carlsten M, Arner ES. The core promoter of human thioredoxin reductase 1: cloning, transcriptional activity, and Oct-1, Sp1, and Sp3 binding reveal a housekeeping-type promoter for the AU-rich element-regulated gene. J Biol Chem. 2001;276:30542–30551. doi: 10.1074/jbc.M101452200. [DOI] [PubMed] [Google Scholar]
- Small-Howard A, Morozova N, Stoytcheva Z, Forry EP, Mansell JB, Harney JW, Carlson BA, Xu XM, Hatfield DL, Berry MJ. Supramolecular complexes mediate selenocysteine incorporation in vivo. Mol Cell Biol. 2006;26:2337–2346. doi: 10.1128/MCB.26.6.2337-2346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Levander OA. High-throughput 96-well microplate assays for determining specific activities of glutathione peroxidase and thioredoxin reductase. Methods Enzymol. 2002;347:113–121. doi: 10.1016/s0076-6879(02)47012-7. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000;74:289–299. doi: 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- Talbot S, Nelson R, Self WT. Arsenic trioxide and auranofin inhibit selenoprotein synthesis: implications for chemotherapy for acute promyelocytic leukaemia. Br J Pharmacol. 2008;154:940–948. doi: 10.1038/bjp.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DJ, Li J, Waters SB, Xing W, Adair BM, Drobna Z, Devesa V, Styblo M. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp Biol Med (Maywood) 2007;232:3–13. [PMC free article] [PubMed] [Google Scholar]
- Thomas DJ, Waters SB, Styblo M. Elucidating the pathway for arsenic methylation. Toxicology and applied pharmacology. 2004;198:319–326. doi: 10.1016/j.taap.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Wang C, Lazarides E. Arsenite-induced changes in methylation of the 70,000 dalton heat shock proteins in chicken embryo fibroblasts. Biochemical and biophysical research communications. 1984;119:735–743. doi: 10.1016/s0006-291x(84)80312-5. [DOI] [PubMed] [Google Scholar]
- Wang TC, Jan KY, Wang AS, Gurr JR. Trivalent arsenicals induce lipid peroxidation, protein carbonylation, and oxidative DNA damage in human urothelial cells. Mutat Res. 2007a;615:75–86. doi: 10.1016/j.mrfmmm.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicology and applied pharmacology. 2007b;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo MH, Xu XM, Carlson BA, Patterson AD, Gladyshev VN, Hatfield DL. Targeting thioredoxin reductase 1 reduction in cancer cells inhibits self-sufficient growth and DNA replication. PLoS ONE. 2007;2:e1112. doi: 10.1371/journal.pone.0001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Svehlikova V, Bao Y, Howie AF, Beckett GJ, Williamson G. Synergy between sulforaphane and selenium in the induction of thioredoxin reductase 1 requires both transcriptional and translational modulation. Carcinogenesis. 2003;24:497–503. doi: 10.1093/carcin/24.3.497. [DOI] [PubMed] [Google Scholar]